
Abstract
The single-tablet regimen of the hepatitis C virus (HCV) NS5A inhibitor ledipasvir and the HCV NS5B polymerase inhibitor sofosbuvir (ledipasvir/sofosbuvir; Harvoni®) was recently approved in the US and the EU. The phase III ION trials included treatment-naive (ION-1 and -3) or treatment-experienced (ION-2) patients with chronic HCV genotype 1 infection (≈20 % of patients in ION-1 and -2 had cirrhosis, whereas no patient in ION-3 had cirrhosis). A sustained virological response 12 weeks’ post-treatment (SVR12) was seen in 99 % of treatment-naive patients receiving ledipasvir/sofosbuvir for 12 weeks in ION-1, with no additional benefit conferred by the addition of ribavirin or extending the treatment duration to 24 weeks. Moreover, in ION-3, an 8-week regimen achieved an SVR12 rate of 94 % overall and 97 % in the subgroup of patients with a baseline HCV RNA level of <6 million IU/mL. SVR12 rates of 94 and 99 % were seen in treatment-experienced patients who received ledipasvir/sofosbuvir for 12 and 24 weeks in ION-2. Data also support the use of ledipasvir/sofosbuvir in chronic HCV genotype 4 infection, in HCV and HIV co-infection and, in combination with ribavirin, in patients with chronic HCV genotype 1 or 4 infection who have decompensated cirrhosis or are liver transplant recipients and in chronic HCV genotype 3 infection. Oral ledipasvir/sofosbuvir was generally well tolerated. In conclusion, ledipasvir/sofosbuvir is an important new single-tablet regimen that represents a significant advance in the treatment of chronic hepatitis C.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Oral single-tablet regimen of the hepatitis C virus (HCV) NS5A inhibitor ledipasvir and the NS5B polymerase inhibitor sofosbuvir |
Achieved a high sustained virological response rate 12 weeks’ post-treatment in treatment-naive and treatment-experienced patients with chronic HCV genotype 1 infection in the ION trials |
Data support its use in chronic HCV genotype 4 infection and in HCV and HIV co-infection |
Data support its use, in combination with ribavirin, in chronic HCV genotype 1 or 4 infection with decompensated cirrhosis or liver transplantation, or in chronic HCV genotype 3 infection |
Generally well tolerated |
1 Introduction
Direct-acting antiviral agents are transforming the treatment of chronic hepatitis C, with interferon-free regimens now a reality. A single-tablet regimen of the hepatitis C virus (HCV) NS5A inhibitor ledipasvir and the NS5B inhibitor sofosbuvir (ledipasvir/sofosbuvir; Harvoni®) was recently approved in the US [1] and the EU [2]. This article reviews the clinical efficacy and tolerability of ledipasvir/sofosbuvir in the treatment of chronic hepatitis C, as well as summarizing its pharmacological properties.
2 Pharmacodynamic Properties
2.1 Mechanism of Action
Ledipasvir inhibits the HCV NS5A protein and sofosbuvir inhibits HCV NS5B RNA-dependent RNA polymerase; both NS5A and NS5B RNA-dependent RNA polymerase are essential for viral replication [1]. Sofosbuvir is a nucleotide prodrug that is metabolized to the pharmacologically active metabolite GS-461203, which is incorporated into HCV RNA by NS5B polymerase where it acts as a chain terminator [1].
2.2 Antiviral Activity
Ledipasvir had potent antiviral activity against HCV genotypes 1a and 1b [50 % effective concentration (EC50) values of 0.031 and 0.004 nmol/L in HCV replicon assays], whereas it had lower activity against HCV genotypes 4a, 4d, 5a and 6a (EC50 values of 0.39, 0.60, 0.15 and 1.1 nmol/L, respectively) and substantially lower activity against HCV genotypes 2a, 2b, 3a and 6e (EC50 values of 21–249, 16–530, 168 and 264 nmol/L, respectively) [2]. Sofosbuvir demonstrated pangenotypic antiviral activity, with EC50 values against HCV genotypes 1a, 1b, 2a, 2b, 3a, 4a, 5a and 6a of 40, 110, 50, 15, 50, 40, 15 and 14 nmol/L, respectively, in HCV replicon assays [3]. No antagonistic effect was seen with the combination of ledipasvir and sofosbuvir in replicon cells [1].
2.3 Resistance
In cell culture, reduced ledipasvir susceptibility was associated with the NS5A amino acid substitutions Y93H in HCV genotypes 1a and 1b and Q30E in HCV genotype 1a [1, 2]. Site-directed mutagenesis of NS5A resistance-associated variants (RAVs) demonstrated that Q30H/R, L311/M/V, P32L and Y93T substitutions in genotype 1a and P58D and Y93S substitutions in genotype 1b were associated with >100- and ≤1000-fold changes in ledipasvir susceptibility [2]. In addition, M28A/G, Q30E/G/K, H58D and Y93C/H/N/S substitutions in genotype 1a and A92K and Y93H substitutions in genotype 1b were associated with >1000-fold changes in ledipasvir susceptibility [2]. In HCV replicons, reduced sofosbuvir susceptibility was associated with the primary NS5B substitution S282T in multiple genotypes [1].
Among patients with chronic HCV genotype 1 infection who received ledipasvir/sofosbuvir in the pivotal, phase III ION trials (see Sect. 4.1.1), 22 of the 29 patients with HCV genotype 1a infection who qualified for resistance testing had at least one NS5A RAV detected at positions K24, M28, Q30, L31, S38 or Y93 (most commonly Q30R, Y93H and L31M) in postbaseline isolates at the time of failure; no NS5A RAVs were detected in the remaining seven patients at failure [2]. In addition, seven of the eight patients with HCV genotype 1b infection who qualified for resistance testing had at least one NS5A RAV detected at positions L31 or Y93 (most commonly Y93H) in postbaseline isolates at the time of failure [2].
Sofosbuvir has a high genetic barrier to resistance; the NS5B S282T variant was not detected during the ION trials in patients with chronic HCV genotype 1 infection who received ledipasvir/sofosbuvir [2, 4–6].
Treatment with ledipasvir/sofosbuvir was associated with high sustained virological response (SVR) rates in patients infected with HCV genotype 1, despite the presence of NS5A or NS5B RAVs at baseline [2, 7, 8]. For example, baseline NS5A RAVs were detected in 16 % of patients with chronic HCV genotype 1 infection, and 92 % of these patients achieved SVR at 12 weeks’ post-treatment (SVR12) with ledipasvir/sofosbuvir with or without ribavirin, according to a pooled analysis of phase II/III trial data [7]. In the ION trials, the NS5B S282T variant was not detected at baseline, and all of the 24 patients with baseline variants associated with resistance to NS5B nucleoside inhibitors achieved SVR [2].
No cross resistance was seen between ledipasvir and sofosbuvir [1]. The sofosbuvir NS5B substitution S282T was fully susceptible to ledipasvir and all ledipasvir resistance-associated NS5A substitutions were fully susceptible to sofosbuvir [1]. Ledipasvir and sofosbuvir were fully active against RAVs associated with direct-acting antiviral agents with different mechanisms of action, including NS5B non-nucleoside inhibitors and NS3 protease inhibitors [1].
2.4 Other Effects
The maximum recommended dose of sofosbuvir (400 mg) and supratherapeutic doses of ledipasvir (120 mg) and sofosbuvir (1200 mg) did not prolong the corrected QT interval to a clinically relevant extent, according to the results of thorough QT studies [1].
Coadministration of ledipasvir/sofosbuvir and amiodarone is not recommended, as cases of symptomatic bradycardia (including fatal cardiac arrest and cases needing pacemaker intervention) have been reported in the postmarketing setting among patients receiving ledipasvir/sofosbuvir and amiodarone [1].
3 Pharmacokinetic Properties
Ledipasvir, sofosbuvir and GS-331007 (the predominant circulating metabolite of sofosbuvir) reached median peak plasma concentrations 4, ≈1 and 4 h, respectively, following oral administration of ledipasvir/sofosbuvir to patients with HCV infection [2]. Ledipasvir exposure was dose proportional over the dose range of 3–100 mg and sofosbuvir and GS-331007 exposures were near dose proportional over the dose range of 200–400 mg [2]. Ledipasvir/sofosbuvir can be administered without regard to food [1, 2]. Plasma protein binding of ledipasvir and sofosbuvir was >99.8 % and 61–65 %; plasma protein binding of GS-331007 was minimal [1, 2].
Ledipasvir undergoes slow oxidative metabolism, although the parent drug still accounted for >98 % of systemic exposure after administration of radiolabelled ledipasvir [1, 2]. Sofosbuvir undergoes extensive hepatic metabolism to the active metabolite GS-461203 [1, 2]. Dephosphorylation results in GS-331007, which lacks anti-HCV activity in vitro and accounted for ≈85 % of total systemic exposure following administration of ledipasvir/sofosbuvir [2].
The main routes of elimination were biliary excretion for ledipasvir and renal clearance for GS-331007 [1, 2]. Following administration of radiolabelled ledipasvir, ≈86 % of the radioactivity was recovered in the faeces, whereas ≈80 and 14 % of the radioactivity was recovered in the urine and faeces following administration of radiolabelled sofosbuvir [1, 2]. The median terminal half-lives of ledipasvir, sofosbuvir and GS-331007 were 47, 0.5 and 27 h, respectively [1, 2].
The ledipasvir/sofosbuvir dosage does not need to be adjusted in patients with mild or moderate renal impairment or mild, moderate or severe hepatic impairment [1, 2, 9]. Although the pharmacokinetics of ledipasvir were not altered to a clinically significant extent in patients with severe renal impairment (estimated glomerular filtration rate of <30 mL/min/1.73 m2) [10], GS-331007 exposure was increased in patients with severe renal impairment or end-stage renal disease, meaning that ledipasvir/sofosbuvir dosage recommendations cannot be made in these patient groups [1, 2].
The pharmacokinetics of ledipasvir, sofosbuvir and GS-331007 were not altered to a clinically significant extent based on race (including in Japanese vs. Caucasian healthy volunteers [11]), gender, age or bodyweight [1, 2].
Although ledipasvir and sofosbuvir are substrates of the transporters P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), neither agent is a substrate for various other transporters or for cytochrome P450 (CYP) and/or uridine diphosphate glucuronosyltransferase (UGT) 1A1 enzymes [1, 2, 12]. In vitro, ledipasvir inhibited P-gp and BCRP and appeared to weakly induce enzymes such as CYP3A4, CYP2C and UGT1A1 [2].
There is potential for clinically relevant interactions between ledipasvir and/or sofosbuvir and potent P-gp inducers [e.g. rifampicin, hypericum (St John’s wort), carbamazepine, phenytoin, tipranavir (ritonavir boosted)], digoxin, dabigatran etexilate, rosuvastatin, simeprevir and tenofovir disoproxil fumarate when used with a pharmacokinetic enhancer (e.g. ritonavir or cobicistat) [1, 2, 12, 13]. Local prescribing information should be consulted for further information regarding the coadminstration of ledipasvir/sofosbuvir and these agents [1, 2].
No clinically significant interactions were seen or would be expected between ledipasvir and/or sofosbuvir and efavirenz/emtricitabine/tenofovir disoproxil fumarate, emtricitabine/rilpivirine/tenofovir disoproxil fumarate, abacavir/lamivudine, atazanavir (ritonavir boosted), darunavir (ritonavir boosted), raltegravir, H2-receptor antagonists (at a dosage not exceeding famotidine 40 mg twice daily), proton pump inhibitors (at a dosage not exceeding omeprazole 20 mg/day), antacids (when the administration of antacids and ledipasvir/sofosbuvir is separated by 4 h), methadone, ciclosporin, tacrolimus and ethinylestradiol/norgestimate [1, 2, 12, 14, 15].
4 Therapeutic Efficacy
A ledipasvir/sofosbuvir dosage of 90/400 mg once daily was administered to patients in the trials discussed in this section. Where specified, ribavirin dosages were 1000 or 1200 mg/day (according to bodyweight) [4–6], 600–1000 mg/day [8] or ≥600 mg/day [16]. In patients with recurrence of HCV genotype 1 or 4 infection post-transplantation, the ribavirin dosage was weight-based in patients with a fibrosis score of F0–F3 or Child-Pugh-Turcotte (CPT) class A cirrhosis, and 600–1200 mg/day in patients with CPT class B or C cirrhosis [17].
4.1 Phase III Trials in Chronic HCV Genotype 1 Infection
The potential of regimens containing ledipasvir and sofosbuvir in the treatment of chronic HCV genotype 1 infection was shown in phase II trials [18, 19]. Given the availability of phase III trials [4–6, 8], these phase II trials [18, 19] are not discussed.
4.1.1 The ION Trials
The main focus of this section is the randomized, open-label, multicentre, phase III ION-1 [4], ION-2 [5] and ION-3 [6] trials examining the efficacy of ledipasvir/sofosbuvir in patients aged ≥18 years with chronic HCV genotype 1 infection; results of the ION-4 trial [20] in patients co-infected with HCV and HIV-1 are discussed in Sect. 4.2.1. Patients in ION-1 [4] and ION-3 [6] were treatment-naive and patients in ION-2 had experienced virological failure after treatment with peginterferon-α plus ribavirin with or without a NS3/4A protease inhibitor [5]. Approximately 20 % of the patients enrolled in ION-1 [4] and ION-2 [5] could have compensated cirrhosis, whereas ION-3 enrolled patients without cirrhosis [6]. Other inclusion criteria included an HCV RNA level of ≥1 × 104 IU/mL, ALT and AST levels of ≤10 × the upper limit of normal (ULN), a platelet count of ≥50,000/mm3 [4, 5] or >90,000/mm3 [6] and a haemoglobin level of ≥11 g/dL (women) or ≥12 g/dL (men).
In terms of factors traditionally considered predictive of poor outcome, 67–80 % of patients across the ION trials had HCV genotype 1a infection, 70–88 % had a non-CC IL28B genotype and 12–19 % were Black. In addition, cirrhosis was present in 16 and 20 % of patients in ION-1 [4] and ION-2 [5].
Patients in ION-1 [4] and ION-2 [5] received ledipasvir/sofosbuvir with or without ribavirin for 12 or 24 weeks, and patients in ION-3 received ledipasvir/sofosbuvir for 8 or 12 weeks or ledipasvir/sofosbuvir plus ribavirin for 8 weeks [6]. The primary endpoint was the SVR12 rate (HCV RNA level of <25 IU/mL) [4–6]. Additional analyses of the ION trials are available as abstracts plus posters or slide presentations [21–24].
Ledipasvir/sofosbuvir was associated with high SVR12 rates in both treatment-naive and treatment-experienced patients with chronic HCV genotype 1 infection [4–6] (Table 1).
In treatment-naive patients in ION-1, SVR12 rates were ≥97 %, regardless of whether or not ledipasvir/sofosbuvir was administered for 12 or 24 weeks or administered with or without ribavirin (Table 1) [4]. SVR12 rates in all four treatment groups were significantly higher than a historical response rate of 60 %. Virological breakthrough occurred in one patient during treatment; the Y93H variant was detected at the time of virological failure. Nonadherence was suspected as plasma concentrations of ledipasvir and GS-331007 were below the level of quantification at weeks 8 and 10 of treatment. An additional two patients experienced virological relapse by weeks 4 and 12 post-treatment [4].
The ION-3 trial demonstrated an SVR12 rate of 94 % in treatment-naive patients without cirrhosis who received ledipasvir/sofosbuvir for 8 weeks (Table 1) [6]. The 8-week regimen of ledipasvir/sofosbuvir was noninferior to the 12-week regimen of ledipasvir/sofosbuvir or to the 8-week regimen of ledipasvir/sofosbuvir plus ribavirin in terms of SVR12 (Table 1). SVR12 rates in all three treatment groups were significantly higher than a historical response rate of 60 %. No patient experienced virological failure during treatment. Virological relapse after the end of therapy occurred in 5, 4 and 1 % of patients receiving ledipasvir/sofosbuvir for 8 weeks, ledipasvir/sofosbuvir plus ribavirin for 8 weeks or ledipasvir/sofosbuvir for 12 weeks, respectively [6].
Post hoc analysis of ION-3 demonstrated that treatment with ledipasvir/sofosbuvir for 8 weeks appeared sufficient in treatment-naive patients without cirrhosis who had a baseline HCV RNA level of <6 million IU/mL [22]. Among patients with a baseline HCV RNA level of <6 and ≥6 million IU/mL, the SVR12 rate was 97 and 90 % in those receiving ledipasvir/sofosbuvir for 8 weeks, compared with 96 and 96 % in those receiving ledipasvir/sofosbuvir for 12 weeks (Table 1) [22].
Among patients who had failed prior peginterferon-α-based treatment, high SVR12 rates were seen with ledipasvir/sofosbuvir administered for 12 or 24 weeks, with or without ribavirin, in the ION-2 trial (Table 1) [5]. All of the patients who achieved SVR12 in ION-2 maintained this response 24 weeks after the end of treatment. SVR12 rates in all four treatment groups were significantly higher than a historical response rate of 25 %. Virological breakthrough occurred during treatment in one patient receiving ledipasvir/sofosbuvir plus ribavirin; nonadherence was suspected as plasma concentrations of ledipasvir and GS-331007 were low to undetectable at weeks 2, 4 and 6 of treatment. Virological relapse occurred in 6 and 4 % of patients receiving ledipasvir/sofosbuvir or ledipasvir/sofosbuvir plus ribavirin for 12 weeks; relapses occurred by weeks 4 or 12 post-treatment. None of the patients receiving the 24-week regimens experienced virological relapse [5].
The finding that none of the patients who achieved SVR12 in the ION trials experienced virological relapse between weeks 12 and 24 post-treatment confirms that SVR12 is an appropriate measure of durable response to ledipasvir/sofosbuvir [21].
High SVR12 rates were seen across various patient subgroups in ION-1 and ION-3, including in patients with traditional predictors of poor response [4, 6]. For example, across the four treatment groups in ION-1, SVR12 rates were 94–100 % in patients with cirrhosis, 97–99 % in patients with chronic HCV genotype 1a infection, 97–99 % in those with non-CC IL28B and 91–100 % in Black patients [4].
In ION-2, ledipasvir/sofosbuvir achieved high SVR12 rates regardless of whether patients had no response to prior treatment or prior virological breakthrough or relapse [5]. For example, in patients receiving ledipasvir/sofosbuvir for 12 or 24 weeks, SVR12 rates were 92 and 98 % in nonresponders and 95 and 100 % in patients with prior breakthrough/relapse. SVR12 rates appeared to vary according to the presence or absence of cirrhosis among treatment-experienced patients receiving 12-week treatment regimens in ION-2. For example, among recipients of ledipasvir/sofosbuvir or ledipasvir/sofosbuvir plus ribavirin for 12 weeks, SVR12 rates were 86 and 82 % in patients with cirrhosis and 95 and 100 % among patients without cirrhosis. By contrast, SVR12 rates were 99–100 % among patients receiving 24-week regimens, regardless of the presence or absence of cirrhosis [5].
Ledipasvir/sofosbuvir was effective regardless of the degree of liver fibrosis or the method of fibrosis determination [23]. Across the various treatment groups in the ION trials, SVR12 rates in patients with a Metavir fibrosis score of F0, F1, F2, F3 and F4 were 89–100, 88–100, 93–100, 86–100 and 81–100 %, respectively, by liver biopsy and 90–100, 94–100, 90–100, 90–100 and 90–100 %, respectively, by FibroTest [23].
In the ION trials, significant (p < 0.001 vs. baseline) improvements in most patient-reported outcomes occurred as early as week 2 of treatment with ledipasvir/sofosbuvir without ribavirin, and were maximized by the end of treatment [25]. Moreover, on the last day of treatment, changes in measures of health-related quality of life, fatigue and work productivity significantly (p < 0.0001) favoured ledipasvir/sofosbuvir recipients versus ledipasvir/sofosbuvir plus ribavirin recipients [25]. Among patients achieving SVR12, improvements in patient-reported outcomes were seen regardless of fibrosis stage (e.g. early or advanced hepatic fibrosis) [24].
4.1.2 Japanese Trial
An open-label, multicentre, phase III Japanese trial (available as an abstract plus poster) included treatment-naive or treatment-experienced patients with chronic HCV genotype 1 infection who received ledipasvir/sofosbuvir (n = 171) or ledipasvir/sofosbuvir plus ribavirin (n = 170) for 12 weeks; up to 40 % of patients could have compensated cirrhosis [8]. The primary endpoint was SVR12. The SVR12 rate was 100 % in ledipasvir/sofosbuvir recipients and 98 % in ledipasvir/sofosbuvir plus ribavirin recipients, with SVR12 rates of 100 and 96 % in treatment-naive patients and 100 and 100 % in treatment-experienced patients [8].
4.2 Other Trials
4.2.1 In HCV and HIV Co-Infection
The open-label, multicentre, phase III ION-4 trial (available as an abstract) examined the efficacy of 12 weeks’ therapy with ledipasvir/sofosbuvir in 335 treatment-naive or -experienced patients co-infected with HCV genotype 1 or 4 and HIV-1 [20]. Patients were receiving stable antiretroviral therapy with emtricitabine/tenofovir disoproxil fumarate in combination with efavirenz, rilpivirine or raltegravir. At baseline, chronic HCV genotype 1a, 1b and 4 infection was present in 75, 23 and 2 % of patients, respectively, 20 % of patients had compensated cirrhosis and 55 % of patients had not responded to prior anti-HCV therapy. The primary endpoint was SVR12 [20].
A 12-week regimen of ledipasvir/sofosbuvir was effective in treatment-naive or -experienced patients co-infected with HCV genotype 1 or 4 and HIV-1, with an overall SVR12 rate of 96 % [20]. Subgroup analysis revealed SVR12 rates of 94 % in treatment-naïve patients, 97 % in treatment-experienced patients, 96 % in patients without cirrhosis and 94 % in patients with cirrhosis [20].
The open-label, single-centre ERADICATE trial included treatment-naive patients without cirrhosis who were co-infected with HCV genotype 1 and HIV [26]. Patients had not received prior treatment for chronic HCV infection and were antiretroviral naive (n = 13) or antiretroviral experienced (permitted regimens included emtricitabine/tenofovir disoproxil fumarate in combination with efavirenz, rilpivirine and/or raltegravir) [n = 37] [26]. Patients received ledipasvir/sofosbuvir for 12 weeks; the primary endpoint was SVR12 [26]. The SVR12 rate was 100 % in antiretroviral-naive patients and 97 % in antiretroviral-experienced patients [26].
4.2.2 In HCV Genotype 1 or 4 Infection Following Liver Transplantation or with Decompensated Cirrhosis
The open-label, multicentre SOLAR-1 trial included a cohort of treatment-naive or treatment-experienced patients with recurrence of HCV genotype 1 or 4 infection following liver transplantation (223 evaluable patients) [17], and a cohort of treatment-naive or treatment-experienced patients with chronic HCV genotype 1 or 4 infection who had decompensated cirrhosis and had not undergone liver transplantation (n = 108) [16]. Both cohorts received ledipasvir/sofosbuvir plus ribavirin for 12 or 24 weeks and the primary efficacy endpoint was SVR12; both analyses are available as abstracts plus slide presentations [16, 17].
Ledipasvir/sofosbuvir plus ribavirin was effective in patients with HCV recurrence post-transplant [17]. SVR12 rates in recipients of ledipasvir/sofosbuvir plus ribavirin for 12 or 24 weeks were 96 and 98 % in patients with a fibrosis score of F0–F3 (n = 111), 96 and 96 % in CPT class A cirrhosis (n = 51), 85 and 83 % in CPT class B cirrhosis (n = 44) and 60 and 67 % in CPT class C cirrhosis (n = 8) [17].
Ledipasvir/sofosbuvir plus ribavirin was also effective in patients with chronic HCV genotype 1 or 4 infection and decompensated cirrhosis [16]. SVR12 rates in recipients of ledipasvir/sofosbuvir plus ribavirin for 12 or 24 weeks were 87 and 89 % overall, 87 and 89 % in CPT class B cirrhosis (n = 57) and 86 and 90 % in CPT class C cirrhosis (n = 42) [16].
A lower SVR12 rate was seen in the open-label, multicentre ELECTRON-2 trial (available as an abstract and slide presentation), which included a treatment arm in which patients with chronic HCV genotype 1 infection and decompensated cirrhosis (CPT class B) received ledipasvir/sofosbuvir without ribavirin for 12 weeks (n = 20) [27]. The SVR12 rate was 65 % [27].
4.2.3 In Chronic HCV Genotype 4 Infection
In the open-label SYNERGY trial (available as an abstract plus slide presentation), treatment-naive or treatment-experienced patients with chronic HCV genotype 4 infection (n = 21) received ledipasvir/sofosbuvir for 12 weeks [28]. SVR12 was seen in 19 of 20 (95 %) evaluable patients [28].
4.2.4 In Chronic HCV Genotype 3 Infection
The ELECTRON-2 trial included treatment arms in which patients with chronic HCV genotype 3 infection who were treatment-naive were randomized to receive ledipasvir/sofosbuvir (n = 25) or ledipasvir/sofosbuvir plus ribavirin (n = 26) for 12 weeks [27] and those who were treatment-experienced received ledipasvir/sofosbuvir plus ribavirin for 12 weeks (n = 50) [analysis available as an abstract plus poster] [29].
In treatment-naive patients, SVR12 was seen in 64 % of patients receiving ledipasvir/sofosbuvir and in 100 % of patients receiving ledipasvir/sofosbuvir plus ribavirin [27]. In treatment-experienced patients, SVR12 was seen in 82 % of patients overall, with an SVR12 rate of 89 % in the subgroup of patients without cirrhosis and 73 % in the subgroup of patients with cirrhosis [29].
5 Tolerability
Oral ledipasvir/sofosbuvir was generally well tolerated in patients with chronic hepatitis C. In a pooled analysis of the ION-1, -2 and -3 trials (available as an abstract and poster) in patients with chronic HCV genotype 1 infection who received ledipasvir/sofosbuvir (n = 1080) or ledipasvir/sofosbuvir plus ribavirin (n = 872), the incidence of adverse events was 74 versus 85 % and the incidence of treatment-related adverse events was 45 versus 71 % [30].
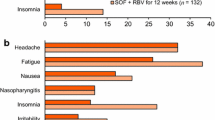
In the ION-1, -2 and -3 trials, the most commonly reported adverse events in patients receiving ledipasvir/sofosbuvir (without ribavirin) for 8–24 weeks were fatigue, headache, nausea, diarrhoea and insomnia (Fig. 1) [30]; the majority of adverse events were of mild to moderate severity [4, 5]. A similar tolerability profile was reported in patients with chronic HCV genotype 3 or 4 infection who received ledipasvir/sofosbuvir [27, 28].

The incidence of adverse events known to be associated with ribavirin (e.g. fatigue, insomnia) was numerically higher in patients receiving ledipasvir/sofosbuvir plus ribavirin than in patients receiving ledipasvir/sofosbuvir alone in the ION-1, -2 and -3 trials (Fig. 1) [30]. Anaemia (haemoglobin <10 g/dL) was reported in 0.09 % of ledipasvir/sofosbuvir recipients versus 7 % of ledipasvir/sofosbuvir plus ribavirin recipients [30].
Across the ION-1, -2 and -3 trials trials, serious adverse events were reported in 0–8 % of patients receiving ledipasvir/sofosbuvir for 8–24 weeks and in 0–3 % of patients receiving ledipasvir/sofosbuvir plus ribavirin for 8–24 weeks [4–6]. Serious treatment-related adverse events were reported in 0.4 % of patients receiving ledipasvir/sofosbuvir and in 0.1 % of patients receiving ledipasvir/sofosbuvir plus ribavirin in the pooled analysis [30]. No deaths were reported during the ION-1, -2 and -3 trials [30].
Few patients (0–3 %) discontinued ledipasvir/sofosbuvir because of adverse events in the ION-1, -2 and -3 trials [4–6]. Dose modification or interruption of treatment because of adverse events occurred in 0.6 % of patients receiving ledipasvir/sofosbuvir versus 14 % of patients receiving ledipasvir/sofosbuvir plus ribavirin [30].
Among patients receiving ledipasvir/sofosbuvir for 8, 12 or 24 weeks, bilirubin levels increased to >1.5 × ULN in 3, <1 and 2 % of patients, respectively, and transient asymptomatic increases in lipase levels to >3 × ULN occurred in <1, 2 and 3 %, respectively, according to a pooled analysis of the ION-1, -2 and -3 trials reported in the US prescribing information [1].
The tolerability profile of ledipasvir/sofosbuvir in patients with compensated cirrhosis appeared similar to that in patients without cirrhosis, according to the results of an integrated safety analysis of phase II/III trials (available as an abstract plus slide presentation) [31].
In SOLAR-1, ledipasvir/sofosbuvir plus ribavirin was generally well tolerated in patients with recurrence of HCV post-transplantation [17] or in those with decompensated cirrhosis [16]. In patients with recurrence of HCV post-transplantation, serious treatment-related adverse events were reported in 4 % of patients [anaemia (n = 4), haemolytic anaemia (n = 2), portal vein thrombosis (n = 1), sick sinus syndrome (n = 1) and sinus arrhythmia (n = 1)] and treatment-emergent death occurred in 2 % of patients [17]. In patients with decompensated cirrhosis, serious treatment-related adverse events were reported in 4 % of patients [anaemia (n = 2), hepatic encephalopathy (n = 1), peritoneal haemorrhage (n = 1)] and treatment-emergent death occurred in 6 % of patients [16].
Ledipasvir/sofosbuvir was generally well tolerated in patients co-infected with HCV and HIV in the ION-4 [20] and ERADICTAE [26] trials, with no discontinuations because of adverse events. In general, there were no clinically significant changes in CD4+ cell counts [26], HIV RNA levels [20, 26] or renal function [26].
6 Dosage and Administration
Ledipasvir/sofosbuvir is approved in the US for the treatment of patients with chronic HCV genotype 1 infection [1] and in the EU for the treatment of patients with chronic HCV genotype 1 or 4 infection and patients with chronic HCV genotype 3 infection who have cirrhosis and/or prior treatment failure [2]. The recommended dosage of oral ledipasvir/sofosbuvir is 90/400 mg once daily [1, 2].
In the US, the recommended duration of ledipasvir/sofosbuvir treatment is 12 weeks in treatment-naive patients with or without cirrhosis; an 8-week course of treatment can be considered in treatment-naive patients without cirrhosis who have a baseline HCV RNA level of <6 million IU/mL [1]. In treatment-experienced patients (i.e. patients who have failed treatment with peginterferon-α plus ribavirin with or without an HCV protease inhibitor), the recommended duration of ledipasvir/sofosbuvir treatment is 12 weeks in patients without cirrhosis and 24 weeks in patients with cirrhosis [1].
In the EU, treatment with ledipasvir/sofosbuvir for 12 weeks is recommended in patients with chronic HCV genotype 1 or 4 infection without cirrhosis; 8 weeks’ treatment may be considered in treatment-naive patients with chronic HCV genotype 1 infection and 24 weeks’ treatment should be considered in treatment-experienced patients with uncertain subsequent treatment options [2]. Treatment with ledipasvir/sofosbuvir for 24 weeks is recommended in patients with chronic HCV genotype 1 or 4 infection and compensated cirrhosis; 12 weeks’ treatment may be considered in patients deemed at low risk of progression who have subsequent retreatment options. Treatment with ledipasvir/sofosbuvir plus ribavirin for 24 weeks is recommended in patients with chronic HCV genotype 1 or 4 infection who have decompensated cirrhosis or are pre- or post-liver transplantation, and for the treatment of patients with chronic HCV genotype 3 infection who have cirrhosis and/or prior treatment failure. These dosage recommendations also apply to patients co-infected with HIV [2].
Local prescribing information should be consulted for contraindications, warnings and precautions related to ledipasvir/sofosbuvir, and for further information regarding dosage modifications and pregnancy warnings in patients receiving concomitant ribavirin.
7 Place of Ledipasvir/Sofosbuvir in the Management of Chronic Hepatitis C
A 12-week regimen of ledipasvir/sofosbuvir is approved for use in treatment-naive patients with chronic HCV genotype 1 infection (Sect. 6). This reflects the results of the ION-1 trial, which showed no additional benefit from extending the duration of treatment to 24 weeks or adding ribavirin in this patient group (Sect. 4.1.1). Subsequently, ION-3 demonstrated that an 8-week course of ledipasvir/sofosbuvir appears sufficient in treatment-naive patients with chronic HCV genotype 1 infection who do not have cirrhosis and who have a baseline HCV RNA level of <6 million IU/mL. In this patient group, extending the duration of treatment to 12 weeks or adding ribavirin to the treatment regimen was not associated with higher SVR12 rates (Sect. 4.1.1).
Recently updated guidelines from the American Association for the Study of Liver Disease (AASLD) and the Infectious Diseases Society of America (IDSA) recommend 12 weeks’ therapy with ledipasvir/sofosbuvir as an option in treatment-naive patients with chronic HCV genotype 1a or 1b infection [32]. AASLD/IDSA guidelines state that shortening the duration of treatment to <12 weeks should be done with caution and at the discretion of the physician [32].
In keeping with the results of the ION-2 trial (Sect. 4.1.1), prescribing information generally recommends a 12-week regimen of ledipasvir/sofosbuvir in treatment-experienced patients with HCV genotype 1 infection without cirrhosis (Sect. 6). AASLD/IDSA guidelines also recommend 12 weeks’ treatment with ledipasvir/sofosbuvir in patients with chronic HCV genotype 1a or 1b infection who do not have cirrhosis and who failed prior treatment with peginterferon-α plus ribavirin, and in patients with chronic HCV genotype 1 infection, regardless of subtype, who are without cirrhosis and have failed prior treatment with peginterferon-α plus ribavirin and an HCV protease inhibitor [32].
Subgroup analyses generally revealed high SVR12 rates across various subgroups in treatment-naive and treatment-experienced patients with chronic HCV genotype 1 infection in the ION trials, including in patients with baseline characteristics traditionally considered to be associated with poor response (Sect. 4.1.1). Thus, factors that traditionally predicted a poor response to interferon-based therapy do not predict response to ledipasvir/sofosbuvir treatment [22].
Historically, response rates have been low in patients with cirrhosis who received interferon-based regimens, including regimens containing HCV protease inhibitors such as telaprevir and boceprevir [4]. However, high SVR12 rates were seen in treatment-naive patients with cirrhosis who received ledipasvir/sofosbuvir for 12 weeks in ION-1. SVR12 rates appeared numerically higher in treatment-experienced patients with cirrhosis who received ledipasvir/sofosbuvir for 24 versus 12 weeks in ION-2, although it should be noted that the study was not powered for intergroup comparisons [5]. Factors predictive of which patients with cirrhosis were most likely to relapse after 12 weeks’ treatment have not yet been identified [5].
Options recommended by AASLD/IDSA guidelines for patients with chronic HCV genotype 1a or 1b infection who have compensated cirrhosis and have failed prior treatment with peginterferon-α plus ribavirin include 24 weeks’ treatment with ledipasvir/sofosbuvir or 12 weeks’ treatment with ledipasvir/sofosbuvir plus ribavirin [32]. Similarly, in patients with chronic HCV genotype 1 infection, regardless of subtype, who have cirrhosis and have failed prior treatment with peginterferon-α plus ribavirin and an HCV protease inhibitor, AASLD/IDSA guidelines recommend a 24-week regimen of ledipasvir/sofosbuvir or a 12-week regimen of ledipasvir/sofosbuvir plus ribavirin [32]. These recommendations are based on the results of an integrated efficacy analysis of phase II/III trial data [31].
Based on emerging data from the SOLAR-1 trial (Sect. 4.2.2), 24 weeks’ treatment with ledipasvir/sofosbuvir plus ribavirin is approved in the EU for use in patients with chronic HCV genotype 1 infection who have decompensated cirrhosis or are pre- or post-liver transplantation (Sect. 6). Ledipasvir/sofosbuvir is also approved in the EU in patients with chronic HCV genotype 4 (with the addition of ribavirin recommended in patients who have decompensated cirrhosis or are pre- or post-liver transplantation), and ledipasvir/sofosbuvir plus ribavirin is also approved for the treatment of patients with chronic HCV genotype 3 infection who have cirrhosis and/or prior treatment failure (Sect. 6).
AASLD/IDSA guidelines recommend 12 weeks’ treatment with ledipasvir/sofosbuvir as an option in patients with chronic HCV genotype 4 infection who are treatment-naive or who have failed prior treatment with peginterferon-α plus ribavirin, with the same regimen recommended in treatment-naive patients with chronic HCV genotype 6 infection who are treatment-naive or who have failed prior treatment [32].
A 12-week regimen of ledipasvir/sofosbuvir was associated with high SVR12 rates in patients co-infected with HCV genotype 1 or 4 and HIV, according to the results of the ION-4 and ERADICATE trials (Sect. 4.2.1), without any clinically significant effects on CD4+ cell counts or HIV RNA levels (Sect. 5). AASLD/IDSA guidelines [32] recommend that patients co-infected with HCV and HIV should receive the same treatment and retreatment as patients without HIV infection, taking into account potential interactions with antiretroviral agents (Sect. 3).
The low pill burden associated with once-daily administration of the single-tablet regimen of ledipasvir/sofosbuvir and shorter treatment durations have the potential to improve treatment adherence [33]. Adding a third direct-acting antiviral agent to ledipasvir/sofosbuvir may further shorten the duration of treatment [34, 35]. For example, among treatment-naive patients with chronic HCV genotype 1 infection (n = 60), SVR12 rates were 100 % in recipients of ledipasvir/sofosbuvir for 12 weeks, 95 % in recipients of ledipasvir/sofosbuvir plus the investigational non-nucleoside NS5B inhibitor GS-9669 for 6 weeks and 95 % in recipients of ledipasvir/sofosbuvir plus the investigational HCV protease inhibitor vedroprevir (GS-9451) for 6 weeks [34].
Treatment with ledipasvir/sofosbuvir may also have potential in patients with chronic HCV genotype 1 infection who failed previous therapy with a sofosbuvir-containing regimen [27, 36, 37]. For example, the open-label SYNERGY trial [36] included a cohort of 14 patients with chronic HCV genotype 1 infection who relapsed after receiving sofosbuvir plus ribavirin for 24 weeks in an earlier trial [38]; these patients received ledipasvir/sofosbuvir for 12 weeks. SVR12 was achieved in all 14 patients, with unquantifiable HCV RNA levels seen from week 4 of treatment onwards [36]. AASLD/IDSA guidelines recommend 24 weeks’ treatment with ledipasvir/sofosbuvir with or without ribavirin in patients with chronic HCV genotype 1 infection and advanced fibrosis in whom a previous sofosbuvir-containing regimen has failed [32].
Oral ledipasvir/sofosbuvir was generally well tolerated in patients with chronic hepatitis C, with the most commonly reported adverse events including fatigue, headache, nausea, diarrhoea and insomnia (Sect. 5). As expected, the addition of ribavirin to ledipasvir/sofosbuvir was associated with numerical increases in the incidence of adverse events known to be associated with ribavirin, although there was no apparent increase in the incidence of serious adverse events.
Ledipasvir/sofosbuvir is an expensive therapy and its cost represents a barrier to treatment [39]. However, results of a US decision-analytic model, conducted from a healthcare payer perspective with a lifetime horizon, predicted that ledipasvir/sofosbuvir would be dominant (i.e. less costly and more effective) or cost effective versus other current treatment regimens in treatment-naive or treatment-experienced patients with chronic HCV genotype 1 infection [40]. It should be noted that this analysis is subject to various limitations [40].
In conclusion, ledipasvir/sofosbuvir is an important new single-tablet regimen that represents a significant advance in the treatment of chronic hepatitis C.
Data selection sources:
Relevant medical literature (including published and unpublished data) on ledipasvir/sofosbuvir was identified by searching databases including MEDLINE (from 1946) and EMBASE (from 1996) [searches last updated 9 March 2015], bibliographies from published literature, clinical trial registries/databases and websites. Additional information was also requested from the company developing the drug.
Search terms: Ledipasvir, GS-5885, sofosbuvir, GS-7977, Sovaldi.
Study selection: Studies in patients with chronic hepatitis C who received ledipasvir/sofosbuvir. When available, large, well designed, comparative trials with appropriate statistical methodology were preferred. Relevant pharmacodynamic and pharmacokinetic data are also included.
References
Gilead Sciences Inc. Harvoni® (ledipasvir and sofosbuvir) tablets for oral use: US prescribing information. 2015. http://www.harvoni.com/. Accessed 30 Mar 2015.
European Medicines Agency. Harvoni (ledipasvir/sofosbuvir) film-coated tablets: EU summary of product characteristics. 2014. http://www.ema.europa.eu/. Accessed 2 Mar 2015.
Hebner C, Lee Y-J, Han B, et al. In vitro pan-genotypic and combination activity of sofosbuvir (GS-7977) in stable replicon cell lines [abstract no. 1875]. Hepatology. 2012;56(4 Suppl):1066A.
Afdhal N, Zeuzem S, Kwo P, et al. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med. 2014;370(20):1889–98.
Afdhal N, Reddy KR, Nelson DR, et al. Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med. 2014;370(16):1483–93.
Kowdley KV, Gordon SC, Reddy KR, et al. Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N Engl J Med. 2014;370(20):1879–88.
Sarrazin C, Dvory-Sobol H, Svarovskaia ES, et al. Baseline and post-baseline resistance analyses of phase 2/3 studies of ledipasvir/sofosbuvir ± RBV [abstract no. 1926 plus poster]. Hepatology. 2014;60(4 Suppl):1128A.
Mizokami M, Takehara T, Yokosuka O, et al. 100 % SVR12 in Japanese patients with chronic genotype 1 hepatitis C virus infection receiving ledipasvir/sofosbuvir fixed dose combination for 12 weeks: results from a multicenter phase 3 study [abstract no. 1929 plus poster]. Hepatology. 2014;60(4 Suppl):1130A.
German P, Mathias A, Yang JC, et al. The pharmacokinetics of ledipasvir, an HCV specific NS5A inhibitor in HCV-uninfected subjects with moderate and severe hepatic impairment [abstract no. 467 plus poster]. Hepatology. 2013;58(4 Suppl 1):432A.
Mogalian E, Mathias A, Yang JC, et al. The pharmacokinetics of ledipasvir, an HCV specific NS5A inhibitor, in HCV-uninfected subjects with severe renal impairment [abstract no. 1952]. Hepatology. 2014;60(4 Suppl):1145A–6A.
Kirby B, German P, Shen G, et al. No differences in the pharmacokinetics of sofosbuvir (GS-7977) and sofosbuvir/ledipasvir (GS-7977/GS-5885) fixed dose combination between Japanese and Caucasians [abstract no. 954]. Hepatol Int. 2013;7(Suppl 1):S375–6.
German P, Pang PS, Fang L, et al. Drug-drug interaction profile of the fixed-dose combination tablet ledipasvir/sofosbuvir [abstract no. 1976 plus poster]. Hepatology. 2014;60(4 Suppl):1162A.
German P, Garrison K, Pang PS, et al. Drug–drug interactions between anti-HCV regimen ledipasvir/sofosbuvir and antiretrovirals [abstract no. 82]. In: 2015 Conference on Retroviruses and Opportunistic Infections. 2015.
German P, Moorehead L, Pang P, et al. Lack of a clinically important pharmacokinetic interaction between sofosbuvir or ledipasvir and hormonal oral contraceptives norgestimate/ethinyl estradiol in HCV-uninfected female subjects. J Clin Pharmacol. 2014;54(11):1290–8.
Mathias A, Cornpropst M, Clemons D, et al. No clinically significant pharmacokinetic drug-drug interactions between sofosbuvir (GS-7977) and the immunosuppressants, cyclosporine A or tacrolimus in healthy volunteers [abstract no. 1869 plus poster]. Hepatology. 2012;56(4 Suppl):1063A–4A.
Flamm SL, Everson GT, Charlton M, et al. Ledipasvir/sofosbuvir with ribavirin for the treatment of HCV in patients with decompensated cirrhosis: preliminary results of a prospective multicenter study [abstract no. 239 plus slide presentation]. Hepatology. 2014;60(4 Suppl):320A–1A.
Reddy KR, Everson GT, Flamm SL, et al. Ledipasvir/sofosbuvir with ribavirin for the treatment of HCV in patients with post-transplant recurrence: preliminary results of a prospective, multicenter study [abstract no. 8 plus slide presentation]. Hepatology. 2014;60(4 Suppl):200A–1A.
Gane EJ, Stedman CA, Hyland RH, et al. Efficacy of nucleotide polymerase inhibitor sofosbuvir plus the NS5A inhibitor ledipasvir or the NS5B non-nucleoside inhibitor GS-9669 against HCV genotype 1 infection. Gastroenterology. 2014;146(3):736–43.
Lawitz E, Poordad FF, Pang PS, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet. 2014;383(9916):515–23.
Naggie S, Cooper C, Saag MS, et al. Ledipasvir/sofosbuvir for 12 weeks in patients coinfected with HCV and HIV-1 [abstract no. 152LB]. In: 2015 Conference on Retroviruses and Opportunistic Infections. 2015.
Bernstein DE, Mangia A, Brau N, et al. Concordance between SVR4, SVR12 and SVR24 in genotype 1 HCV-infected patients who received all oral fixed-dose combination ledipasvir/sofosbuvir with or without ribavirin in phase 3 clinical trials [abstract no. 1947 plus slide presentation]. Hepatology. 2014;60(4 Suppl):1142A–3A.
Jacobson IM, Kwo PY, Kowdley KV, et al. Virologic response rates to all oral fixed-dose combination ledipasvir/sofosbuvir regimens are similar in patients with and without traditional negative predictive factors in phase 3 clinical trials [abstract no. 1945 plus poster]. Hepatology. 2014;60 (4 Suppl):1141A–2A.
Gordon SC, Fried MW, Kwo PY, et al. No differences in the efficacy of fixed-dose combination ledipasvir/sofosbuvir in patients according to fibrosis stage determined by liver biopsy or laboratory biomarker in phase 3 clinical trials [abstract no. 1958 plus poster]. Hepatology. 2014;60(4 Suppl):1149A–50A.
Younossi Z, Stepanova M, Marcellin P, et al. Sustained virologic response with ledipasvir (LDV) and sofosbuvir (SOF) regimens lead to substantial improvement in patient-reported outcomes (PROs) among chronic hepatitis C (CHC) patients with early hepatic fibrosis as well as those with advanced hepatic fibrosis [abstract no. 1445 plus poster]. Hepatology. 2014;60(4 Suppl):892A–3A.
Younossi ZM, Stepanova M, Marcellin P, et al. Treatment with ledipasvir and sofosbuvir improves patient-reported outcomes: results from the ION-1, 2 and 3 clinical trials. Hepatology. 2015. doi:10.1002/hep.27724.
Osinusi A, Townsend K, Kohli A, et al. Virologic response following combined ledipasvir and sofosbuvir administration in patients with HCV genotype 1 and HIV co-infection. JAMA. 2015. doi:10.1001/jama.2015.1373.
Gane E, Hyland RH, Pang P, et al. Sofosbuvir/ledipasvir fixed dose combination is safe and effective in HCV infected populations including decompensated patients and patients with prior sofosbuvir treatment experience [abstract no. 238 plus slide presentation]. Gastroenterology. 2014;146(5 Suppl 1):S904–S5.
Kapoor R, Kohil A, Sidharthan S, et al. All oral treatment for genotype 4 chronic hepatitis C infection with sofosbuvir and ledipasvir: interim results from the NIAID SYNERGY trial [abstract no. 240 plus slide presentation]. Hepatology. 2014;60(4 Suppl):321A.
Gane EJ, Hyland RH, An D, et al. High efficacy of LDV/SOF regimens for 12 weeks for patients with HCV genotype 3 or 6 infection [abstract no. LB-11 plus poster]. Hepatology. 2014;60(1 Suppl).
Alqahtani S, Afdhal NH, Zeuzem S, et al. Safety of ledipasvir/sofosbuvir with and without ribavirin for the treatment of patients with chronic HCV genotype 1 infection: an analysis of the phase 3 ION trials [abstract no. 1944 plus poster]. Hepatology. 2014;60 (4 Suppl):1140A–1A.
Bourlière M, Sulkowski MS, Omata M, et al. An integrated safety and efficacy analysis of >500 patients with compensated cirrhosis treated with ledipasvir/sofosbuvir with or without ribavirin [abstract no. 82 plus slide presentation]. Hepatology. 2014;60(4 Suppl):239A.
American Association for the Study of Liver Diseases and Infectious Diseases Society of America. Recommendations for testing, managing, and treating hepatitis C (December 19). 2014. http://www.hcvguidelines.org/full-report-view. Accessed 2 Mar 2015.
Petersen T, Gordon LA, Townsend K, et al. Pill burden and treatment length reduce adherence to IFN-free hepatitis C therapy in an urban cohort [abstract no. 667]. Top Antivir Med. 2014;22(e-1):331–2.
Kohli A, Osinusi A, Sims Z, et al. Virological response after 6 week triple-drug regimens for hepatitis C: a proof-of-concept phase 2A cohort study. Lancet. 2015. doi:10.1016/S0140-6736(14)61228-9.
Lawitz E, Poordad F, Hyland RH, et al. High rates of SVR in patients with genotype 1 HCV infection and cirrhosis after treatment with ledipasvir/sofosbuvir + ribavirin or ledipasvir/sofosbuvir + GS-9669 for 8 weeks [abstract no. 1948]. Hepatology. 2014;60(4 Suppl):1143A.
Osinusi A, Kohli A, Marti MM, et al. Re-treatment of chronic hepatitis C virus genotype 1 infection after relapse: an open-label pilot study. Ann Intern Med. 2014;161(9):634–8.
Wyles DL, Pockros PJ, Yang JC, et al. Retreatment of patients who failed prior sofosbuvir-based regimens with all oral fixed-dose combination ledipasvir/sofosbuvir plus ribavirin for 12 weeks [abstract no. 235 plus slide presentation]. Hepatology. 2014;60(4 Suppl):317A–8A.
Osinusi A, Meissner EG, Lee YJ, et al. Sofosbuvir and ribavirin for hepatitis C genotype 1 in patients with unfavorable treatment characteristics: a randomized clinical trial. JAMA. 2013;310(8):804–11.
Hoofnagle JH, Sherker AH. Therapy for hepatitis C: the costs of success. N Engl J Med. 2014;370(16):1552–3.
Younossi ZM, Park H, Saab S, et al. Cost-effectiveness of all-oral ledipasvir/sofosbuvir regimens in patients with chronic hepatitis C virus genotype 1 infection. Aliment Pharmacol Ther. 2015;41(6):544–63.
Disclosure
The preparation of this review was not supported by any external funding. Gillian Keating is a salaried employee of Adis/Springer. During the peer review process, the manufacturer of the agent under review was offered an opportunity to comment on this article. Changes resulting from comments received were made by the author on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Additional information
The manuscript was reviewed by: M. Buti, Department of Hepatology, Hospital General Universitari Vall d’Hebron, Barcelona, Spain; M. Romero-Gómez, Unit for Clinical Management of Digestive Diseases and CIBERehd, Valme University Hospital, University of Seville, Seville, Spain; C. A. Stedman, University of Otago Christchurch and Department of Gastroenterology, Christchurch Hospital, Christchurch, New Zealand.
Rights and permissions
About this article

Cite this article
Keating, G.M. Ledipasvir/Sofosbuvir: A Review of Its Use in Chronic Hepatitis C. Drugs 75, 675–685 (2015). https://doi.org/10.1007/s40265-015-0381-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-015-0381-2