
Abstract
The fixed-dose combination tablet of ledipasvir (LDV), an HCV NS5A inhibitor, and sofosbuvir (SOF), an HCV nucleotide analog NS5B polymerase inhibitor, was the first all-oral (one-pill once daily), interferon-free and ribavirin-free regimen approved for the treatment of patients with chronic hepatitis C. With over 5,900 HCV-infected patients enrolled in LDV/SOF clinical trials through late 2017, the accelerated clinical development program was able to generate safety and efficacy data across a broad range of patient populations. The initial registration trials demonstrated that 12 weeks of treatment with LDV/SOF resulted in high cure rates of over 95% in HCV genotype 1 patients regardless of historical negative treatment predictors including cirrhosis or prior treatment history. The program subsequently expanded to include other HCV genotypes and special populations with significant unmet medical need including but not limited to decompensated liver disease, HIV/HCV coinfection, posttransplantation, and children. With favorable pharmacokinetic properties, good safety profile, and high efficacy rates, the approval of LDV/SOF (Harvoni®) ushered in a new era of treatment and management for the millions of HCV-infected patients globally.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Direct-acting antivirals
- HCV genotype 1 infection
- Hepatitis C virus
- NS5A inhibitors
- NS5B nucleotide inhibitors
1 Introduction
Globally, chronic hepatitis C virus (HCV) infection remains a significant public health challenge, with over 80 million persons infected [1]. In 2011, the first class of direct-acting antivirals (DAAs), namely, HCV nonstructural protein (NS) 3/4A protease inhibitors, was approved [2] . While these treatments were successful in up to 75% in specific populations such as treatment-naive patients, these regimens were less effective in treatment-experienced patients, especially in those with advanced liver disease [3]. In addition, these treatments were given in combination with pegylated interferon (Peg-IFN) and ribavirin (RBV) for up to 48 weeks and were associated with additional serious side effects including anemia, rash, serious infection, decompensation, high discontinuation rates, and complicated response-guided therapy algorithms with high pill burdens. It was estimated that up to 50% of patients with HCV infection were not eligible for these treatments due to relative or absolute contraindications to Peg-IFN [4].
There remained a significant unmet medical need for simplified treatment regimens that were more effective with improved safety and tolerability profiles. As such, the goal of the LDV/SOF clinical development program was to develop an IFN-free, RBV-free all-oral regimen for the treatment of chronic HCV infection by combining two potent DAAs, ledipasvir (LDV), an NS5A inhibitor, and sofosbuvir (SOF), a pangenotypic nucleotide NS5B polymerase inhibitor, into a fixed-dose combination (FDC) tablet. This regimen would obviate toxicity, tolerability, as well as contraindications, associated with Peg-IFN and/or RBV which were components of approved therapy.
The clinical development program was initially focused on genotype 1 HCV infection which represents the majority of all cases of chronic HCV infection in the United States and Europe [5]; however, the program was expanded rapidly to include all HCV genotypes and distinct patient populations across the globe with a significant unmet medical need.
2 Clinical Pharmacology of Sofosbuvir, Ledipasvir, and Ledipasvir/Sofosbuvir
Clinical pharmacology studies were conducted with either LDV/SOF or the components, SOF and LDV, as individual agents, alone or in combination, including coadministration with other DAAs. The SOF and LDV clinical development programs run in parallel with the development of the LDV/SOF fixed-dose combination (FDC) tablet prior to initiation of the LDV/SOF phase 3 clinical studies.
2.1 Clinical Pharmacology of Sofosbuvir
The pharmacokinetic (PK) properties of sofosbuvir (SOF) were evaluated extensively in healthy adult subjects and in patients with chronic hepatitis C infection [6, 7]. Following oral administration, SOF is absorbed quickly, and peak plasma concentration is observed ~0.5–2 h post-dose, regardless of dose level. SOF is extensively metabolized in the liver to form the pharmacologically active nucleoside analog triphosphate GS-461203 which undergoes subsequent dephosphorylation to yield GS-331007, the pharmacologically inactive, primary circulating nucleoside metabolite responsible for over 85% of systemic drug exposure. Peak plasma concentration of GS-331007 is observed between 2 and 4 h post-dose. The terminal half-life is 0.4 h for SOF and 27 h for GS-331007 which supports once-daily dosing. Consumption of a moderate- or high-fat meal increases SOF AUC by 2-fold and C max by 1.3-fold, while the exposure of GS-331007 is not altered. These observed increases in SOF levels are not considered clinically meaningful, and SOF can be administered without regard to food [8]. Sofosbuvir is approximately 61–65% bound to human plasma proteins, while GS-331007 has minimal binding.
Population pharmacokinetic analysis in HCV-infected patients demonstrated that race, gender, age, and baseline body mass index (BMI) had no clinically relevant effect on the exposure of SOF or GS-331007. Due to the hepatic metabolism of SOF and concerns around the use of DAAs with hepatic impairment, a PK study was conducted in HCV-infected patients with hepatic impairment. This study showed C max and AUC values of SOF were ~80% and 130% higher in cirrhotic compared to non-cirrhotic patients; however, these differences were not considered clinically significant. As such, no dose adjustment of sofosbuvir is recommended for patients with mild, moderate, and severe hepatic impairment [9]. Renal clearance is the major elimination pathway for GS-331007, and a PK study was conducted in HCV-negative subjects with renal impairment. This study showed that the AUC of GS-331007 was increased 55%, 88%, and 451% in subjects with mild, moderate, and severe renal impairment, respectively. Based on these findings, dose adjustment for patients with mild or moderate renal impairment is not required; however, sofosbuvir is currently not approved for use in patients with severe renal impairment [10, 11]. There are ongoing studies evaluating the safety and efficacy of LDV/SOF in HCV-infected patients with end-stage renal disease on dialysis.
SOF has limited clinically relevant drug-drug interactions as both SOF and GS-331007 are not substrates, inducers, or inhibitors of drug-metabolizing enzymes such as cytochrome P450 (CYP) or UDP-glucuronosyltransferase (UGT) A1 enzymes. In PK studies, coadministration of SOF with several drugs that impact CYP enzymes such as contraceptives, methadone, immunosuppressants, and antiretrovirals have not been shown to have clinically relevant effects on PK parameters. However, SOF is a substrate of drug transporters P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), while GS-331007 is not; therefore, inhibitors or inducers of these transporters may alter plasma concentrations of SOF. For example, coadministration with P-gp inducers such as rifampin and St John’s wort can decrease SOF concentration affecting efficacy and should be avoided [12, 13].
2.2 Clinical Pharmacology of Ledipasvir/Sofosbuvir
2.2.1 Clinical Pharmacology of LDV/SOF in Adults
Pharmacokinetic properties of ledipasvir, sofosbuvir in combination, and GS-331007 were studied in healthy adult subjects and patients with chronic hepatitis C infection [14]. While AUC and C max of SOF and GS-331007 were similar in healthy subjects and those with chronic HCV infection, AUC and C max of LDV were 24% and 32% lower, respectively, in HCV-infected patients. Following oral administration of LDV/SOF, median peak concentration is observed 4–4.5 h post-dose. LDV concentrations are not affected by food supporting the recommendation that LDV/SOF can be administered without regard to food. Ledipasvir is minimally metabolized by the liver, highly protein-bound (more than 99%), and is primarily eliminated in the feces as an unchanged drug through biliary excretion. Studies conducted in subjects with renal insufficiency or hepatic impairment demonstrated that no dose adjustment is required in patients with end-stage renal disease or those with severe hepatic impairment. Population pharmacokinetic analysis in HCV-infected patients indicated that race, gender, and age had no clinically relevant effect on the exposure of LDV, similar to observations with SOF.
Ledipasvir is not a substrate, inducers, or inhibitors of traditional drug-metabolizing enzymes, e.g., CYP- or UGT1A1-mediated drug-drug interactions. In vitro, LDV is a substrate and an inhibitor of P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) and as such may alter the absorption of substrates of these transporters. Several drug interaction studies were conducted with LDV/SOF to assess for clinically meaningful drug interactions. Potent inducers of P-gp such as rifampin, St. John’s wort, and carbamazepine will reduce plasma concentrations of SOF and/or LDV and should be avoided. Immunosuppressants such as cyclosporine and tacrolimus, opiate substitution therapy, and oral contraceptives can be safely coadministered with LDV/SOF. Tipranavir, an antiretroviral can lead to reduced SOF and LDV levels and should be avoided with LDV/SOF coadministration. Rosuvastatin exposure can increase with LDV/SOF coadministration, potentially increasing the risk of rhabdomyolysis, and is contraindicated [14].
Ledipasvir solubility decreases as pH increases; therefore, medications that increase gastric pH may result in a decreased concentration of LDV. This effect can be managed with specific dosing instructions: antacid dosing should be separated by at least 4 h from LDV/SOF; H2-receptor antagonists should be given simultaneously or staggered apart from LDV/SOF at a dose no higher than famotidine 40 mg twice daily or equivalent; and proton-pump inhibitors at doses comparable to omeprazole 20 mg or lower can be administered simultaneously.
2.2.2 Clinical Pharmacology of LDV/SOF in Adolescent Patients
The pharmacokinetic properties of SOF, GS-331007, and LDV were evaluated in adolescent patients (12 to <18 years of age) in study GS-US-337-1116 (Group 1), who received the adult dose of LDV/SOF (90/400 mg) [15]. No clinically relevant differences in the exposure of SOF, GS-331007, and LDV were observed in the adolescent population compared with adult patients in the phase 2 and 3 studies, confirming the appropriateness of LDV/SOF (90/400 mg) for use in adolescents ages 12 to <18 years.
3 Dose Selection of Sofosbuvir, Ledipasvir, and Ledipasvir/Sofosbuvir
3.1 Dose Selection of Sofosbuvir
Data from dose-ranging phase 1b and phase 2 studies of SOF conducted within the SOF development program as either monotherapy or combination therapy with Peg-IFN and RBV revealed exposure-response relationships that supported the dose selection of SOF 400 mg for the treatment of HCV infection. The phase 1b study P7851-1102 assessed once-daily doses GS-9851 from 50 to 400 mg (also known as PSI-7851) 50:50 diasteromeric mixture of SOF and GS-491241 (also known as PSI-7976) (an HCV RNA NS5B inhibitor) administered for 3 consecutive days to treatment-naive patients with chronic genotype 1 HCV infection. When GS-9851 enters into the liver cell, both GS-491241 and SOF molecules rapidly get converted into the same active triphosphate. Mean maximal decreases from baseline in HCV RNA were 0.09, 0.49, 0.56, 1.15, and 1.95 log10 IU/mL for placebo and GS-9851 50, 100, 200, and 400 mg doses, respectively [13]. The GS-9851 400 mg dose had the earliest and most potent antiviral effect in the greatest percentage of patients, with the majority having a continued reduction in HCV RNA (≥1.0 log10) 2 days after the last dose of GS-9851 (Fig. 1).

Median log10 HCV RNA change from baseline following multiple doses of GS-9851: phase 1 study P7851-1102. GS-9851 (also known as PSI-7851) is a mixture of two phosphate diastereoisomers (GS-491241 (also known as PSI-7976) and SOF)
Subsequent data from two phase 2 dose-finding studies P7977-0221 and P7977-0422 (PROTON ) confirmed the selection of the SOF 400 mg dose. In study P7977-0221, a total of 64 treatment-naive patients with genotype 1 HCV infection were randomized to receive SOF doses of 100, 200, or 400 mg or matching placebo for 27 days in combination with Peg-IFN + RBV for 48 weeks [16]. From days 0 to 27 (SOF/placebo treatment period), mean HCV RNA levels rapidly declined with all doses of SOF with Peg-IFN + RBV compared with placebo given with Peg-IFN + RBV. The sustained virologic response 12 weeks after treatment completion (SVR12) rates were higher in the SOF 200 mg and 400 mg groups (72% and 80%, respectively) compared to the SOF 100 mg and placebo groups (56% and 50%, respectively). Based on the lower rates of virologic failure, SOF 200 and 400 mg were the therapeutic doses selected for further evaluation in the phase 2 study P7977-0422 (PROTON).
In study P7977-0422, 122 treatment-naive patients with genotype 1 HCV infection were randomized to receive SOF 200 mg, SOF 400 mg, or matching placebo once daily with Peg-IFN + RBV for 12 weeks [17]. In addition, 25 treatment-naive genotype 2 or 3 HCV-infected patients received open-label SOF 400 mg once daily in combination with Peg-IFN + RBV for 12 weeks. Sofosbuvir 200 and 400 mg in combination with Peg-IFN + RBV for 12 weeks led to high SVR12 rates of 90–92% in patients with genotype 1, 2, or 3 HCV infection. On-treatment failures occurred in the SOF 200 mg group but not in the SOF 400 mg group.
In both phase 2 studies, all doses of SOF were well tolerated. The adverse event and laboratory profiles were similar for Peg-IFN + RBV, SOF 200 mg with Peg-IFN + RBV, and SOF 400 mg with Peg-IFN + RBV, consistent with the expected safety profile of Peg-IFN and RBV. There were no new adverse events or laboratory abnormalities attributable to SOF. Based on the totality of the safety, pharmacokinetic, and antiviral activity, the SOF 400 mg dose was selected to move forward for phase 3 evaluation in the SOF clinical development program and in combination with LDV in phase 2 trials in the LDV clinical development program.
3.2 LDV/SOF Dose Selection
The phase 1 study GS-US-256-0102 assessed once-daily doses of LDV from 1 to 90 mg administered for 3 consecutive days in treatment-naive patients with chronic genotype 1 HCV infection [18]. A dose-dependent response was observed for LDV doses of 3 mg through 30 mg. Ledipasvir resulted in rapid reductions in plasma HCV RNA of ≥2 log10 IU/mL as early as 8 h and reductions >3 log10 IU/mL on day 2 (36 h) following administration of 3 through 90 mg (Fig. 2). Similar and maximal antiviral responses (median approximately 3 log reduction) were observed following LDV doses of 10, 30, or 90 mg. Maximum effect (E max) modeling indicated that exposures achieved following administration of LDV doses ≥30 mg provided >95% of maximal antiviral responses in genotype 1a HCV-infected patients. There was no evidence of additional antiviral activity at the 90 mg dose based on median reductions in HCV RNA; however, HCV RNA suppression was sustained for a longer period compared with the 30 mg dose. Based on these data, LDV 30 mg and 90 mg once-daily doses were selected for clinical evaluation in phase 2 trials.

Median log 10 HCV RNA change from baseline following administration of LDV (phase 1 study GS-US-256-0102). Patients received LDV or placebo once daily for 3 days. Arrows indicate time of dosing
Study GS-US-248-0120 was a phase 2, dose-finding trial that evaluated the safety, tolerability, and antiviral efficacy of LDV 30 mg or 90 mg, administered in combination with the NS3 protease inhibitor vedoprevir, non-nucleoside NS5B inhibitor tegobuvir, and RBV in 234 treatment-naive patients with chronic genotype 1 HCV infection. Treatment with LDV 90 mg in combination with other DAAs and RBV for 12 or 24 weeks resulted in higher SVR24 rates compared to LDV 30 mg in combination with DAAs and RBV (58.5% vs 47.8%). In addition, the incidence of virologic breakthrough was lower in the LDV 90 mg group compared to the LDV 30 mg group (10.6% vs 19.6%, respectively).
Treatment with LDV in combination with other DAAs and RBV was generally well tolerated, and increasing the LDV dose did not alter the safety profile of the regimens in terms of overall frequency or severity of AEs or laboratory abnormalities. Based on efficacy and the favorable safety and tolerability profile, the 90 mg dose of LDV was selected for coformulation in the LDV/SOF fixed-dose combination.
4 Safety and Efficacy of Ledipasvir/Sofosbuvir in Phase 2 Trials
The LDV/SOF clinical development program was initiated once phase 2 clinical data became available showing that SOF in combination with RBV ± Peg-IFN resulted in high efficacy across all HCV genotypes and were well tolerated as compared to available standard of care [17, 19, 20] (Table 1). The goal of the phase 2 trials in the LDV/SOF clinical development program was to evaluate the combination of SOF 400 mg with LDV 90 mg or LDV/SOF (90 mg/400 mg) FDC with or without RBV in a broad population of HCV patients irrespective of treatment history and fibrosis status. Safety, efficacy, and pharmacokinetic data generated from these studies are described below.
4.1 Study P7977-0523 (ELECTRON)
Study P7977-0523 (ELECTRON) was a phase 2 multicenter, open-label trial to evaluate the safety, tolerability, and antiviral efficacy of SOF-containing treatment regimens in HCV patients [21, 22]. This study had multiple arms and was the first to evaluate the use of sofosbuvir in combination with ledipasvir. The latter arms of the study utilized the fixed-dose combination tablet of LDV/SOF. The study was conducted at two sites in New Zealand and commenced enrollment of patients in June 2012; only results relevant to the combination of SOF and LDV or LDV/SOF are described below (Table 2).
Part 4 (Groups 12 and 13) of the study enrolled nine patients with genotype 1 HCV infection who had documented null response following previous treatment with Peg-IFN and RBV for ≥12 weeks and 25 treatment-naive patients with genotype 1 HCV infection who received SOF 400 mg once daily + LDV 90 mg once daily and weight-based RBV (1,000–1,200 mg/day divided twice daily) for 12 weeks. The latter arms of the study included Part 6 (Groups 16 and 17) which randomized 19 patients with genotype 1 HCV infection who were prior null responders to Peg-IFN therapy with cirrhosis to receive either LDV/SOF (90 mg/400 mg) once daily or LDV/SOF once daily in combination with weight-based RBV for 12 weeks; Part 16 (Group 18) enrolled ten non-cirrhotic treatment-naive patients with genotype 2 or 3 HCV infection to receive LDV/SOF once daily for 12 weeks; Group 20 enrolled 14 patients with genotype 1 HCV infection and hemophilia to receive LDV/SOF once daily in combination with weight-based RBV for 12 weeks, while Group 21 enrolled 25 treatment-naive patients with genotype 1 HCV infection to receive LDV/SOF once daily in combination with weight-based RBV for 6 weeks.
This study showed that in treatment-naive and null-responder patients with genotype 1 HCV infection, treatment with SOF (400 mg) with LDV (90 mg) and RBV for 12 weeks provided a high virologic response rate with an SVR12 rate of 100% compared with patients who received only SOF + RBV, where 84% and 10% of treatment-naive and null-responder patients, respectively, achieved SVR12. In patients who were null responders with genotype 1 HCV infection and cirrhosis, treatment with LDV/SOF or LDV/SOF + RBV for 12 weeks led to high virologic responses with SVR12 rates of 70% and 100%, respectively. Patients with multiple negative predictors of response such as prior null response and cirrhosis achieved high SVR rates with LDV/SOF with or without RBV. Treatment-naive patients with genotype 1 HCV infection who received LDV/SOF with RBV for 6 weeks had a lower SVR12 rate of 68% compared with 100% in those who received 12 weeks of LDV/SOF with RBV, indicating that 6 weeks of LDV/SOF with RBV was likely to be too short a duration of treatment to achieve an optimal response rate.
The most common reported adverse events were headache, fatigue, and nausea. Most of the adverse events were mild in severity. Overall, five patients experienced severe adverse events; of these, the only severe event considered related to treatment was grade 3 hemolytic anemia, a known side effect of RBV. One patient discontinued treatment after 7 weeks due to an adverse event (spontaneous perforation of a colonic diverticulum, assessed as not related to treatment); this patient went on to achieve SVR. The RBV-free groups had lower rates of adverse events and laboratory abnormalities compared to the RBV-containing groups.
4.2 Study GS-US-337-0118 (LONESTAR)
Study GS-US-337-0118 (LONESTAR) was a phase 2, randomized open-label trial to evaluate LDV/SOF with or without RBV in patients with HCV genotype 1 infection [23]. The study was conducted at a single center in the United States and enrolled patients from Nov 2012 to Dec 2012 (Table 3).
In Cohort A, 60 non-cirrhotic, treatment-naive patients were randomly assigned (1:1:1; stratified by HCV genotype [1a vs 1b]) to receive LDV/SOF once daily for 8 weeks (Group 1), LDV/SOF and weight-based ribavirin for 8 weeks (Group 2), or LDV/SOF for 12 weeks (Group 3). In Cohort B, 40 patients with a history of virological failure after receiving a protease inhibitor regimen were randomly allocated (1:1; stratified by genotype and presence or absence of cirrhosis) to receive LDV/SOF for 12 weeks (Group 4) or LDV/SOF and weight-based ribavirin for 12 weeks (Group 5).
This study showed that 8 or 12 weeks of LDV/SOF with or without RBV in patients with genotype 1 HCV infection (including approximately 50% with compensated cirrhosis) resulted in an overall SVR12 rate of 97%. The SVR12 rate was 100% in treatment-naive patients who received LDV/SOF + RBV for 8 weeks and 95% in patients who received LDV/SOF for 8 or 12 weeks. In treatment-experienced patients, SVR12 was achieved by 100% and 95% of patients who received LDV/SOF with or without RBV for 12 weeks, respectively.
Patients who were receiving LDV/SOF + RBV had the higher rates of adverse events compared to those receiving LDV/SOF. The most common adverse events were nausea, anemia, upper respiratory tract infection, and headache, with most of these events assessed as mild in severity. Anemia was noted only in patients receiving RBV with eight patients requiring RBV dose reductions to manage anemia; all eight achieved SVR12. No patient discontinued treatment because of an adverse event. Four patients had serious adverse events, of which anemia was the only serious adverse event considered related to study treatment. The only grade 3 or 4 hematological abnormality that occurred during treatment was decreased hemoglobin in four patients, all of whom had received RBV.
4.3 Study GS-US-337-012 (ELECTRON-2)
Study GS-US-337-0122 was a phase 2 multicenter, open-label trial to evaluate the efficacy and safety of sofosbuvir-containing regimens for the treatment of chronic HCV infection [24]. This multi-cohort study was the first to evaluate the use of LDV/SOF in non-genotype 1 HCV infection (Table 4).
In Cohort 2 (Groups 3 and 4), 51 treatment-naive, HCV genotype 3 patients were randomly assigned to receive either LDV/SOF once daily for 12 weeks (Group 3) or LDV/SOF and weight-based ribavirin for 12 weeks (Group 4). Cohort 2 (Group 5) evaluated the safety and efficacy of LDV/SOF in 25 patients with genotype 6 HCV infection. Additional details in patients with HCV genotype 3 and 6 infection are presented in Sects. 6.2.2 and 6.2.5 respectively.
4.4 Safety of Ledipasvir/Sofosbuvir in Phase 2 Trials
Across the phase 2 clinical trials, treatment with LDV/SOF with or without RBV was safe and well tolerated. Importantly, a higher proportion of patients in the RBV-containing treatment groups had adverse events, treatment-related adverse events, or adverse events leading to dose modification or interruption of any study drug than patients in the RBV-free treatment groups. The groups receiving RBV also had a higher incidence of laboratory abnormalities that were consistent with the expected toxicity profile of ribavirin, namely, decreases in hemoglobin and lymphocytes and increases in total bilirubin.
5 Safety and Efficacy of Ledipasvir/Sofosbuvir in Phase 3 Registrational Trials
The LDV/SOF phase 3 clinical development program was designed to further evaluate the safety and efficacy of LDV/SOF in a diverse population of patients with HCV genotype 1 infection irrespective of baseline and demographic characteristics. In late 2012, there were several ongoing phase 3 studies in the SOF clinical development program; however, the standard of care in patients with HCV genotype 1 infection was still an HCV protease inhibitor (boceprevir or telaprevir) combined with Peg-IFN and RBV for 24–48 weeks [2, 3]. Based on the existing medical need and the safety data generated from over 1,000 patients treated with LDV in combination with other DAAs, the LDV/SOF phase 3 program was initiated. The phase 3 registrational trials had innovative study designs that helped to significantly accelerate the clinical development program. In the LDV/SOF phase 3 program supporting initial registration, three large multicenter studies (two in treatment naive and one in treatment experienced) were conducted and are described below.
5.1 Efficacy of Ledipasvir/Sofosbuvir in Treatment-Naive Genotype 1 Patients
5.1.1 ION-1 (Study GS-US-337-0102)
The ION-1 trial was designed to assess the efficacy and safety of 12 or 24 weeks of the fixed-dose combination of LDV/SOF with or without RBV in previously untreated patients with chronic HCV genotype 1 infection, including those with compensated cirrhosis [25].
This was a multicenter, randomized, open-label trial that enrolled patients at 99 sites in the United States and Europe from October 17, 2012, to May 17, 2013. Eligible patients had chronic HCV genotype 1 infection and had not received treatment for HCV infection previously. All patients received LDV/SOF. Ribavirin was administered orally twice daily, with the dose determined according to body weight (1,000 mg daily in patients with a body weight <75 kg and 1,200 mg daily in patients with a body weight ≥75 kg). Patients were randomly assigned in a 1:1:1:1 ratio to one of four treatment groups: LDV/SOF for 12 weeks, LDV/SOF plus RBV for 12 weeks, LDV/SOF for 24 weeks, or LDV/SOF plus RBV for 24 weeks. Randomization was stratified according to HCV genotype 1 subtype (1a or 1b) and the presence or absence of cirrhosis.
Subject enrollment occurred in two parts. Part A enrolled and randomized approximately 200 patients (50 per treatment group), and enrollment was halted in all four treatment groups once Part A was fully enrolled. After patients in 12-week treatment groups reached posttreatment Week 4, the data monitoring committee (DMC) reviewed safety and SVR4 efficacy data from the first 12 weeks of dosing for all patients in the 12-week treatment group. Futility in the 12-week treatment groups was assessed using an interim futility stopping procedure that utilized a conditional power approach under the observed trend. Stopping for futility was triggered when the conditional power was less than 5% (which was equivalent to an observed response rate of 60% or less). If the predefined interim futility criteria were met, the 12-week treatment groups were to be discontinued. As futility criteria were not met, the study was continued as planned. Part B commenced enrollment after this interim futility analysis was complete. Approximately 600 additional patients (approximately 150 per group) were enrolled in Part B.
In the final analysis, a total of 870 patients were randomized, of which 865 received at least 1 dose of study drug. Of the 865 randomized and treated patients, 27 (3.1%) prematurely discontinued study treatment. In general, patients were representative of a treatment-naive population, and demographics and baseline characteristics were generally balanced across the four treatment groups. Overall, 67% of the patients had HCV genotype 1a infection, 12% were black, 70% had the non-CC IL28B genotype, and 16% had cirrhosis.
The SVR12 rates observed in all four treatment groups were superior to the historical rate of 60% (P < 0.001 for all comparisons) (Table 5). The SVR12 rates were high across all treatment groups (LDV/SOF 12-week group, 99%; LDV/SOF + RBV 12-week group, 97%; LDV/SOF 24-week group, 98%; and LDV/SOF + RBV 24-week group, 99%). Of the 865 patients who were treated, only 3 had virologic failure (1 virologic breakthrough and 2 relapses). The addition of RBV or extending the treatment duration of LDV/SOF from 12 to 24 weeks did not significantly improve the SVR12 rates. High response rates were observed in all patient subgroups, including patients with characteristics historically associated with a poor response to treatment including older age, cirrhosis, high BMI, high HCV RNA levels, and IL-28B non-CC genotype.
Population and deep sequencing of the HCV NS5A and NS5B genes were performed from pretreatment samples and from posttreatment samples from all patients with virologic failure. The prevalence of pretreatment NS5A resistance-associated variants (RAVs) detected with a 1% cutoff was 16% (140/861) overall, of which 135 (96%) achieved SVR12, suggesting that the presence of NS5A RAVs did not impact treatment outcome. The two patients that relapsed had preexisting NS5A RAVs at baseline.
The innovative study design and built-in futility analysis of the ION-1 study were crucial in saving considerable amounts of time in the development program and bringing the drug to the market at a much earlier date than initially envisioned.
5.1.2 ION-3 (Study GS-US-337-0109)
The ION-3 trial was conducted primarily to explore the feasibility of shortening treatment duration in previously untreated patients with HCV genotype 1 infection without cirrhosis [26].
This was a multicenter, randomized, open-label trial that enrolled patients at 58 sites in the United States from May 20, 2013, to June 19, 2013. Eligible patients had chronic HCV genotype 1 infection without cirrhosis and had not received treatment for HCV infection previously. All patients received LDV/SOF with or without weight-based RBV (1,000 or 1,200 mg divided twice daily). Patients were randomly assigned in a 1:1:1 ratio to one of three treatment groups: LDV/SOF for 8 weeks, LDV/SOF plus RBV for 8 weeks or LDV/SOF for 12 weeks. Randomization was stratified according to HCV genotype (1a or 1b).
A total of 677 patients were randomized or which 8 (1.3%) prematurely discontinued study treatment. The population was representative of the population of patients with HCV infection in the United States. Demographic and baseline characteristics were generally balanced across the three treatment groups. Overall, 80% had HCV genotype 1a infection, 19% were black, 6% were Hispanic, and 74% had a non-CC IL28B genotype.
The SVR12 rates observed in all three treatment groups were superior to the adjusted historical rate of 60% (P < 0.001 for all comparisons) (Table 6). The SVR12 rate was 94% with 8 weeks of LDV/SOF, 93% with 8 weeks of LDV/SOF + RBV, and 95% with 12 weeks of LDV/SOF. Importantly, the SVR12 rate in patients who received 8 weeks of LDV/SOF without ribavirin was noninferior to the response rates in the other two treatment groups. This showed that the addition of RBV to the 8-week regimen of LDV/SOF or extending the treatment duration to 12 weeks in genotype 1, non-cirrhotic patients did not result in improved SVR rates. Furthermore, once again the SVR rates did not vary significantly according to patients’ demographic or clinical characteristics, including those historically associated with a poor response to IFN-based treatment.
Population and deep sequencing of the HCV NS5A and NS5B genes were performed from pretreatment samples and from posttreatment samples from all patients with virologic failure.
Overall, 116 of 647 (17.9%) patients were identified as having at least one baseline NS5A RAV with a 1% assay cutoff. Of these, 104 (89.7%) patients with baseline NS5A RAVs achieved SVR12 following treatment. Importantly of the 116 patients with baseline NS5A RAVs, 80 (69%) patients had at least 1 NS5A RAV conferring >100-fold reduced susceptibility to LDV in vitro. Despite the presence of these NS5A RAVs, 69 of these 80 (86.3%) patients achieved SVR12.
5.2 Efficacy of Ledipasvir/Sofosbuvir in Treatment-Experienced Genotype 1 Patients
5.2.1 ION-2 (Study GS-US-337-0108)
The ION-2 trial was designed to assess the efficacy and safety of 12 or 24 weeks of LDV/SOF with or without RBV in patients with chronic HCV genotype 1 infection who had been previously treated, including those with compensated cirrhosis [27].
This was a multicenter, randomized, open-label trial that enrolled patients at 64 sites in the United States from January 3, 2013, to February 26, 2013. Eligible patients had chronic HCV genotype 1 infection and had failed prior treatment with either Peg-IFN and RBV or an NS3/4A protease inhibitor combined with Peg-IFN and RBV. All patients received LDV/SOF with or without weight-based RBV (1,000 or 1,200 mg divided twice daily).
Patients were randomly assigned in a 1:1:1:1 ratio to one of four treatment groups: LDV/SOF for 12 weeks, LDV/SOF plus RBV for 12 weeks, LDV/SOF for 24 weeks, or LDV/SOF plus RBV for 24 weeks. Randomization was stratified according to genotype (1a vs 1b), presence or absence of cirrhosis, and response to prior therapy (relapse or virologic breakthrough vs no response.
A total of 441 patients were randomized, of which 440 received at least 1 dose of study drug. In general, patients were representative of a treatment-experienced population: 88% had the non-CC IL28B genotype, 52% had received prior treatment with a protease inhibitor regimen, and 20% had cirrhosis. Demographic and baseline characteristics were generally well balanced across the four treatment groups.
The SVR12 rates observed in all four treatment groups were superior to the adjusted historical rate of 25% (P < 0.001 for all comparisons) (Table 7). The SVR12 rates were high across all treatment groups (LDV/SOF 12-week group, 93.6%; LDV/SOF + RBV 12-week group, 96.4%; LDV/SOF 24-week group, 99.1%; and LDV/SOF + RBV 24-week group, 99.1%). The addition of RBV to or extending the treatment duration of LDV/SOF from 12 to 24 weeks did not appreciably enhance the observed SVR12 rates. High response rates were observed in all patient subgroups, including patients with characteristics historically associated with a poor response to treatment including older age, cirrhosis, high BMI, high HCV RNA levels, and IL-28B non-CC genotype.
A total of 62 of 439 (14.1%) patients with successful NS5A sequencing were identified as having baseline NS5A RAVs. Of these, 54 (87.1%) patients with baseline NS5A RAVs achieved SVR12. Variants associated with resistance to NS3/4A protease inhibitors were detected at baseline in 163 of the 228 patients (71%) who underwent successful sequencing and had received prior treatment with a protease inhibitor regimen. Of these 159 (98%) patients with baseline NS3/4A RAVs achieved SVR12.
5.3 Safety of Ledipasvir/Sofosbuvir in Phase 3 Registrational Trials
Treatment with LDV/SOF with or without RBV was safe and well tolerated across the phase 3 program [25,26,27]. There were no placebo-controlled regimens; however, the safety profile observed was generally similar to that observed in the placebo group of a prior trial with SOF and RBV in HCV-infected patients [28].
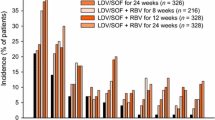
The integrated phase 3 safety population of 1,952 patients provided a large safety dataset to support the safety of LDV/SOF (Table 8). The most frequently reported adverse events were fatigue (29.3%), headache (23.1%), and nausea (13.5%), which were all reported more commonly in the patients receiving RBV-containing regimens. The majority of adverse events were mild, with <5% (91 patients) experiencing a grade 3 or 4 adverse event.
Thirteen (0.7%) patients receiving LDV/SOF with or without RBV had an adverse event leading to discontinuation of LDV/SOF. A total of 51 (2.6%) patients had at least 1 serious adverse event (SAE), with only 5 (0.3%) patients experiencing a treatment-related serious adverse event (anemia, factor VIII inhibition, mesenteric vein thrombosis, salpingitis, and headache). One patient died of liver failure secondary to HCV infection and alcohol use 121 days after treatment completion.
Importantly, the difference in the adverse event profile of RBV-free (LDV/SOF) and RBV-containing (LDV/SOF + RBV) treatment groups was also evaluated. The addition of RBV to the treatment regimen was associated with an increase in the total incidence of adverse events, treatment-related adverse events, and serious adverse events compared to patients receiving RBV-free regimens for all treatment durations. Consistent with the frequent need for RBV dose modification, a higher proportion of patients in the RBV-containing (LDV/SOF + RBV) treatment groups (13.5%) had AEs leading to dose modification or interruption of any study drug than patients in the RBV-free (LDV/SOF) treatment groups (0.6%).
In the integrated phase 3 safety population, approximately 75% of patients had at least one laboratory abnormality with the majority (66.8%) being only grade 1 or 2 laboratory abnormalities. The percentage of patients receiving LDV/SOF + RBV who had a grade 3 laboratory abnormality (11.4%) was approximately twofold higher than patients receiving LDV/SOF (5.4%). Few patients had grade 4 laboratory abnormalities. The groups receiving RBV had a higher incidence of laboratory abnormalities that are consistent with the expected toxicity profile of ribavirin, namely, decreases in hemoglobin and lymphocytes and increases in total bilirubin.
5.4 Summary of Phase 3 Data Supporting Initial Registration of Ledipasvir/Sofosbuvir
LDV/SOF (Harvoni®) was the first all-oral, single-tablet, IFN-free and RBV-free treatment approved for the vast majority of patients infected with HCV. Across the phase 3 registrational trials, treatment with LDV/SOF offered a short, simple, well-tolerated regimen with significantly shorter treatment durations without the need for response-guided treatment algorithms compared with up to 48 weeks of standard of care treatment algorithms.
The phase 3 program showed that 12 weeks of LDV/SOF was a highly effective treatment for patients with HCV genotype 1 infection across a broad range of demographic and baseline characteristics. LDV/SOF was the first Peg-IFN-free, RBV-free treatment to demonstrate SVR rates >90% in genotype 1 HCV-infected patients who had failed the current standard of care. In addition, factors that had been traditionally associated with relapse (e.g., age ≥65 years, black or African-American race, Hispanic or Latino ethnicity, high BMI, genotype 1a, high viral load, non-CC IL28B allele) had no notable impact on SVR12 rates nor did the presence of baseline LDV-associated NS5A RAVs in a subset of patients.
No additional benefit appeared to be associated with the addition of ribavirin or with extension of the duration of treatment to 24 weeks. In addition, the 8-week regimen of LDV/SOF was highly efficacious among non-cirrhotic genotype 1 patients who had not been treated previously. At the time of approval, emerging data indicated that in patients who experience relapse with SOF treatment, retreatment with LDV/SOF would be a viable option.
Treatment with LDV/SOF with or without RBV was generally well tolerated, with no treatment-emergent deaths and few permanent discontinuations of study drug due to AEs, SAEs, grade 3 or 4 AEs, or grade 3 or 4 laboratory abnormalities. Importantly, the exclusion of RBV significantly reduces the incidence of AEs and clinically significant laboratory abnormalities experienced by patients. It was observed that increasing treatment duration from 8 to 12 weeks resulted in small but consistent increases in the incidence of AEs but did not change the observed AE profile.
These data and others presented in Sect. 6 supported the approval of LDV/SOF (Harvoni®) in the United States on October 10, 2014, as the first all-oral, single-tablet, IFN-free and RBV-free treatment for HCV genotype 1, 4, 5, and 6 infection in patients with or without cirrhosis. By the end of 2017, Harvoni had been approved in over 80 countries in North and South America, Europe, Asia, Africa, and Australia.
6 Safety and Efficacy of Ledipasvir/Sofosbuvir in Other Patient Populations
The LDV/SOF clinical development program included additional pivotal phase 2 and 3 trials that were designed to assess the efficacy and safety of LDV/SOF in key patient populations with an unmet medical need. These included but were not limited to populations with non-genotype 1 HCV infection, HCV/HIV coinfection, decompensated liver disease, post-liver and kidney transplantation, and children.
6.1 LDV/SOF in Patients with Compensated Cirrhosis Who Failed Prior IFN-Based Treatment
Based on the data from the ION-2 study, a 24-week regimen of LDV/SOF was approved for the treatment of genotype 1 subjects with compensated cirrhosis who had failed prior IFN-based therapy [27]. In that study, 24 weeks of treatment with LDV/SOF ± RBV resulted in a numerically higher SVR rate (100%, 44/44) than 12 weeks of treatment with LDV/SOF ± RBV (84%, 37/44); although it was acknowledged that this difference in SVR was based on a small number of subjects, in the absence of additional data, a conservative duration of 24 weeks was recommended in the initial approval of LDV/SOF. The SIRIUS (GS-US-337-0121) study was conducted to evaluate the potential of shortening treatment duration in this population.
6.1.1 GS-US-337-0121 (SIRIUS)
GS-US-337-0121 was a double-blind, placebo-controlled trial conducted in France, in which 155 cirrhotic subjects were randomized 1:1 to one of two groups: Group 1 (n = 77), LDV/SOF once daily for 24 weeks + matched RBV placebo, or Group 2 (n = 78), deferred treatment group, matched LDV/SOF placebo once daily and matched RBV placebo (divided dose) for 12 weeks followed by LDV/SOF once daily and RBV for 12 weeks [29]. Randomization was stratified by HCV genotype and response to prior HCV therapy.
In this study, all subjects had prior virologic failure despite prior treatment, with Peg-IFN + RBV, or Peg-IFN + RBV and a protease inhibitor regimen. All subjects had cirrhosis with the exception of one subject randomized to the LDV/SOF + RBV 12-week group. The majority of subjects were male (73.5%) and white (97.4%) with non-CC IL28B alleles (93.5%).
Out of the 155 subjects randomized in this study, 1 subject discontinued treatment due to an AE while taking placebo. Of the remaining 154 subjects, a total of 149 achieved SVR12 across both treatment groups; 97.4% of subjects in the LDV/SOF 24-week group and 96.1% of subjects in the LDV/SOF + RBV 12-week group achieved SVR12 (Table 9). All five subjects who did not achieve SVR12 relapsed, and no subjects experienced on-treatment virologic failure.
Among the 30 subjects with NS5A RAVs at baseline, all 15 subjects (100%) treated with LDV/SOF + RBV achieved SVR12, while 13 of 15 subjects (86.7%) in the LDV/SOF 24-week group achieved SVR12. Both relapse subjects treated with LDV/SOF for 24 weeks had pretreatment NS5A RAVs that were maintained or enriched posttreatment.
LDV/SOF for 24 weeks and LDV/SOF + RBV for 12 weeks were both well tolerated with no subjects discontinuing treatment due to AEs. Comparing these two regimens overall, a higher frequency of AEs and treatment-related AEs were observed with LDV/SOF + RBV for 12 weeks compared with LDV/SOF for 24 weeks. This difference was attributable to a higher incidence in RBV-associated AEs such as pruritus and dyspnea. Importantly, when comparing the three 12-week treatment periods, similar percentages of subjects with any AE were observed during treatment with LDV/SOF (84.6%), placebo (81.8%), and LDV/SOF + RBV (86.8%) suggesting that there is a high background rate of symptoms in HCV-infected patients. Specifically, the only AEs reported more commonly (with an increase in frequency >10%) than placebo were headache and fatigue for LDV/SOF 12 week.
The comparable efficacy of LDV/SOF + RBV for 12 weeks and LDV/SOF for 24 weeks showed that a shorter course of LDV/SOF, when given with RBV, does not compromise the ability of treatment-experienced patients with cirrhosis to achieve SVR. This data led to the approval of LDV/SOF+ RBV for 12 weeks in previously treated adults with compensated cirrhosis who have failed on Peg-IFN ± ribavirin ± protease inhibitor on November 12, 2015.
6.2 LDV/SOF in Patients with Non-genotype 1 Infection
The most common HCV genotype in the United States and in Europe is genotype 1, while genotypes 2 and 3 HCV infection represent the majority of the remaining cases of chronic HCV infection in United States and in Europe. Genotype 4, 5, and 6 HCV infections are most prevalent in the Middle East, South Africa, and Southeast Asia, respectively [5, 30,31,32].
At initiation of many of the trials described in Sect. 6.1, there was no approved all-oral, IFN-free, RBV-free therapy for non-genotype 1 HCV infection. The only approved regimen for the treatment of non-genotype 1 HCV infection was SOF + RBV with or without Peg-IFN for 12–24 weeks [21, 31, 33]. While this combination resulted in high SVR rates >90%, there remained a need for simpler, better-tolerated RBV-free regimens given the significant toxicity, tolerability, and adherence issues associated with Peg-IFN and RBV. In patients in whom RBV was relatively or absolutely contraindicated (e.g., cardiac disease, sickle cell disease, thalassemia), there was a critical medical need for a RBV-free regimen. Furthermore, the in vitro activity of LDV across multiple genotypes provided the opportunity to conduct these studies.
6.2.1 Efficacy of LDV/SOF in Patients with Genotype 2 HCV Infection
The efficacy and safety of LDV/SOF in patients with HCV genotype 2 infection was evaluated in two pivotal studies, namely, GS-US-337-1468 and GS-US-337-1903.
6.2.1.1 GS-US-337-1468 (LEPTON)
Study GS-US-337-1468 was a phase 2 multicenter, open-label trial to evaluate the efficacy and safety of oral regimens for the treatment of HCV infection [34]. Patients were enrolled and treated at two sites in New Zealand from August 2014 through April 2015. In Cohort 2, Group 1, 26 patients with genotype 2 HCV infection received LDV/SOF (90/400 mg) once daily for 12 weeks.
Overall 68% of patients were male, 85% were white, and 77% were HCV treatment naive. Two patients had cirrhosis. A total of 25 patients (96.2%) achieved SVR12 (Table 10). No patients experienced on-treatment virologic failure or virologic relapse. The only patient who did not achieve SVR12 withdrew consent and prematurely discontinued from the study after receiving a single dose of LDV/SOF.
Pretreatment NS5A were detected in 16 patients (64%) using a 15% assay cutoff, with L31 M present in 13 of 16 patients. All patients with pretreatment NS5A RAVs achieved SVR12. The NS5B RAV M289I was detected in two genotype 2b patients using a 15% assay cutoff, and both patients achieved SVR12.
6.2.1.2 GS-US-337-1903
Study GS-US-337-1903 was a phase 3, randomized, multicenter, open-label trial conducted at 40 sites in Japan [35]. This was important in the context of genotype 2 infections accounting for 25–30% of HCV infections in Japan. A total of 239 patients were randomized 1:1 to receive either LDV/SOF 12 weeks or SOF + RBV 12 weeks (Cohort 1). Twenty-five patients who were ineligible for or intolerant of RBV therapy were treated with LDV/SOF for 12 weeks (Cohort 2).
The median age for the study population was 63 years (range 20–82), although in Cohort 2 the median age was 77 years with a range of 59–82 years. Overall, 34% of patients were treatment experienced, 21% had a non-CC IL28B genotype, and 14% had cirrhosis. In Cohort 1, SVR12 rates were 96% with LDV/SOF and 95% with SOF + RBV, thus achieving non-inferiority (Table 10). Among RBV intolerant/ineligible patients in Cohort 2, SVR12 was 96%.
Overall, 92% (118 of 129) of patients treated with LDV/SOF had pretreatment NS5A RAVs using a 15% assay cutoff. SVR12 was achieved in 114/118 (97%) of these patients. A total of six patients (5%) had baseline NS5B NI RAVs, of which one patient relapsed following 12 weeks treatment with LDV/SOF. The high rates of SVR12 in patients with pretreatment NS5A RAVs or NS5B NI RAVs suggest there is little utility of pretreatment resistance testing for patients with genotype 2 HCV infection.
6.2.2 LDV/SOF in Patients with Genotype 3 HCV Infection
Genotype 3 HCV infections account for approximately 20% of all HCV infections globally and 40% of infections in Asia [1, 32]. More recently, genotype 3 HCV infection has been associated with greater risk of steatosis, fibrosis progression, hepatocellular carcinoma, and all-cause mortality [36, 37]. The efficacy of LDV/SOF in patients with HCV genotype 3 infection was evaluated in two pivotal studies, namely, GS-US-337-0122 and GS-US-337-1701.
6.2.2.1 Study GS-US-337-0122 (ELECTRON-2)
The use of LDV/SOF in genotype 3 HCV infection was evaluated in ELECTRON-2, a phase 2 multicenter, open-label trial to evaluate the safety, tolerability, and antiviral efficacy of SOF-containing treatment regimens in HCV patients. In this study, Cohort 2 (Groups 3 and 4), 51 treatment-naive genotype 3 patients were enrolled to receive LDV/SOF once daily with or without weight-based RBV for 12 weeks; and 50 treatment-experienced patients received LDV/SOF for 12 weeks [24].
Overall, 84% of patients were white and 63% were male. The presence of cirrhosis was more common among treatment-experienced (44%) than treatment-naive (20%) patients. Among treatment-naive patients, the SVR12 results were higher in the LDV/SOF + RBV 12-week treatment group (100%; 26 of 26 patients) compared with the LDV/SOF 12-week treatment group (64%; 16 of 25 patients) (Table 10). Of the nine treatment-naive patients (36%) who did not achieve SVR12 in the LDV/SOF treatment group, eight relapsed and one patient discontinued study treatment. Of the 50 treatment-experienced patients with genotype 3 HCV receiving LDV/SOF + RBV, 41 (82%) achieved SVR12. In those with and without cirrhosis, the SVR12 was 73% and 89%, respectively. Of the nine treatment-experienced patients who did not achieve SVR12, one experienced virologic breakthrough and eight had virologic relapse.
Common NS5A RAVs detected at baseline included 30A/V/S/R/T. The RAVs L31M and Y93H were observed in only 1 (1%) and 8 (8%) of baseline samples, respectively. Of these only one patient with Y93H RAV at baseline experienced relapse.
6.2.2.2 Study GS-US-337-1701
Study GS-US-337-1701 was a phase 2 open-label trial of LDV/SOF with RBV in patients with genotype 3 HCV infection [38]. This study was conducted in Canada at 15 sites. A total of 111 patients received LDV/SOF + RBV for 12 weeks, of which 35.1% of patients had cirrhosis at screening.
The majority of patients had genotype 3a HCV infection (94.6%) and non-CC (CT or TT) IL28B alleles (62.2%), and 35.1% had cirrhosis. The study showed that LDV/SOF + RBV in treatment-naive patients with genotype 3 HCV infection resulted in 89.2% of patients achieving SVR12 (Table 10). Overall, 12 patients (10.8%) did not achieve SVR12. Of these, 8 patients (7.2%) relapsed, 3 patients (2.7%) were lost to follow-up, and 1 patient (0.9%) died. No patients had on-treatment virologic failure (i.e., breakthrough, rebound, or nonresponse). Among patients with cirrhosis, the SVR12 rates were lower (79.5%) compared with patients without cirrhosis (94.3%).
Baseline NS5A RAVs were detected in 15 of 106 patients (14.2%), of these 13 patients (86.7%) with baseline NS5A RAVs achieved SVR12. NS5B NI RAVs were detected in 10 of 104 patients (9.6%) with successful NS5B deep sequencing. All patients with baseline NS5B NI RAVs achieved SVR. A total of eight patients experienced viral relapse, of which two had Y93H at baseline (1.0% and 18% of viral population). Y93H was no longer detectable at virologic failure in both patients. Three other patients with Y93H at baseline achieved SVR12. No other NS5A RAVs or NS5B NI RAVs were detected in patients with relapse at baseline or virologic failure. A high percentage of patients achieved SVR12 regardless of the presence of NS5A RAVs at baseline, suggesting a minor impact of these on the treatment outcome. Furthermore, all patients with baseline NS5B NI RAVs achieved SVR12. This suggests that there is little utility of pretreatment resistance testing for patients with genotype 3 HCV infection considering LDV/SOF therapy.
6.2.3 LDV/SOF in Patients with Genotype 4 HCV Infection
Genotype 4 hepatitis C virus (HCV) accounts for an estimated 13% of patients with HCV globally. In several countries in sub-Saharan Africa and the Middle East, genotype 4 accounts for more than half of HCV infections [1, 39]. Historically, genotype 4 HCV has been considered difficult to treat because of its low rate of response to Peg-IFN and RBV [40]. At initiation of these trials, SOF and the protease inhibitor simeprevir in combination with Peg-IFN and RBV for 12 weeks were approved and had been shown to substantially improve SVR rates in patients with HCV genotype 4 [33]; however, due to the safety profile of Peg-IFN and RBV, there remained a need for IFN- and RBV-free therapy. The efficacy of LDV/SOF in patients with HCV genotype 4 infection was evaluated in two pivotal studies, namely, SYNERGY and GS-US-337-1119.
6.2.3.1 SYNERGY Trial
The SYNERGY trial (NCT01805882) was a single-center, open-label cohort, nonrandomized phase 2a trial in HCV-infected patients conducted in the United States [41]. In this study, 21 HCV genotype 4 patients were enrolled to receive LDV/SOF once daily for 12 weeks.
Overall 60% were treatment naive and 43% had advanced fibrosis. One patient took the first dose and then withdrew consent. Among the 20 patients who completed treatment, all achieved SVR12 (Table 10).
6.2.3.2 Study GS-US-337-1119
Study GS-US-337-1119 was a phase 2 multicenter, open-label trial to evaluate the efficacy and safety of LDV/SOF in patients with HCV genotype 4 or 5 infection conducted in France [42]. A total of 44 genotype 4 patients were enrolled to receive LDV/SOF once daily for 12 weeks.
The majority of patients were white (82%) or male (64%). Among treatment-experienced patients, 41% had cirrhosis, while only 5% of treatment-naive patients had cirrhosis. The SVR12 rate was 93.2% (41 of 44) for genotype 4 patients (Table 10). For genotype 4 patients, 21 (95.5%) treatment-naive and 20 (90.9%) treatment-experienced patients achieved SVR12. No patients had on-treatment virologic failure. Each of the three genotype 4 patients who did not achieve SVR12 relapsed.
Pretreatment NS5A RAVs were detected in all 44 patients (100%) with genotype 4 HCV infection. Of the 44 patients with genotype 4 HCV infection and NS5A RAVs, 41 (93%) achieved SVR12. Three of ten patients with genotype 4 HCV infection with triple NS5A RAVs pretreatment had virologic relapse, while all patients with genotype 4 HCV infection with double or single NS5A RAVs pretreatment achieved SVR12. A global prevalence study of NS5A RAVs across 454 patients with genotype 4 HCV infection showed that these specific triple RAVs associated with reduced susceptibility to LDV are found in less than 2.7% (12 of 454) of patients with genotype 4 HCV infection.
6.2.4 LDV/SOF in Genotype 5 HCV Infection
At initiation of study GS-US-337-1119, there was also no approved all-oral, IFN-free, RBV-free therapy for patients with genotype 5 HCV infection. For patients with genotype 5 HCV infection, the only treatment option approved was SOF + Peg-IFN + RBV for 12 weeks or SOF + RBV for 24 weeks in patients ineligible or intolerant to Peg-IFN [33]. Thus, there was an unmet medical need for IFN- and RBV-free treatment regimens, given the significant toxicity, tolerability, and adherence issues associated with these compounds.
6.2.4.1 Study GS-US-337-1119
The use of LDV/SOF in genotype 5 HCV infection was evaluated in study GS-US-337-1119, a phase 2 multicenter, open-label trial to evaluate the efficacy and safety of LDV/SOF in patients with HCV genotype 4 or 5 infection [43]. A total of 41 genotype 5 patients were enrolled to receive LDV/SOF once daily for 12 weeks.
All patients were white and 51% were male. Among treatment-experienced patients, 30% had cirrhosis, while only 13% of treatment-naive patients had cirrhosis. The SVR12 rate was 92.7% in genotype 5 patients (Table 10). Overall, 19 (90.5%) treatment-naive and 19 (95%) treatment-experienced patients achieved SVR12. No patients had on-treatment virologic failure. Two genotype 5 patients relapsed. One genotype 5 patient who did not achieve SVR12 had HCV RNA < LLOQ at their last on-treatment visit.
Baseline NS5A sequencing was successful and analyzed in 39 of 41 patients. Baseline NS5A RASs were observed in 4 of these 39 patients (10.3%). Following treatment with LDV/SOF for 12 weeks, SVR12 was achieved in three of four patients with baseline NS5A RASs.
6.2.5 LDV/SOF in Genotype 6 HCV Infection
Genotype 6 HCV constitutes about 1% of HCV infections globally and is found mainly in Southeast Asia and Southern China [1]. Genotype 6 HCV is genetically diverse, with 23 subtypes, many of which have not been cloned, limiting in vitro testing of antiviral agents. Due to its genetic diversity and relatively low prevalence, genotype 6 HCV infection was not as well characterized as the other genotypes, but long-term infection appears to be associated with the similar risk of cirrhosis and hepatocellular carcinoma as genotype 1 HCV infection.
6.2.5.1 GS-US-337-0122 (ELECTRON-2)
The use of LDV/SOF in genotype 6 HCV infection was evaluated in ELECTRON-2 (GS-US-337-0122), a phase 2 multicenter, open-label trial to evaluate the safety, tolerability, and antiviral efficacy of SOF-containing treatment regimens in HCV patients [24]. In this study, Cohort 2 (Group 5) evaluated the safety and efficacy of LDV/SOF in treatment-naive or treatment-experienced patients with genotype 6 HCV infection. A total of 25 patients were enrolled to receive LDV/SOF once daily for 12 weeks [24].
Overall, 84% of patients were Asian and 64% were male. Among treatment-naive and treatment-experienced patients with genotype 6 HCV infection, 24 patients (96%) achieved SVR12 (Table 10). One patient (4.0%), who had discontinued treatment after 8 weeks, relapsed and discontinued the study due to withdrawal of consent.
Importantly, baseline NS5A RAVs were observed in 23 (92%) patients with genotype 6 HCV infection. Following treatment with LDV/SOF for 12 weeks, SVR12 was achieved in 22 of 23 patients with NS5A RAVs. The one patient with NS5A RAVs who did not achieve SVR discontinued treatment early at 8 weeks.
6.2.6 Safety of LDV/SOF in Patients with Non-genotype 1 Infection
Treatment with LDV/SOF was generally safe and well tolerated, and the adverse event profile was similar across the different HCV genotypes. The safety profile associated with the use of LDV/SOF ± RBV in non-genotype 1 HCV infection did not differ, as expected, from the safety profile observed in patients with genotype 1 HCV infection with no new safety signal observed.
These studies supported the use of LDV/SOF (Harvoni®) for the treatment of patients with genotype 4, 5, or 6 HCV infection which was first approved in the United States on November 12, 2015. In addition, LDV/SOF has been approved for the treatment of genotype 2 or 3 HCV infection in certain regions including Canada and the European Union.
6.3 LDV/SOF in Patients with HCV/HIV Coinfection
Globally, it is estimated that 4 to 5 million persons are chronically infected with both human immunodeficiency virus type 1 (HIV-1) and HCV [44]. It has been shown that patients with HCV/HIV coinfection have higher rates of cirrhosis, hepatocellular carcinoma, and hepatic decompensation than patients with HCV monoinfection [45, 46]. However, uptake of HCV treatment in the IFN era was lower in the HCV-/HIV-coinfected population owing to historically lower SVR rates, patient comorbidities, patient and practitioner perceptions, high rates of treatment-related cytopenias, and complex interactions with concomitant antiretroviral drugs [47]. The first DAAs approved, namely, the NS3/4A protease inhibitors boceprevir and telaprevir, were not approved by the Food and Drug Administration for patients with HCV/HIV coinfection. The efficacy of LDV/SOF in patients with HCV/HIV coinfection was evaluated in two studies, namely, ERADICATE and GS-US-337-0115.
6.3.1 Study CO-US-337-0115 (ERADICATE)
The ERADICATE trial (NCT01878799) was a single-center, open-label cohort, phase 2b pilot study of previously untreated, non-cirrhotic patients with HCV genotype 1 and HIV coinfection conducted from June 2013 to September 2014 [48]. Eligible patients included those with HCV genotype 1 infection receiving antiretroviral therapy (ART) with HIV RNA values of 50 copies/mL or fewer and a CD4 T-lymphocyte count of ≥100 cells/mL or patients with untreated HIV infection with a CD4 T-lymphocyte count of ≥500 cells/mL.
Fifty patients with HCV/HIV coinfection were enrolled and received LDV/SOF once daily for 12 weeks. Of the 50 enrolled, 37 were receiving ART, and 13 were not receiving antiretroviral treatment. Patients were predominantly African-American (84%), men (74%), IL28B non-CC genotype (84%), and with genotype 1a infection (74%). Median baseline CD4 count was 576 cells/mm3 for patients receiving ART and 687 cells/mm3 for patients not receiving antiretroviral treatment.
Forty-nine of 50 participants (98%) achieved SVR12 and 1 patient experienced relapse (Table 11). Deep sequencing was carried out on one patient who experienced relapse, which showed enrichment of the Y93H mutation (NS5A RAV) that was present at baseline.
6.3.2 Study GS-US-337-0115 (ION-4)
Study GS-US-337-0115 (ION-4) was a phase 3, open-label, multicenter trial that assessed the antiviral efficacy, safety, and tolerability of LDV/SOF administered for 12 weeks in HCV treatment-naive and treatment-experienced (including treatment intolerant) patients with chronic genotype 1 or 4 HCV infection who were coinfected with HIV-1 [49].
This was a multicenter, randomized, open-label trial that enrolled patients between March 2014 and June 2014 and was conducted at 60 sites in the United States, Puerto Rico, Canada, and New Zealand. Eligible patients had chronic HCV genotype 1 or 4 and HIV-1 coinfection including those with compensated cirrhosis and/or prior treatment failure. On the basis of drug-interaction data in healthy volunteers that was available at study initiation, the antiretroviral drugs allowed in the study included emtricitabine and tenofovir disoproxil fumarate plus efavirenz, raltegravir, or rilpivirine. All patients received LDV/SOF for 12 weeks.
A total of 335 patients were enrolled and received at least 1 dose of study drug. In general, patients were representative of the HIV-infected population. Overall, 75% of the patients had HCV genotype 1a infection, 34% were black, 55% were treatment experienced, and 20% had cirrhosis.
Overall, 322 patients (96%) achieved SVR12 (Table 11). Of the 13 patients who did not achieve SVR, 10 patients relapsed, and 2 patients had on-treatment virologic failure (both in the setting of noncompliance). High SVR12 rates were observed in most subgroups, including patients who were treatment-experienced with cirrhosis (97.9%). High and similar SVR12 rates were also observed irrespective of ARV regimen. In this study, there were 13 treatment-experienced patients enrolled who had failed a SOF + RBV regimen. All 13 of these patients achieved SVR12 and are further described in Sect. 6.5.
Pretreatment NS5A and NS5B deep sequencing data was obtained for all 335 patients enrolled in study GS-US-337-0115 (ION-4). Baseline analyses of NS5A RAVs and NS5B NI RAVs were conducted with a 15% cutoff.
Of 325 patients, 34 (10.5%) had pretreatment NS5A RAVs, of which 31 (91.2%) achieved SVR12. The two patients who experienced on-treatment virologic failure had no NS5A RAVs at baseline and developed NS5A RAVs at the time of virologic failure. Four of the ten patients who experienced virologic relapse had pretreatment NS5A RAVs, and eight of the ten patients who relapsed had posttreatment NS5A RAVs.
6.3.3 Safety of LDV/SOF in Patients with HIV/HCV Coinfection
The use of LDV/SOF in HCV-/HIV-coinfected patients was safe and well tolerated with no discontinuations due to adverse events. The adverse event profile was similar to that observed in HCV-monoinfected patients. There were no clinically significant changes in CD4 cell counts or HIV RNA levels observed. In addition, no renal adverse event signal or trends suggestive of renal toxicity regardless of ARV regimen were identified with intensive renal laboratory monitoring. However, due to the elevated levels of TFV with TDF-containing regimens in the presence of LDV/SOF in patients who have preexisting renal disease, it is recommended that such patients are monitored according to TDF prescribing information.
These studies supported the supplemental indication in the United States for LDV/SOF (Harvoni®) for the treatment of coinfected patients, granted on November 12, 2015.
6.4 LDV/SOF in Patients Who Are Posttransplant or with Decompensated Liver Disease
Prior to 2012, for posttransplantation patients with compensated liver disease, recurrence of HCV infection following transplantation was essentially universal and was associated with poorer graft and patient survival compared with patients undergoing liver transplantation for other causes [50,51,52,53].
In patients with decompensated liver disease, the 1-year mortality for patients with Child-Pugh-Turcotte (CPT) B decompensated cirrhosis was approximately 20%, while the 1-year mortality for patients with CPT C decompensated cirrhosis was >50% [54].
When studies in these populations were initiated, there were no approved therapies for the treatment of HCV patients with decompensated liver disease. The poor adverse event profile of IFN-based regimens had limited their use in this sick patient population to specialized centers and clinical trials [52]. Instead, the mainstay of treatment in the United States for patients with decompensated liver disease due to HCV had been liver transplantation. Unfortunately, less than 5% of patients with decompensated liver disease due to HCV in the United States were listed in a given year for transplantation, and <2% received liver transplantations annually [55]. As such, posttransplantation patients with compensated liver disease as well as patients with decompensated liver disease regardless of transplantation status remained populations with a high unmet medical need for treatment. Two studies, namely, GS-US-337-0123 (SOLAR-1) and GS-US-337-0124 (SOLAR-2), were designed to determine the efficacy and safety of LDV/SOF in combination with ribavirin in patients with advanced liver disease including patients who have undergone liver transplantation.
6.4.1 Studies GS-US-337-0123 (SOLAR-1) and GS-US-337-0124 (SOLAR-2)
GS-US-337-0123 and GS-US-337-0124 were phase 2, multicenter, randomized, open-label trials that were conducted at 63 sites in the United States, Europe, Canada, Australia, and New Zealand with patients enrolled between September 2013 and August 2014 [56, 57].
These two studies were identical in study design, including eligibility criteria. A total of 670 patients were enrolled in two cohorts. Cohort A consisted of two groups of patients with advanced cirrhosis Child-Pugh class B and C who had not undergone liver transplantation (Groups 1 and 2, respectively). Cohort B consisted of five groups of patients, all of whom had undergone liver transplantation previously (Group 3, non-cirrhotic; Group 4, compensated cirrhosis (CPT-1); Group 5, Child-Pugh class B; Group 6, Child-Pugh class C; and Group 7, fibrosing cholestatic hepatitis). Patients in each of the seven groups were randomized in a 1:1 ratio to receive either 12 or 24 weeks of LDV/SOF once daily plus RBV. Groups 3, 4, and 7 received weight-based RBV (1,000 mg/day in patients with a body weight of <75 kg and 1,200 mg/day in patients with a body weight ≥75 kg), while in groups 1, 2, 5, and 6, RBV was administered at a starting dose of 600 mg in a divided daily dose and titrated upward as tolerated.
A total of 455 of 670 patients (67.9%) were posttransplantation, while 329 patients (49.1%) had decompensated cirrhosis, regardless of transplantation status. Of these, 78 patients (23.7%) had a MELD score >15. Across all groups, the majority of patients were male (76.9%) and white (91.5%) with a mean age of 58 years (range, 21–81) and a mean (SD) BMI of 27.9 (4.96) kg/m2.
The SVR12 and relapse rates presented below (Table 12) are from pooled analysis of both studies. Overall, 92.7% (569 of 614) patients with genotype 1 HCV infection and 82.5% (33 of 40) patients with genotype 4 HCV infection achieved SVR12. There were 13 patients (12 genotype 1 HCV infection and 1 genotype 4 HCV infection) who were transplanted prior to their posttreatment Week 12 visit and were excluded from the analysis. Overall, a small number of patients relapsed: 20 of 589 patients (3.4%) with genotype 1 HCV infection and 3 of 36 patients (8.3%) with genotype 4 HCV infection relapsed. In decompensated patients with genotype 1 HCV infection, irrespective of transplantation status, the relapse rates were 8.1% and 4.3% in patients who received LDV/SOF + RBV for 12 or 24 weeks, respectively, resulting in a numerical difference in relapse rates of 3.8% which was not clinically significant (95% CI, −2.1 to 10.2%).
Pretreatment resistance analysis was performed for 622 patients who received LDV/SOF + RBV with NS5A sequencing (587 patients with genotype 1 infection and 35 patients with genotype 4 infection) and for 619 patients with NS5B sequences (586 patients with genotype 1 infection and 33 patients with genotype 4 infection). For patients with genotype 1 infection treated for 12 weeks, the SVR rates were 89.4% versus 96.4% in patients with or without NS5A RAVs (1% cutoff), respectively. Among post-liver transplantation patients with compensated liver disease (Groups 3, 4, and 7), the presence of pretreatment NS5A RAVs had minimal, if any, impact on relapse. Among 586 patients with genotype 1 and full-length NS5B sequence, 28 had NS5B NI RAVs (4.8%), of which 27 achieved SVR12 (96.4%; 27 of 28). The lack of significant associations between the presence of either NS5A or NS5B pretreatment RAVs with SVR rates in patients with genotype 1 HCV infection who are posttransplant or have decompensated cirrhosis is consistent with the results of virologic analyses for patients with genotype 1 HCV infection and compensated disease. For genotype 4 patients, there were no patients without RAVs at 1% cutoff.
In this population, it was important to also understand the effects of successful treatment on hepatic outcomes such as CPT and MELD scores. Among the patients who had CPT C at baseline, 61.3% improved to CPT B at posttreatment Week 12, and of the patients who had CPT B at baseline, 33.1% improved to CPT A by posttreatment Week 12. These improvements in CPT score were driven largely by improvements in albumin and bilirubin (64% and 43.6%). Among patients with MELD scores ≥15 at baseline, 63.2% had a MELD score <15 at posttreatment Week 12. Conversely, among patients with MELD scores <15 at baseline, 5.8% had a MELD score ≥15 at posttreatment Week 12. Overall improvements in CPT and MELD scores were observed in the majority of patients who achieved SVR12 (66.9% and 59.8%, respectively) suggesting short-term clinical improvements with HCV eradication.
6.4.2 Safety of LDV/SOF in Patients Who Are Posttransplant or with Decompensated Liver Disease
As expected in a patient population with decompensated liver disease and/or patients who were post-liver transplantation, high percentages of patients experienced AEs, Grade 3 or 4 AEs, and serious adverse events were observed. However, few patients (3.0%) experienced treatment-related SAEs or adverse events that led to discontinuation of LDV/SOF. Twenty treatment-emergent deaths were reported; none were considered related to LDV/SOF. The most commonly reported AEs were fatigue (42.5%), anemia (33.6%), and headache (27.3%). Longer treatment with LDV/SOF + RBV for 24 weeks compared with 12 weeks was not associated with an increased safety burden. Additional analyses demonstrated a similar AE profile among patients with decompensated cirrhosis, regardless of transplantation status.
For posttransplantation patients with compensated liver disease, treatment with LDV/SOF + RBV was safe and well tolerated. None of the four treatment-emergent deaths (multifocal leukoencephalitis, myocardial infarction, infection [food poisoning/pneumonia], and graft rejection) were considered related to LDV/SOF. The most clinically relevant safety finding was anemia, a known effect of RBV therapy which was likely exacerbated by preexisting disease. Hemoglobin reductions were appropriately addressed through monitoring and toxicity management. Furthermore, the decreased hemoglobin levels observed during LDV/SOF + RBV treatment resolved in nearly all patients by posttreatment Week 4, demonstrating the reversibility of the anemia following RBV discontinuation. Despite the lack of drug-drug interactions between LDV/SOF + RBV and common immunosuppressants, it was observed that a common reason for adjustment in the dose or frequency of administration of immunosuppressive agents was improved hepatic function, likely as the result of the suppression of HCV viremia.
For patients with decompensated liver disease, regardless of transplantation status, treatment with LDV/SOF + RBV was also safe and tolerable. All 16 treatment-emergent deaths that occurred were associated with the clinical progression of end-stage liver disease, in some cases potentially exacerbated by immunosuppression (i.e., sepsis, septic shock, multi-organ failure); none were considered to be drug related. Similar to posttransplantation patients with compensated liver disease, anemia was the most clinically relevant safety finding.
The data above led to the approval of LDV/SOF in patients who are posttransplant or with decompensated liver disease on February 12, 2016.
6.5 LDV/SOF in Adolescent Patients
The prevalence of HCV in children varies globally, with estimates of 0.05–0.36% in the United States and Europe and up to 5.8% in regions of Africa [58, 59]. Despite the overall more favorable prognosis compared to adults, approximately 4–6% of children with chronic HCV infection have evidence of advanced fibrosis or cirrhosis, and some children eventually require liver transplantation for end-stage liver disease as a consequence of HCV infection [60, 61].
While there was a transformation in the treatment of HCV infection with the development of DAAs in adults, the standard of care in adolescent patients (12 to <18 years old) was IFN or Peg-IFN with weight-based RBV. Patient acceptance was very low given the requirement for subcutaneous injections for Peg-IFN and the substantial adverse events associated with therapy, including concerns for growth and development in this age group [62]. As such there was a need to address this unmet medical need in the pediatric population.
6.5.1 Study GS-US-337-1116
Study GS-US-337-1116 was a phase 2, multicenter, open-label trial conducted at 24 sites in Europe and the United States, United Kingdom, and Australia from November 2014 to October 2015 [15]. Eligible patients were 12 to <18 years old and had chronic infection with HCV genotype 1.
A total of 100 patients were enrolled and received LDV/SOF once daily for 12 weeks. The median age of patients was 15 years, and the majority were HCV treatment naive (80%); 84% were infected through perinatal transmission. In this study, 63% of patients were female, 90% were white, 76% had a non-CC IL-28B genotype, and 81% had HCV genotype 1a infection. Treatment with LDV/SOF for 12 weeks resulted in a high SVR12 rate of 97% (Table 13). This rate was similar to the SVR12 rates observed in adult patients treated with LDV/SOF in other clinical studies. Of note, the only patient with known cirrhosis achieved SVR12. A total of three patients (3.0%) did not achieve SVR12 and had “other” virologic outcome (due to reasons such as lost to follow-up). No patients experienced virologic failure.
Virologic analyses were performed for the 97 patients who had a posttreatment virologic outcome. NS5A RAVs were detected at baseline in 8.2% and 5.2% of patients with a 1% and 15% detection assay cutoff, respectively. The presence of NS5A and NS5B RAVs did not impact treatment outcome; all patients with RAVs achieved SVR12.
6.5.2 Safety of LDV/SOF in Adolescent Patients
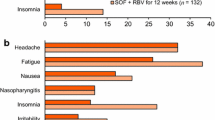
Treatment with LDV/SOF was safe and well tolerated in HCV-infected adolescents, and no new safety signal was detected. The most commonly reported adverse events were headache (27% of patients), diarrhea (14%), and fatigue (13%). No patient experienced serious adverse events or discontinued treatment because of an adverse event. All adverse events were mild or moderate in intensity; no patient experienced grade 3 or 4 adverse events. Most laboratory abnormalities were mild in severity.
In addition, effects on short-term development and growth were evaluated. No clinically relevant effects on development as assessed by changes from baseline in Tanner pubertal stages or growth as assessed by changes from baseline in body height, body height percentiles, body weight, or body weight percentiles to posttreatment were observed. In addition, no clinically relevant changes from baseline in BMI or BMI percentiles were observed.
Treatment with LDV/SOF demonstrated a favorable safety profile, comparable PK exposure, and high efficacy in adolescent patients 12 to <18 years of age. The safety profile of LDV/SOF in adolescents was consistent with that observed for adults treated with LDV/SOF in previous studies in adults ≥18 years of age. The data above led to the approval of LDV/SOF in adolescents 12 to <18 years of age on April 7, 2017.
6.6 LDV/SOF in Retreatment of Patients Who Failed Prior SOF Regimens
At the time these studies were developed, there was no approved therapy for patients with HCV infection who had failed a SOF + RBV ± Peg-IFN regimen. This was a growing problem due to the extensive number of patients being treated with these regimens at that time. The available evidence suggested that the primary mode of resistance to SOF is the development of the S282 T mutation. This mutation develops rarely and is rapidly overgrown by wild-type virus, suggesting that patients who have failed SOF can be retreated with a SOF-containing regimen.
Overall, 90 patients who had previously failed on a SOF-based treatment regimen received at least 1 dose of LDV/SOF in the clinical studies described in Table 14 [49, 63, 64]. Among the patients, with or without cirrhosis, who had previously failed a SOF-containing regimen and were treated with LDV/SOF with or without RBV for 12 weeks across these four clinical studies, 56 had failed SOF + RBV, 25 had failed SOF + Peg-IFN + RBV, 8 had failed a prior LDV/SOF + RBV 6-week regimen, and 1 had failed a prior SOF + GS-9669 + RBV 12-week regimen. SVR12 was achieved in all (100%) patients, irrespective of whether these treatment-experienced patients received 12 weeks of LDV/SOF or LDV/SOF + RBV. This rate was comparable with the rates observed in prior Peg-IFN + RBV ± PI failures in the ION studies, where 100% of patients achieved SVR12.
The presence of pretreatment NS3 RAVs, NS5A RAVs, or NS5B NI RAVs had no clinical impact on whether a patient with genotype 1 infection achieved SVR12 as all patients achieved SVR12. The single patient who relapsed was shown to have genotype 3a infection. The safety profile for patients who had previously failed on a SOF-based treatment was consistent with the expected safety profile of LDV/SOF in the previous phase 3 studies. This data led to the approval of LDV/SOF in previously treated adults who have failed on sofosbuvir + ribavirin ± Peg-IFN on April 23, 2017.
6.7 LDV/SOF in Other Key Populations
The clinical development program of LDV/SOF has generated safety and efficacy data in over 5,900 HCV-infected patients from phase 2 and 3 trials through late 2017. This comprehensive program has included studies in special patient populations [22, 65,66,67,68,69,70,71] as well as global, regional, and local studies to support the registration of LDV/SOF worldwide [72,73,74,75,76,77]. Additional trials in key populations are summarized in Table 15.
7 Conclusion
The development of LDV/SOF (Harvoni®) revolutionized the treatment and management of HCV-infected patients globally. The once-daily, single-tablet regimen of LDV/SOF has been shown to be a highly effective, safe, and tolerable treatment option for patients with chronic HCV across a broad range of characterictics and situations. The pace of the initial clinical development program was unprecedented in its speed due to the widespread recognition from patients, providers, and regulators of the unmet medical need for a safe, simple, and effective all-oral treatment for HCV. In addition a significant number of clinical trials have been conducted in the development program since the first approval of LDV/SOF, with consistent results showing that LDV/SOF is safe and effective across unique populations. This has set a new standard for inclusion of vulnerable groups and special populations in clinical trials in a more timely and comprehensive fashion.
Abbreviations
- DAAs:
-
Direct-acting antivirals
- FDC:
-
Fixed-dose combination
- HCV:
-
Hepatitis C virus
- HIV-1:
-
Human immunodeficiency virus type 1
- IFN:
-
Interferon
- LDV:
-
Ledipasvir
- Peg-IFN:
-
Pegylated interferon
- RBV:
-
Ribavirin
- SOF:
-
Sofosbuvir
- SVR:
-
Sustained virologic response
References
Gower E et al (2014) Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol 61(1 Suppl):S45–S57
Jacobson IM et al (2011) Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 364(25):2405–2416
Bacon BR et al (2011) Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 364(13):1207–1217
Chen EY et al (2013) A small percentage of patients with hepatitis C receive triple therapy with boceprevir or telaprevir. Clin Gastroenterol Hepatol 11(8):1014–20.e1–2
Alter MJ et al (1999) The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N Engl J Med 341(8):556–562
Sovaldi (sofosbuvir) tablets: US prescribing information (2013) Gilead Sciences, Foster City. http://www.gilead.com/~/media/Files/pdfs/medicines/liver-disease/sovaldi/sovaldi_pi.pdf
Kirby BJ et al (2015) Pharmacokinetic, pharmacodynamic, and drug-interaction profile of the hepatitis C virus NS5B polymerase inhibitor sofosbuvir. Clin Pharmacokinet 54(7):677–690
German P, Yang J, West S et al (2014) Effect of food and acid reducing agents on the relative bioavailability and pharmacokinetics of ledipasvir/sofosbuvir fixed-dose combination tablet. Presentation at the 15th international workshop on clinical pharmacology of HIV and hepatitis therapy, Washington, 19–21 May 2014
Lawitz E, Rodríguez-Torres M, Cornpropst MT et al (2012) The effect of hepatic impairment on the pharmacokinetics and antiviral activity of PSI-7977 in hepatitis C infected subjects treated for seven days. J Hepatol 56(suppl 2):S445–S446
Cornpropst M, Denning J, Clemons D et al (2012) The effect of renal impairment and end stage renal disease on the single-dose pharmacokinetics of GS-7977. Presented at 47th annual meeting of the European Association for the Study of the Liver, Barcelona
Gane E, Robson RA, Bonacini M et al (2014) Safety, antiviral efficacy, and pharmacokinetics of sofosbuvir in patients with severe renal impairment. Presented at 65th annual meeting of the American Association for the Study of Liver Diseases, Boston
Denning J et al (2013) Pharmacokinetics, safety, and tolerability of GS-9851, a nucleotide analog polymerase inhibitor for hepatitis C virus, following single ascending doses in healthy subjects. Antimicrob Agents Chemother 57(3):1201–1208
Lawitz E et al (2013) Pharmacokinetics, pharmacodynamics, and tolerability of GS-9851, a nucleotide analog polymerase inhibitor, following multiple ascending doses in patients with chronic hepatitis C infection. Antimicrob Agents Chemother 57(3):1209–1217
Harvoni (ledipasvir/sofosbuvir) tablets: US prescribing information (2014) Gilead Sciences, Foster City. http://www.gilead.com/~/media/Files/pdfs/medicines/liver-disease/harvoni/harvoni_pi.pdf
Balistreri WF et al (2017) The safety and effectiveness of ledipasvir-sofosbuvir in adolescents 12-17 years old with hepatitis C virus genotype 1 infection. Hepatology 66(2):371–378
Lawitz E, Lalezari J, Rodriguez-Torres M et al (2010) High rapid virologic response (RVR) with PSI-7977 QD plus PEG-IFN/RBV in a 28-day phase 2a trial. Hepatology 52(suppl):706
Lawitz E et al (2013) Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double-blind, phase 2 trial. Lancet Infect Dis 13(5):401–408
Lawitz EJ et al (2012) A phase 1, randomized, placebo-controlled, 3-day, dose-ranging study of GS-5885, an NS5A inhibitor, in patients with genotype 1 hepatitis C. J Hepatol 57(1):24–31
Kowdley KV et al (2013) Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet 381(9883):2100–2107
Gane EJ et al (2013) Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 368(1):34–44
Gane EJ et al (2014) Efficacy of nucleotide polymerase inhibitor sofosbuvir plus the NS5A inhibitor ledipasvir or the NS5B non-nucleoside inhibitor GS-9669 against HCV genotype 1 infection. Gastroenterology 146(3):736–743.e1
Stedman CA et al (2016) Once daily ledipasvir/sofosbuvir fixed-dose combination with ribavirin in patients with inherited bleeding disorders and hepatitis C genotype 1 infection. Haemophilia 22:214–217
Lawitz E et al (2014) Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet 383(9916):515–523
Gane EJ et al (2015) Efficacy of ledipasvir and sofosbuvir, with or without ribavirin, for 12 weeks in patients with HCV genotype 3 or 6 infection. Gastroenterology 149(6):1454–1461.e1
Afdhal N et al (2014) Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 370(20):1889–1898
Kowdley KV et al (2014) Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N Engl J Med 370(20):1879–1888
Afdhal N et al (2014) Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med 370(16):1483–1493
Jacobson IM et al (2013) Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med 368(20):1867–1877
Bourliere M et al (2015) Ledipasvir-sofosbuvir with or without ribavirin to treat patients with HCV genotype 1 infection and cirrhosis non-responsive to previous protease-inhibitor therapy: a randomised, double-blind, phase 2 trial (SIRIUS). Lancet Infect Dis 15(4):397–404
Armstrong GL et al (2006) The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 144(10):705–714
Nguyen MH, Keeffe EB (2005) Prevalence and treatment of hepatitis C virus genotypes 4, 5, and 6. Clin Gastroenterol Hepatol 3(10 Suppl 2):S97–S101
Fattovich G et al (2001) Hepatitis C virus genotypes: distribution and clinical significance in patients with cirrhosis type C seen at tertiary referral centres in Europe. J Viral Hepat 8(3):206–216
Lawitz E et al (2013) Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 368(20):1878–1887
Gane EJ et al (2017) Efficacy of ledipasvir plus sofosbuvir for 8 or 12 weeks in patients with hepatitis C virus genotype 2 infection. Gastroenterology 152(6):1366–1371
Asahina Y et al (2018) Ledipasvir-sofosbuvir for treating Japanese patients with chronic hepatitis C virus genotype 2 infection. Liver Int. 3 Jan 2018. https://doi.org/10.1111/liv.13685
Bochud PY et al (2009) Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol 51(4):655–666
van der Meer AJ et al (2012) Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. JAMA 308(24):2584–2593
Feld JJ et al (2017) Ledipasvir-sofosbuvir plus ribavirin in treatment-naive patients with hepatitis C virus genotype 3 infection: an open-label study. Clin Infect Dis 65(1):13–19
Breban R et al (2013) Towards realistic estimates of HCV incidence in Egypt. J Viral Hepat 20(4):294–296
Roulot D et al (2007) Epidemiological characteristics and response to peginterferon plus ribavirin treatment of hepatitis C virus genotype 4 infection. J Viral Hepat 14(7):460–467
Kohli A et al (2015) Ledipasvir and sofosbuvir for hepatitis C genotype 4: a proof-of-concept, single-centre, open-label phase 2a cohort study. Lancet Infect Dis 15(9):1049–1054
Abergel A et al (2016) Ledipasvir plus sofosbuvir for 12 weeks in patients with hepatitis C genotype 4 infection. Hepatology 64(4):1049–1056
Abergel A et al (2016) Ledipasvir-sofosbuvir in patients with hepatitis C virus genotype 5 infection: an open-label, multicentre, single-arm, phase 2 study. Lancet Infect Dis 16(4):459–464
Alter MJ (2006) Epidemiology of viral hepatitis and HIV co-infection. J Hepatol 44(1 Suppl):S6–S9
Lo Re 3rd V et al (2014) Hepatic decompensation in antiretroviral-treated patients co-infected with HIV and hepatitis C virus compared with hepatitis C virus-monoinfected patients: a cohort study. Ann Intern Med 160(6):369–379
Thomas DL (2008) The challenge of hepatitis C in the HIV-infected person. Annu Rev Med 59:473–485
Chung RT et al (2004) Peginterferon Alfa-2a plus ribavirin versus interferon alfa-2a plus ribavirin for chronic hepatitis C in HIV-coinfected persons. N Engl J Med 351(5):451–459
Osinusi A et al (2015) Virologic response following combined ledipasvir and sofosbuvir administration in patients with HCV genotype 1 and HIV co-infection. JAMA 313(12):1232–1239
Naggie S et al (2015) Ledipasvir and sofosbuvir for HCV in patients coinfected with HIV-1. N Engl J Med 373(8):705–713
Berenguer M (2002) Natural history of recurrent hepatitis C. Liver Transpl 8(10 Suppl 1):S14–S18
Malkan G et al (2001) Natural history of recurrent hepatitis C after liver transplantation. Transplant Proc 33(1–2):1468
Navasa M, Forns X (2007) Antiviral therapy in HCV decompensated cirrhosis: to treat or not to treat? J Hepatol 46(2):185–188
Wright TL et al (1992) Recurrent and acquired hepatitis C viral infection in liver transplant recipients. Gastroenterology 103(1):317–322
D’Amico G, Garcia-Tsao G, Pagliaro L (2006) Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol 44(1):217–231
Kim WR et al (2015) OPTN/SRTR 2013 annual data report: liver. Am J Transplant 15(Suppl 2):1–28
Charlton M et al (2015) Ledipasvir and sofosbuvir plus ribavirin for treatment of HCV infection in patients with advanced liver disease. Gastroenterology 149(3):649–659
Manns M et al (2016) Ledipasvir and sofosbuvir plus ribavirin in patients with genotype 1 or 4 hepatitis C virus infection and advanced liver disease: a multicentre, open-label, randomised, phase 2 trial. Lancet Infect Dis 16(6):685–697
El-Shabrawi MH, Kamal NM (2013) Burden of pediatric hepatitis C. World J Gastroenterol 19(44):7880–7888
Suryaprasad AG et al (2014) Emerging epidemic of hepatitis C virus infections among young nonurban persons who inject drugs in the United States, 2006-2012. Clin Infect Dis 59(10):1411–1419
Barshes NR et al (2006) The natural history of hepatitis C virus in pediatric liver transplant recipients. Liver Transpl 12(7):1119–1123
Bortolotti F et al (2008) Long-term course of chronic hepatitis C in children: from viral clearance to end-stage liver disease. Gastroenterology 134(7):1900–1907
Wirth S (2012) Current treatment options and response rates in children with chronic hepatitis C. World J Gastroenterol 18(2):99–104
Osinusi A et al (2014) Re-treatment of chronic hepatitis C virus genotype 1 infection after relapse: an open-label pilot study. Ann Intern Med 161(9):634–638
Wyles D et al (2015) Ledipasvir-sofosbuvir plus ribavirin for patients with genotype 1 hepatitis C virus previously treated in clinical trials of sofosbuvir regimens. Hepatology 61(6):1793–1797
Moon J et al (2017) Efficacy and safety of ledipasvir/sofosbuvir for the treatment of chronic hepatitis C in persons with sickle cell disease. Clin Infect Dis 65(5):864–866
Colombo M et al (2017) Treatment with ledipasvir-sofosbuvir for 12 or 24 weeks in kidney transplant recipients with chronic hepatitis C virus genotype 1 or 4 infection: a randomized trial. Ann Intern Med 166(2):109–117
Levitsky J et al (2016) Perioperative ledipasvir-sofosbuvir for HCV in liver-transplant recipients. N Engl J Med 375(21):2106–2108
Liu CJ et al (2018) Efficacy of ledipasvir and sofosbuvir treatment of HCV infection in patients coinfected with HBV. Gastroenterology 154(4):989–997
Deterding K et al (2017) Ledipasvir plus sofosbuvir fixed-dose combination for 6 weeks in patients with acute hepatitis C virus genotype 1 monoinfection (HepNet Acute HCV IV): an open-label, single-arm, phase 2 study. Lancet Infect Dis 17(2):215–222
Rockstroh JK et al (2017) Ledipasvir-sofosbuvir for 6 weeks to treat acute hepatitis C virus genotype 1 or 4 infection in patients with HIV coinfection: an open-label, single-arm trial. Lancet Gastroenterol Hepatol 2(5):347–353
Lawitz E, Landis CS, Maliakkal BJ et al (2017) Safety and efficacy of treatment with once-daily ledipasvir/sofosbuvir (90/400 mg) for 12 weeks in genotype 1 HCV-infected patients with severe renal impairment [Abstract 1587]. In: The Liver Meeting® 2017 – the 68th annual meeting of the American Association for the Study of Liver Diseases (AASLD), Washington, 20–24 Oct 2017
Mizokami M et al (2015) Ledipasvir and sofosbuvir fixed-dose combination with and without ribavirin for 12 weeks in treatment-naive and previously treated Japanese patients with genotype 1 hepatitis C: an open-label, randomised, phase 3 trial. Lancet Infect Dis 15(6):645–653
Lim YS et al (2016) A phase IIIb study of ledipasvir/sofosbuvir fixed-dose combination tablet in treatment-naive and treatment-experienced Korean patients chronically infected with genotype 1 hepatitis C virus. Hepatol Int 10(6):947–955
Chuang WL et al (2016) Ledipasvir/sofosbuvir fixed-dose combination tablet in Taiwanese patients with chronic genotype 1 hepatitis C virus. J Gastroenterol Hepatol 31(7):1323–1329
Wei L, Xie Q, Hou JL et al (2017) Safety and efficacy of ledipasvir/sofosbuvir in a genotype 1 HCV infected Chinese population: results from a phase 3 clinical trial. In: The Liver Meeting® 2017 – the 68th annual meeting of the American Association for the Study of Liver Diseases (AASLD), Washington, 20–24 Oct 2017
Shiha G, Waked I, Soliman R et al (2017) Ledipasvir/sofosbuvir for 8 or 12 weeks with or without ribavirin in HCV genotype 4 patients in Egypt [Abstract OP158]. In: Asian Pacific Association for the Study of the Liver (APASL), Shanghai, 15–19 Feb 2017
Isakov V et al (2018) Ledipasvir-sofosbuvir for 8 weeks in non-cirrhotic patients with previously untreated genotype 1 HCV infection +/− HIV-1 co-infection. Clin Drug Investig 38:239–247
Acknowledgements
The authors would like to thank the patients and their families as well as study site staff who participated in the clinical trials of Sovaldi and Harvoni.
Compliance with Ethical Standards
Conflict of Interest Anu Osinusi and John G. McHutchison are employees of Gilead Sciences, Inc.
Ethical Approval
All procedures performed in the studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Osinusi, A., McHutchison, J.G. (2019). The Clinical Development of Ledipasvir/Sofosbuvir (LDV/SOF, Harvoni®). In: Sofia, M. (eds) HCV: The Journey from Discovery to a Cure. Topics in Medicinal Chemistry, vol 32. Springer, Cham. https://doi.org/10.1007/7355_2018_48
Download citation
DOI: https://doi.org/10.1007/7355_2018_48
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-28399-5
Online ISBN: 978-3-030-28400-8
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)