
Abstract
Adoptive T cell therapy (ACT) is a safe and effective personalized cancer immunotherapy that can comprise naturally occurring ex vivo expanded cells (e.g., tumor-infiltrating lymphocytes [TIL]) or T cells genetically engineered to confer antigen specificity (T-cell receptor [TCR] or chimeric antigen receptor [CAR] engineered T cells) to mediate cancer rejection. In recent years, some ACTs have produced unprecedented breakthrough responses: TIL therapy has moved from melanoma to solid tumor applications, TCR-engineered cells are developed for hematologic and solid tumors, and CAR-engineered T cells have received Food and Drug Administration (FDA) approval for the treatment of patients with certain B-cell malignancies. Although results are encouraging, to date, only a small percentage of patients with advanced malignancies can benefit from ACT. Besides ACT availability and accessibility, treatment-related toxicities represent a major hurdle in the widespread implementation of this therapeutic modality. The large variety of observed toxicities is caused by the infused cell product or as side effects of accompanying medication and chemotherapy. Toxicities can occur immediately or can be delayed. In order to render those highly promising therapeutic approaches safe enough for a wider pool of patients outside of clinical trials, an international consensus for toxicity management needs to be established.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Our work provides an overview of important toxicities of adoptive cellular immunotherapy. |
We discuss the concepts of tumor infiltrating lymphocyte (TIL), T-cell receptor (TCR), and chimeric antigen receptor (CAR) adoptive T-cell therapy (ACT). |
We discuss toxicity pathomechanisms and review up-to-date treatment strategies. |
1 Introduction
T cells harbor great potential to treat cancer [1]. So-called adoptive cell immunotherapy (ACT) uses T cells isolated from patients’ tumors (tumor-infiltrating lymphocytes or TILs) or genetically engineered with T-cell receptors (TCRs) or chimeric antigen receptors (CARs) [2].
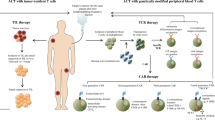
TIL therapy employs naturally occurring T cells and has been established over decades with very promising results largely in melanoma [3, 4], but also in ovarian cancer [5,6,7] and colorectal cancer [8, 9]. In contrast to TILs, gene transfer-based T-cell therapy strategies have been developed to confer new target specificity to peripheral blood T cells (Fig. 1), and new generations of CARs provide increased functionality to overcome tumor-specific immune tolerance. TCRs recognize peptides derived from tumor-associated antigens (TAAs) presented in the context of human leukocyte antigen (MHC)-restricted antigen peptides [2, 10, 11]. CARs are antibody recognition domains linked to TCR and other costimulatory signaling molecules (Fig. 2b) [12,13,14]. Both have been shown effective in refractory tumors [10, 15].

Basic principles of adoptive cellular immunotherapy. Please note that high-dose IL-2 is administered in TIL ACT and administration is optional in TCR T-cell ACT. Lymphodepletive chemotherapy consisting of cyclophosphamide and fludarabine is administered in TIL and TCR-T ACT and optional in CAR-T ACT. ACT Adoptive T-cell therapy, CAR chimeric antigen receptor, HD-IL-2 high-dose interleukin 2, PBL peripheral blood lymphocyte, TCR T-cell receptor, TIL tumor-infiltrating lymphocytes

TCR and CAR ACT. a Schematic overview over the process of T-cell receptor engineering for TCR ACT. The upper row of boxes describes the steps in the engineering process; the boxes below describe safety measures that are applied in parallel in order to limit clinical toxicity. Adapted from Kapanen et al. 2015 [130]. b Schematic of components of endogenous TCR, genetically engineered TCR and CAR. Adapted from June et al. 2015 [13]. ACT adoptive cellular therapy, CAR chimeric antigen receptor, TCR T-cell receptor
For all three ACT approaches discussed in this review, T cells are expanded ex vivo and re-infused in large numbers into a lymphodepleted cancer patient (Fig. 1) [2], although TCR-ACT has been effective without lymphodepletion as well [16]. Lymphodepleting chemotherapy before TIL infusion reduces regulatory T cells (Treg) and resident tumor microenvironment cells competing for T-cell homeostatic cytokines, increases the levels of Toll-like receptor ligands [17, 18], and favors the proliferation of the infused T cells through homeostatic expansion. This translates into a markedly improved T-cell survival and response rate and duration in melanoma [19, 20].
After treatment with TIL and in most TCR-ACT clinical trials, patients receive high-dose interleukin 2 (IL-2) in order to bolster T-cell division expansion within the host (Fig. 1).
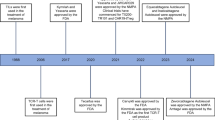
TIL and engineered T cells (TCR and CAR) are currently applied mainly within clinical trials at highly specialized centers. The landscape is nonetheless rapidly evolving. In August 2017, the FDA approved the first anti-CD19 CAR T-cell product, tisagenlecleucel (Kymriah, Novartis, Basel, Switzerland), for the treatment of pediatric and young adult patients with relapsed and/or refractory B-cell precursor acute lymphoblastic leukemia [21]. In October 2017, a second anti-CD19 CAR, axicabtagene ciloleucel (Yescarta, Kite Pharma, Santa Monica, CA, USA), was approved by the FDA for the treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy. Both products received approval in Europe in 2018. Despite great promise and rapid development of ACT, treatment-related toxicities remain an important issue. Preventing or managing unwanted toxicity has therefore emerged as a key component in the successful clinical application of these technologies. This article will review the treatment principles and toxicities of the three most prominent classes of ACT: TILs, TCR-engineered T cells, and CAR T cells.
2 Tumor-Infiltrating Lymphocytes (TILs)
2.1 Treatment Principle
TIL therapy consists of the administration of autologous ex vivo expanded T cells that naturally infiltrate tumors. Figure 1 illustrates the general sequence of TIL ACT: following surgical resection of a suitable lesion, TILs are isolated, cultured and expanded ex vivo in the presence of IL-2 to generate a T-cell product of predominantly T-effector-memory cells [22]. Before TIL reinfusion, patients undergo lymphodepleting chemotherapy. High doses of recombinant IL-2 are administered after TIL reinfusion to facilitate TIL expansion and engraftment in vivo. Response rates appear higher in patients treated with minimally cultured ‘young’ TIL that retain a higher proliferative potential and a higher lytic activity [23].
Most lymphodepleting regimens consist of fludarabine and cyclophosphamide; as an example, the NIH Surgery Branch regimen combines 5 days of fludarabine 25 mg/m2 (D-7 to D-3) and 2 days of cyclophosphamide 60 mg/kg (D-7, D-6; Fig. 1) [9]. After completion of TIL infusion, patients are treated with IL-2, usually administered as bolus infusions; the largest dataset originates from high-dose (HD) IL-2 (typically boluses of ≥ 600,000 IU/kg), although low-dose regimens (≤ 600,000 IU/kg) have been described [4, 19, 24,25,26,27]. Most patients treated receive between two and ten doses of HD IL-2, with a median of approximately six doses [4, 25,26,27,28,29,30]. Usually, an interval of 8 h is chosen between doses.
2.2 Clinical Results and Applications
While most TIL clinical trials from 1988 to date have been for metastatic melanoma [4, 25,26,27,28,29,30], they have also been reported for renal cell carcinoma, breast cancer, and colorectal cancer [8, 9]. Overall response rates in melanoma patients, mostly highly pretreated, ranged from 27.5 to 57%, and complete response rates from 6.4 to 22% [24,25,26, 28,29,30,31]. Median overall survival was either not reached at the time of publication or rather variable and ranged from 8.5 months [31] to 16.4 months [28] in treated and evaluable patients. Two-year overall survival was reported as 40% in two studies [25, 31]. It should be mentioned that most studies have not reported results as an intent-to-treat analysis, with 20–40% [19, 24,25,26, 28] of patients being taken off study due to rapid disease progression before TIL reinfusion, or unsuccessful TIL expansion [28]. Furthermore, few studies have comprehensively described toxicity, the majority reporting only on grade 3 or higher adverse events.
2.3 Toxicities
TIL therapy requires an inpatient hospital stay with a median duration of 20 days [26, 28]; patients are discharged upon hematologic and other systems recovery. Non-myeloablative lymphodepleting chemotherapy causes both hematological and non-hematological toxicities. Transient cytopenia including neutropenia, lymphopenia as well as prolonged depression of CD4 + T cells are observed in virtually all patients [4, 9, 24,25,26,27,28,29,30,31,32]. Neutropenic fever occurs in 37–51% of patients [24, 26]. Granulocyte-colony stimulating factor (G-CSF) as well as blood product support is routinely required, with a median of five red blood cell transfusions and 30 units of platelets [4, 9, 24,25,26,27,28,29,30,31,32] (Table 1). Side effects are managed according to standards of good clinical practice [33]. In the absence of prophylaxis, a minority of patients experience opportunistic infections, including Pneumocystis jirovecii pneumonia (e.g., 6% in [26]) or Herpes zoster reactivation (e.g., 9% in [26]), thus mandating routine prophylaxis for a minimum duration of 6 months post-chemotherapy. Non-hematological high-grade toxicities include diarrhea (e.g., 12% in [28]), hyperbilirubinemia (e.g., 14% in [28]) and fludarabine-induced neurotoxicity [25, 26, 28]. The overall mortality from this regimen is < 1% [25, 27].
High-grade toxicity attributed to the TIL infusion product itself is exceedingly uncommon. Immediate infusion reactions to TILs are rare and mainly low grade [2, 9, 26, 27, 32, 34,35,36,37,38,39], and they are difficult to discriminate from early reactions to low residual levels of IL-2 remaining in the TIL product after ex vivo culture. Allergic reactions include acute release of cytokines with fever, skin reaction and dyspnea or delayed symptom onset. Management is symptomatic and use of corticosteroids discouraged. In case of a side effect related to TIL infusion, every effort should be taken not to stop the cell infusion, if the clinical symptoms allow it [2, 40, 41] (Table 1). Autoimmune melanocyte destruction, manifesting as vitiligo or uveitis, may occur in approximately 35% and 15% of patients, respectively [26].
High-dose IL-2 is associated with transient and typical dose-limiting toxicity. Both efficacy and toxicity are dose- and schedule-dependent (reviewed in [42,43,44,45]). Although high-dose IL-2 is associated with significant morbidity, the incidence and severity of toxicities have decreased overall as more patients have been treated worldwide and clinicians have gained experience in the prevention and management of side effects [4, 26, 32, 45, 46] (Table 1). The implementation of lymphodepleting chemotherapy greatly limits the immediate IL-2 toxicity relative to an immunocompetent host, as it eliminates resident lymphocytes as a source of cytokines contributing to IL-2 side effects [19].
Generally, toxicities associated with high-dose IL-2 therapy are transient and can be managed using standard interventions [43]. Infusion of IL-2 requires an adequate hospitalization setting offering hemodynamic and respiratory monitoring as well as personnel to conduct frequent physical examination, blood tests, and radiological imaging when required, according to protocol. IL-2 toxicity can manifest in multiple organ systems, most significantly the heart, lungs, kidneys, and central nervous system. The most common manifestation is capillary leak syndrome, resulting in a hypovolemic state and extravascular fluid accumulation. Most patients become tachycardic and hypotensive 4–6 h after IL-2 administration, mimicking a sepsis-like pathophysiology. Usual management includes cautious crystalloid fluid boluses (max 1000–1500 mL/day) and careful avoidance of fluid overload, which can precipitate pulmonary edema due to capillary leak. Systolic arterial blood pressure can usually be stabilized to a new baseline of approximately 80–90 mmHg. Heart rate must generally return below 100 beats/min before administering the next IL-2 dose [43]. Cardiac arrhythmias happen rarely. In case of transient atrial flutter or fibrillation, IL-2 continuation is possible if rhythm returns to normal sinus rate. In case of ventricular arrhythmia, definitive discontinuation of IL-2 is mandatory. Capillary leak syndrome can contribute significantly to the development of oliguria, cardiac ischemia, dyspnea from pulmonary congestion (3–47%, [25, 28]) requiring intubation in 6–9% of patients [24, 26] and mental status changes (confusion). Treatment is mainly supportive. In addition, thyroid dysfunction is a relatively common sequel of IL-2 therapy, with 9% of patients presenting hypothyroidism requiring hormone replacement, and 7% of patients presenting hyperthyroidism [47]. In rare severe cases, vasopressors, intubation or continuous hemofiltration may be indicated. Safe administration of high-dose IL-2 depends on the experience of the caring team, adherence to standards of IL-2 administration and patient assessment guidelines, and that patient-eligibility criteria are respected. Further, it is important to carefully assess vital parameters prior to each high-dose IL-2 administration and strictly recognize and avoid contraindications as determined by the clinical study protocol [42, 43, 45, 46] (Table 1). Toxicities accompanying TIL therapy are mostly low grade, transient and manageable by standard supportive care but patients should only be treated in specialized centers.
3 T-Cell Receptor (TCR)-Transduced T Cells
3.1 Treatment Principle
Antigen specificity of T cells is endowed by their TCR, which binds a cognate ligand consisting of a peptide presented in the major histocompatibility complex (MHC), the so-called pMHC complex. TCR-ACT consists of autologous T lymphocytes engineered ex vivo to express an exogenous cancer-specific TCR (as described in Figs. 1 and 2a); this redirects autologous peripheral T cells to recognize a specific cancer antigen processed and presented in the context of the patient’s MHC. The use of peripheral T cells obviates the need to harvest and expand natural lymphocyte clones. A crucial determinant of both efficacy and safety is the affinity of the chosen TCR for its target pMHC. TCRs in the upper end of natural affinity are associated with higher efficacy but affinity thresholds have been reported beyond which T-cell activity levels drop, and cross-reactivity becomes an important risk [48]. The ideal pMHC target of a candidate TCR comprises a peptide from a tumor antigen that is exclusively expressed by cancer cells (expression in non-essential normal tissues may be tolerable), that is essential for cancer cell survival to reduce the risk of tumor escape through downregulation, and that is presented on frequent MHC molecules (reviewed in [49]). TCRs selected for gene modification are usually obtained from naturally occurring tumor-reactive T cell clones, although TCRs have also been isolated from mice transgenic for human HLAs that have been vaccinated with the targeted human antigen. In addition, the affinity of natural TCRs can be optimized by structure-based rational design [50] as well as by phage display screening technology [51, 52] (Fig. 2a). Although successful in enhancing the performance of the transduced T cells against cancer, non-natural TCRs may also carry a higher risk of ‘off-tumor, on-target’ toxicity (recognition of the pMHC expressed at low levels in normal tissues), or ‘off-tumor, off-target’ toxicity (cross-reactivity with a different pMHC expressed in normal tissues). Notably, the mispairing of introduced TCR subunits with endogenous TCR subunits can generate autoreactive T cells [53, 54], but this can be minimized by optimal transgene design or gene editing [55].
For TCR T-cell ACT, peripheral T lymphocytes are activated and gene-modified to express the TCR, and then expanded in culture. As described in the TIL section, patients are usually pretreated with lymphodepleting chemotherapy, and high-dose IL-2 may be administered after cell transfer. In sharp contrast to autologous TIL therapy, the genetic engineering of the T cells to express specific TCRs may lead to a high rate of toxicity mediated by the cell product itself due to autoreactivity, as discussed above. Rigorous pre-clinical testing is performed in order to negatively select autoreactive TCRs [49].
3.2 Clinical Results and Applications
Several TCR-ACT clinical trials have been conducted (Table 2) in patients with melanoma [10, 56,57,58], colorectal cancer [59], esophageal cancer [16, 60, 61], other carcinomas [60, 62], advanced multiple myeloma [63], acute myeloid leukemia, and myelodysplastic syndrome [64]. While the targeting of tissue differentiation antigens such as MART-1 has had limited success, TCR against cancer-germline antigens such as melanoma-associated antigen (MAGE)-A3 [58, 60, 61], and New York esophageal squamous cell carcinoma (NY-ESO)-1 [63, 65] have demonstrated high response rates between 23 and 80% with rare durable and complete responses.
3.3 Toxicities
Toxicities resulting from lymphodepleting chemotherapy and high-dose IL-2 were described in the TIL section. The infused T cells can cause acute cytokine release syndrome (CRS), as well as tissue-directed autoimmune reactions [66]. Cytokine release is triggered by the engagement of infused T cells with the targeted tumor cells (Table 1 and CAR section), and its severity depends on the number and fitness of infused T cells, their avidity for the tumor antigen and the tumor bulk. The resulting clinical picture is a systemic inflammatory response syndrome, characterized by fever, tachycardia, hypotension, vasodilation, and capillary leak [66]. Severe forms of CRS can progress to shock and fatal multi-organ failure. Management is similar to that for responding to side effects of high-dose IL-2, described above (Table 1) and which has been reviewed extensively in the literature [42, 43, 46]. Mild forms of CRS can be treated with non-steroidal anti-inflammatory drugs and anti-pyretic drugs.
The nature of autoimmune toxicity is largely dependent on the target antigen of the TCR. For example, severe adverse events have been reported when TCR-ACT was directed against lineage antigens; that is, antigens overexpressed in tumors but also expressed at low levels by the normal tissue of origin. For example, high-grade on-target colitis was reported upon administration of TCR-transduced T cells targeting carcinoembryonic antigen (CEA), expressed highly in gastrointestinal cancers but also at low levels in the normal intestine [59], while on-target skin reactions were observed with TCRs against melanoma-specific antigens MART-1 and gp100 [56, 57], also expressed by normal melanocytes.
Careful selection of the target and the TCR mitigates the risk of excess toxicity during clinical development (Fig. 2a). Commonly targeted and potentially safe antigens for TCR ACT include oncoviral antigens, cancer germline (testis) antigens such as NY-ESO-1, and tumor neo-antigens [67]. Oncoviral antigens are highly immunogenic, but only present in 10–15% of all malignancies; TIL specific to Epstein-Barr virus (EBV) epitopes resulted in high response rates with durable responses in patients with EBV-associated nasopharyngeal carcinoma [36], and anti-human papillomavirus (HPV)-specific TIL administered in metastatic cervical cancer evoked durable complete responses [38].
Cancer germline antigens are normally expressed in gonads and the thymus but some exhibit cancer-specific expression and are shared among many tumor types [65, 68]. MAGE-A3 and NY-ESO1 have been targeted in metastatic melanoma, metastatic synovial sarcoma, or multiple myeloma [61, 65]. Since the affinity of the wild-type TCRs to these targets is usually weak, affinity-enhanced TCRs have been generated to increase anti-tumor activity, bearing the risk of losing strict specificity and generating cross-reactivity with other self-antigens. Thus far, anti-NY-ESO1 TCR T cells have demonstrated a clinical benefit without toxicity [63, 65]. However, treatment of melanoma patients with an HLA-A*0201 restricted TCR directed against the germline antigen MAGE-A3 produced lethal neurotoxicity; deep characterization of the molecular basis for the toxicity revealed that the TCR also recognized HLA-A*0201 epitopes in MAGE-A9 and A12, and that MAGE-A12 was expressed in the human brain (in addition to possibly MAGE-A9) [61]. Furthermore, lethal off-target cardiotoxicity was observed in patients receiving ACT with an HLA-A*01 restricted TCR against MAGE-A3 due to unexpected cross-reactivity of the TCR with a titin epitope in the HLA-A*01 background, exclusively expressed in the heart in beating cardiomyocytes [58, 69].
In order to limit on-target toxicity for oncoviral and germ-line antigens, their absence from panels of healthy tissue is tested in silico, using online databases (Human Protein Atlas, CGA database), and in vitro using polymerase chain reaction (PCR) cDNA libraries and immunohistochemistry (IHC) in tissue panels [49, 70]. TCRs are tested against random epitopes and allogeneic MHC molecules using, for example, lymphoblastoid B-cell lines with various MHC allotypes [71, 72]. Further testing for self-avidity and efficient cellular processing and presentation is recommended [49] as well as screening against a combinatorial peptide library and additional cell subsets to detect off-target toxicity due to cross-reactivity [73]. Various techniques to reduce the risk of mispairing [74], including siRNA-induced silencing of endogenous TCR [75] have been described.
Neo-antigens resulting from somatic DNA alterations in cancer cells are by definition tumor-specific and are potentially recognized by a high-affinity T-cell repertoire, and as such represent attractive targets for immunotherapy both for their safety and efficacy [76, 77]. Neo-antigens are mostly patient-specific (i.e., with very few being shared among patients); their utilization, however, requires high-throughput methods for neo-epitope and TCR identification [76, 78, 79]. The rapid development of whole genome sequencing approaches might help to find neo-antigen targets for ACT from circulating tumor DNA (reviewed in [80]). Very recent developments in molecular-genetic methodology like CRISPR/CAS9 genetic engineering could be useful for supporting the development of personalized TCR-ACT, and there is currently a first trial recruiting at the National Institutes of Health (NIH) using individual TCRs (ClinicalTrials.gov Identifier: NCT03412877).
Management of toxicities depends on the organ system involved as well as the type of toxicity. In reported clinical trials, side effects resulting from on-target toxicity as reported after the MAGE-3 TCR study were managed using symptomatic therapy (e.g., for seizure control) and immunosuppression using corticosteroids [61]. Efforts to limit toxicity by inducible T-cell suicide are discussed in chapter 5 below.
4 CAR T Cells
4.1 Treatment Principle
A CAR combines an extracellular antigen-binding domain, which typically comprises a single-chain variable fragment (scFv) from a monoclonal antibody, or a natural ligand [81] that confers recognition of a tumor-associated antigen, with an intracellular domain carrying signaling motifs capable of T-cell activation and costimulation [12]. Currently, the most common method of ex vivo genetic engineering of T cells is via lentiviral and gamma-retroviral vector-based transduction methods [82,83,84,85]. These allow for stable integration of the desired transgene(s). Alternative non-viral delivery technologies include electroporation for transient expression [86], and transposon/transposase delivery systems that allow larger gene cargo [87, 88].
In contrast to TCRs, CARs can recognize any molecule present on the surface of target tumor cells in a non-MHC restricted manner. MHC-independent antigen recognition enables CAR-modified T cells to treat any patient whose tumor expresses the target antigen, and thus, unlike TCR-ACT, CAR-ACT permits the treatment of tumors that have acquired defects in antigen processing and MHC presentation [89]. While antigen recognition by CARs occurs by engagement of larger epitopes, imparting less risk of cross-reactivity [90], solid tumors remain an important challenge for CAR therapy as there exist few bona fide tumor antigens, thus running the risk of on-target/off-tumor toxicities (Fig. 2b).
4.2 Clinical Results and Applications
Administration of CAR-modified T cells that target the B-cell lineage differentiation antigen CD19 (CAR19) has led to impressive clinical responses in patients with acute B-cell leukemia, chronic lymphocytic leukemia, diffuse large B-cell lymphoma, and other non-Hodgkin lymphomas (NHLs) [15, 91,92,93,94,95,96,97], which led to their regulatory approval. CAR19 has therefore entered the mainstream and is a valuable therapeutic option for patients with hematologic malignancies.
4.3 Toxicities
Toxicities arising from CAR therapy include toxicity from lymphodepleting chemotherapy, as described in the TIL section, CRS and CAR T-cell-related encephalopathy syndrome (CRES), and auto-immune events. CRS is the most commonly observed toxicity. While in the majority of cases CRS presents as a mild, flu-like disorder with fever, malaise, headache, tachycardia, and myalgias, in a proportion of patients it can rapidly evolve into a sepsis-like symptomatology, with vascular leak, hypotension, rash, pulmonary edema, systemic coagulopathy, and multi-organ failure [98]. The severity of CRS correlates with tumor burden [21]. Most toxicities are grade 1–2 and manageable [99]. Some predictive biomarkers for the occurrence of CRS like the dose of infused CAR T cells, disease burden or preexisting endothelial activation have been established but warrant further clinical trials for their validation [100].
Since algorithms for accurate and consistent grading and management of the toxicities were lacking, a CARTOX (CAR T-cell therapy-associated toxicity) working group has been formed and guidelines for diagnostic, grading, and treatment of toxicities have been published in 2018 [99]. This review also includes a list of lethal events observed to date in CAR T-cell trials. The same working group presented CAR treatment guidelines for pediatric patients [101]. The magnitude and timing of the toxicities associated with CAR T-cell therapy vary considerably across different CAR T-cell constructs and across different diseases (acute lymphocytic leukemia [ALL] versus NHL) [102]. For example, in the pivotal multicenter ZUMA‑1 trial of a CAR19 bearing the CD28/CD3ζ (28/ζ) endodomain in 101 patients with refractory aggressive B-cell NHL, the rates of grade ≥ 3 CRS and neurological toxicities were 13% and 28%, respectively [103]. Conversely, in an interim analysis of the JULIET trial of a CAR19 bearing the 4-1BB/CD3ζ (BB/ζ) endodomain in 51 patients with relapsed or refractory diffuse large B-cell lymphoma, these rates were 26% and 13% [104].
Symptoms of CRS can be graded according to Lee et al. [105]. Rarely, CRS can develop into a fulminant hemophagocytic lymphohistiocytosis (HLH), characterized by hepatosplenomegaly, hepatotoxicity, jaundice, and diffuse lymphoadenopathy, or macrophage activation syndrome (MAS) with high fever, hepatosplenomegaly, hepatotoxicity, jaundice, coagulopathy, hypofibrinogenemia, cytopenia, hypertriglyceridemia, and extreme hyperferritinemia. Plasma levels of IL-6, IL-10, and interferon-gamma (IFNγ) have been found to be very high during CRS [106], and they also correlate closely with the expansion and persistence of CAR T cells [107]. Although IFNγ is likely produced directly by CAR T cells, IL-6 is contributed largely by activated macrophages, which must persist despite chemotherapy according to recent preclinical studies [108, 109]. Given the potential key role of macrophages in CRS induced by CAR T cells, it has been recommended that candidate patients be screened for hereditary mutations predisposing to HLH, including PRF1, MUNC13-4, STXBP2, and STX11 [98].
Intensive monitoring and prompt management of toxicities are essential to minimize the morbidity and mortality associated with this potentially curative therapeutic approach (Table 2). Table 1 shows CRS treatment options according to Neelapu et al. [99]. Corticosteroids have been part of the management. The potential to attenuate the clinical efficacy of CAR T cells is a concern, although short-term steroid treatment did not appear to limit the efficacy of CAR T-cells [91, 106]. Blockade of IL-6 receptor (IL-6R) with the commercially available, FDA-approved antibody tocilizumab, along with anti-tumor necrosis factor alpha (TNFα) antibody etanercept, produced prompt resolution of the symptomatology without affecting the expansion or efficacy of CAR T cells [106]. Effective IL-6 blockade can also be achieved through siltuximab, a commercially available IL-6 blocking antibody [98]. IL-6 blockade is recommended to be administered early in case of CRS [99, 110]. Recent preclinical research shows that IL-1 is also required to trigger CRS [108, 109], indicating that IL-1 blockade might be useful in the management of CAR therapy toxicity.
The second most worrisome CAR-specific side effect is acute onset neurotoxicity (CRES), which can occur in association or independently of CRS. CRES is described as a biphasic phenomenon with a first phase that can occur together with CRS and is responsive to tocilizumab treatment, followed by a second phase that is not responsive to IL-6R blockade [99]. Early signs of CRES include decreased attention, coordination problems, agitation or delirium with preserved alertness, headache, and language deficits. In the majority of cases, symptoms resolve within 4 weeks, in more severe cases, cerebral edema, seizures, focal deficits, and diminished consciousness including coma can occur. In 133 patients receiving an anti-CD19 CAR bearing the BBz endodomain, neurologic adverse events (AEs—any) were recorded in 40%, presenting a median of 4 days after CAR T-cell infusion [111]. The highest grade neurotoxicity evolved within a median of 1 day from the onset of neurotoxicity, while the duration of reversible neurologic AEs was < 4 weeks (median 5 days) in all but one patient. There were four deaths due to CRES: two from acute cerebral edema, one from disseminated intravascular coagulation and multifocal brainstem hemorrhage, and one from cortical laminar necrosis and coma. These largely occurred during the dose-escalation phase of the study, in patients who received a dose of CAR T cells subsequently determined to be above the maximally tolerated dose. In > 90% of patients, neurologic AE presented in the presence of CRS, and patients without CRS only presented grade transient 1 neurotoxicity. In addition to CRS, the severity of neurotoxicity correlated with CAR T-cell expansion in vivo, higher disease burden, higher dose of CAR T cells, and a fludarabine-containing chemotherapy preparative regimen. Severe CRS was a major risk factor for grade ≥ 3 CRES, and plasma IL-6 levels > 500 pg/mL within 6 days of CAR T-cell infusion were associated with grade ≥ 4 neurotoxicity in 100% of patients [111].
The pathophysiology mechanisms of CRES are under investigation. A careful review of clinical, laboratory, and autopsy data from the above patients suggested that brain endothelial cell activation is an early event in CRES, which leads to breakdown of the blood–brain endothelial barrier and entry of inflammatory cytokines and CAR T cells in the brain, leading in severe cases to local severe inflammation, cerebral edema, hemorrhage, and infarctions [111]. Mouse models have revealed that CRES is largely driven by activation of endogenous macrophages, recruited and activated by CAR T cells. Such monocytes produce IL-1 and nitric oxide, which drive the neurotoxicity, and monocyte depletion in the mice prevented CRES. Tocilizumab could prevent systemic CRS but not the delayed-onset lethal CRES, while the IL-1 receptor antagonist anakinra could effectively reverse CRES in mice without affecting the anti-leukemia efficacy of CAR T cells [108, 109].
The CARTOX working group developed algorithms for grading and management of CRES [99]. Treatment is symptom dependent. Anti-IL-6R therapy can be considered to relieve systemic toxicity of CRS. However, based on the recent mouse evidence, the use of IL-1 antagonist anakinra should be evaluated in the clinic. In higher grade CRES, administration of corticosteroids should also be considered [99].
Severe immune-mediated adverse events, which can be on-target [80] or off-target (as explained in more detail in the TCR section) following CAR T-cell infusion, have been appreciated. In order to limit on-target toxicity, careful selection of the target antigen is key, as discussed already in the TCR section of this article. Therapy with CAR T cells against carbonic anhydrase-9 (CAIX), for example, delivered to 12 patients with CAIX-expressing metastatic renal cell carcinoma had to be stopped because of G2–G4 liver toxicity due to CAIX expression in the bile duct epithelium [112].
Several attempts have been made to limit toxicity from CAR-ACT through engineering solutions [113]. For example, the so-called split-signaling CARs entail the co-transfection of T cells with two distinct CARs, one (zeta-CAR) that provides the main antigen binding ectodomain and a CD3ζ endodomain and a second (costimulatory-CAR) that recognizes a second antigen on target tumor cells with a different ectodomain linked to a costimulatory endodomain. Engagement of the zeta-CAR drives suboptimal activation of T cells upon antigen recognition, while engagement of the costimulatory-CAR boosts T-cell activation upon recognition of the second antigen. This combinatorial strategy therefore requires the simultaneous expression of the two antigens to fully activate CAR T cells, which occurs on the tumor, and avoids the CAR T-cell activation against normal tissues, which may express only one of the two antigens [114].
5 Management of Adoptively Transferred T cells to Reduce Autoimmune Toxicity
Agents suppressing effector T cells could be useful in the management of acute TCR-ACT autoimmune toxicities. Corticosteroids are most readily used, such as pulse corticosteroids (methylprednisolone) followed by a taper. Clinicians must also familiarize themselves with drugs used in acute allotransplant rejection as further means to control acute autoimmunity, including rabbit anti-thymocyte globulin (rATG-thymoglobulin), mycophenolate, tacrolimus, and/or anti-CD52 antibody alemtuzumab [115].
Additional safety strategy approaches include suicide genes that can eliminate CAR-T or TCR T cells on command [116]. For example, T cells transfected with the herpes simplex virus thymidine kinase (HSV-TK) can be subsequently eliminated by the use of the prodrug ganciclovir, which induces apoptosis specifically in HSV-tk transfected CAR T cells. This has been successfully tested in clinical trials in order to avoid graft versus host disease after allogenic hematopoietic stem cell transplantation [117,118,119]. Another strategy employs an inducible caspase 9 suicide gene, integrated in the delivered transgene [120,121,122]. This particular suicide gene can be selectively activated by a chemical inducer of dimerization (CID) small molecule, which has been shown to increase safety in an allogenic stem cell transplantation setting [120] and is about to be tested in CAR T cells in several phase I/II clinical trials (e.g., NCT03639844).
Beside suicide gene engineering, T-cell death can be achieved using antibody-dependent cell-mediated cytotoxicity (ADCC). A pre-clinically validated suicide strategy is retroviral delivery of the CD20 molecule into T cells, which allows targeting transduced T cells in vivo with anti-CD20 monoclonal antibody [123]. An alternative approach has combined epitopes from CD34 and CD20, enabling CD34 selection, cell tracking, as well as deletion after anti-CD20 monoclonal antibody administration [124]. Another approach has introduced a 10-amino acid tag of c-myc protein into the TCR sequence allowing elimination with anti-myc tag monoclonal antibody administration [125]. Finally, another approach has used truncated human epidermal growth factor receptor (EGFR) polypeptide/anti-EGFR monoclonal antibody [126]. The above methods rely on elimination of transduced T cells through ADCC, which can be slow, especially following high-dose chemotherapy, and are unable to control a rapidly expanding T cell population in the lymphodepleted host.
6 Conclusions
ACT immunotherapy shows great promise for treating and eradicating advanced metastatic cancers, but clinicians must familiarize themselves with its potential side effects. Except for CAR19, which is approved for B-cell malignancies in the US and Europe, all ACT is administered within clinical trials in specialized centers. Adverse events may be immediate or delayed, and although usually mild, they can be severe and persist for the duration of the genetically modified T-cell lifespan [127]. Unique to T-cell therapies is the potential for extraordinary long-term persistence of transferred T cells for up to 10 years or longer [128, 129]. This persistence extends the promise for long-term surveillance of residual tumor cells and possible elimination and definitive cure of tumors, but also increases the timeline of potential toxicities far beyond those of chemotherapy or antibody-based therapies.
The rapidly growing knowledge regarding the interaction between the immune system and tumors, together with rapid advances in technology, will support the development of TIL, TCR, and CAR-T ACT to move toward the goal of treating cancer with high degree of safety, high efficacy, and low cost. The CARTOX working group treatment algorithms for toxicity management in adults and pediatric patients provide guidelines for building the medical practice of CAR19 T-cell therapy and offer a solid framework for establishing standardized and safe practices in the development of adoptive cell therapy with further CARs, TCRs, and TILs.
References
Harris DT, Kranz DM. Adoptive T cell therapies: a comparison of T cell receptors and chimeric antigen receptors. Trends Pharmacol Sci. 2016;37(3):220–30. https://doi.org/10.1016/j.tips.2015.11.004.
Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62–8. https://doi.org/10.1126/science.aaa4967.
Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4 + T cells in human melanoma. Nat Med. 2015;21(1):81–5. https://doi.org/10.1038/nm.3773.
Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–7. https://doi.org/10.1158/1078-0432.CCR-11-0116.
Aoki Y, Takakuwa K, Kodama S, Tanaka K, Takahashi M, Tokunaga A, et al. Use of adoptive transfer of tumor-infiltrating lymphocytes alone or in combination with cisplatin-containing chemotherapy in patients with epithelial ovarian cancer. Cancer Res. 1991;51(7):1934–9.
Freedman RS, Edwards CL, Kavanagh JJ, Kudelka AP, Katz RL, Carrasco CH, et al. Intraperitoneal adoptive immunotherapy of ovarian carcinoma with tumor-infiltrating lymphocytes and low-dose recombinant interleukin-2: a pilot trial. J Immunother Emphas Tumor Immunol. 1994;16(3):198–210.
Fujita K, Ikarashi H, Takakuwa K, Kodama S, Tokunaga A, Takahashi T, et al. Prolonged disease-free period in patients with advanced epithelial ovarian cancer after adoptive transfer of tumor-infiltrating lymphocytes. Clin Cancer Res. 1995;1(5):501–7.
Fabbri M, Ridolfi R, Maltoni R, Ridolfi L, Riccobon A, Flamini E, et al. Tumor infiltrating lymphocytes and continuous infusion interleukin-2 after metastasectomy in 61 patients with melanoma, colorectal and renal carcinoma. Tumori. 2000;86(1):46–52.
Topalian SL, Solomon D, Avis FP, Chang AE, Freerksen DL, Linehan WM, et al. Immunotherapy of patients with advanced cancer using tumor-infiltrating lymphocytes and recombinant interleukin-2: a pilot study. J Clin Oncol. 1988;6(5):839–53. https://doi.org/10.1200/JCO.1988.6.5.839.
Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–9. https://doi.org/10.1126/science.1129003.
Schumacher TN. T-cell-receptor gene therapy. Nat Rev Immunol. 2002;2(7):512–9. https://doi.org/10.1038/nri841.
Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. 1993;90(2):720–4.
June CH, Levine BL. T cell engineering as therapy for cancer and HIV: our synthetic future. Philos Trans R Soc Lond B Biol Sci. 2015;370(1680):20140374. https://doi.org/10.1098/rstb.2014.0374.
Pinthus JH, Waks T, Kaufman-Francis K, Schindler DG, Harmelin A, Kanety H, et al. Immuno-gene therapy of established prostate tumors using chimeric receptor-redirected human lymphocytes. Cancer Res. 2003;63(10):2470–6.
Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–33. https://doi.org/10.1056/NEJMoa1103849.
Kageyama S, Ikeda H, Miyahara Y, Imai N, Ishihara M, Saito K, et al. Adoptive transfer of MAGE-A4 T-cell receptor gene-transduced lymphocytes in patients with recurrent esophageal cancer. Clin Cancer Res. 2015;21(10):2268–77. https://doi.org/10.1158/1078-0432.CCR-14-1559.
Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR, et al. CD8 + T cell immunity against a tumor/self-antigen is augmented by CD4 + T helper cells and hindered by naturally occurring T regulatory cells. J Immunol. 2005;174(5):2591–601.
Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005;26(2):111–7. https://doi.org/10.1016/j.it.2004.12.003.
Dudley ME, Wunderlich JR, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother. 2002;25(3):243–51.
Muranski P, Boni A, Wrzesinski C, Citrin DE, Rosenberg SA, Childs R, et al. Increased intensity lymphodepletion and adoptive immunotherapy—how far can we go? Nat Clin Pract Oncol. 2006;3(12):668–81. https://doi.org/10.1038/ncponc0666.
June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379(1):64–73. https://doi.org/10.1056/NEJMra1706169.
Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003;26(4):332–42.
Rosenberg SA, Yannelli JR, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, et al. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst. 1994;86(15):1159–66.
Dudley ME, Gross CA, Langhan MM, Garcia MR, Sherry RM, Yang JC, et al. CD8 + enriched “young” tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin Cancer Res. 2010;16(24):6122–31. https://doi.org/10.1158/1078-0432.CCR-10-1297.
Dudley ME, Gross CA, Somerville RP, Hong Y, Schaub NP, Rosati SF, et al. Randomized selection design trial evaluating CD8 + -enriched versus unselected tumor-infiltrating lymphocytes for adoptive cell therapy for patients with melanoma. J Clin Oncol. 2013;31(17):2152–9. https://doi.org/10.1200/JCO.2012.46.6441.
Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23(10):2346–57. https://doi.org/10.1200/JCO.2005.00.240.
Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26(32):5233–9. https://doi.org/10.1200/JCO.2008.16.5449.
Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res. 2013;19(17):4792–800. https://doi.org/10.1158/1078-0432.CCR-13-0380.
Itzhaki O, Hovav E, Ziporen Y, Levy D, Kubi A, Zikich D, et al. Establishment and large-scale expansion of minimally cultured “young” tumor infiltrating lymphocytes for adoptive transfer therapy. J Immunother. 2011;34(2):212–20. https://doi.org/10.1097/CJI.0b013e318209c94c.
Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res. 2012;18(24):6758–70. https://doi.org/10.1158/1078-0432.CCR-12-1177.
Hong JJ, Rosenberg SA, Dudley ME, Yang JC, White DE, Butman JA, et al. Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res. 2010;16(19):4892–8. https://doi.org/10.1158/1078-0432.CCR-10-1507.
Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319(25):1676–80. https://doi.org/10.1056/nejm198812223192527.
Haanen J, Carbonnel F, Robert C, Kerr KM, Peters S, Larkin J et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2017;28(suppl_4):iv119–iv42. https://doi.org/10.1093/annonc/mdx225.
Chandran SS, Somerville RPT, Yang JC, Sherry RM, Klebanoff CA, Goff SL, et al. Treatment of metastatic uveal melanoma with adoptive transfer of tumour-infiltrating lymphocytes: a single-centre, two-stage, single-arm, phase 2 study. Lancet Oncol. 2017;18(6):792–802. https://doi.org/10.1016/S1470-2045(17)30251-6.
Deniger DC, Kwong ML, Pasetto A, Dudley ME, Wunderlich JR, Langhan MM, et al. A pilot trial of the combination of vemurafenib with adoptive cell therapy in patients with metastatic melanoma. Clin Cancer Res. 2017;23(2):351–62. https://doi.org/10.1158/1078-0432.CCR-16-0906.
Li J, Chen QY, He J, Li ZL, Tang XF, Chen SP, et al. Phase I trial of adoptively transferred tumor-infiltrating lymphocyte immunotherapy following concurrent chemoradiotherapy in patients with locoregionally advanced nasopharyngeal carcinoma. Oncoimmunology. 2015;4(2):e976507. https://doi.org/10.4161/23723556.2014.976507.
Rosenberg SA, Lotze MT, Yang JC, Aebersold PM, Linehan WM, Seipp CA, et al. Experience with the use of high-dose interleukin-2 in the treatment of 652 cancer patients. Ann Surg. 1989;210(4):474–84 (discussion 84–5).
Stevanovic S, Draper LM, Langhan MM, Campbell TE, Kwong ML, Wunderlich JR, et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J Clin Oncol. 2015;33(14):1543–50. https://doi.org/10.1200/JCO.2014.58.9093.
Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4 + T cells in a patient with epithelial cancer. Science. 2014;344(6184):641–5. https://doi.org/10.1126/science.1251102.
Libert C, Dejager L. How steroids steer T cells. Cell Rep. 2014;7(4):938–9. https://doi.org/10.1016/j.celrep.2014.04.041.
Olnes MJ, Kotliarov Y, Biancotto A, Cheung F, Chen J, Shi R, et al. Effects of systemically administered hydrocortisone on the human immunome. Sci Rep. 2016;6:23002. https://doi.org/10.1038/srep23002.
Schwartzentruber DJ. Interleukin-2: Clinical applications—principles of administration and management of side effects. In: Rosenberg AZ, editor. Principles and Practice of the Biologic Therapy of Cancer. Philadelphia: Lippincott, Williams & Wilkins; 2000. p. 32–50.
Dutcher JP, Schwartzentruber DJ, Kaufman HL, Agarwala SS, Tarhini AA, Lowder JN, et al. High dose interleukin-2 (Aldesleukin)—expert consensus on best management practices-2014. J ImmunoTher Cancer. 2014;2:26.
Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105–16. https://doi.org/10.1200/JCO.1999.17.7.2105.
Schwartz RN, Stover L, Dutcher JP. Managing toxicities of high-dose interleukin-2. Oncology (Williston Park). 2002;16(11 Suppl 13):11–20.
Schwartzentruber DJ. Guidelines for the safe administration of high-dose interleukin-2. J Immunother. 2001;24(4):287–93.
Krouse RS, Royal RE, Heywood G, Weintraub BD, White DE, Steinberg SM, et al. Thyroid dysfunction in 281 patients with metastatic melanoma or renal carcinoma treated with interleukin-2 alone. J Immunother Emphas Tumor Immunol. 1995;18(4):272–8.
Irving M, Zoete V, Hebeisen M, Schmid D, Baumgartner P, Guillaume P, et al. Interplay between T cell receptor binding kinetics and the level of cognate peptide presented by major histocompatibility complexes governs CD8 + T cell responsiveness. J Biol Chem. 2012;287(27):23068–78. https://doi.org/10.1074/jbc.M112.357673.
Kunert A, Obenaus M, Lamers CHJ, Blankenstein T, Debets R. T-cell receptors for clinical therapy: in vitro assessment of toxicity risk. Clin Cancer Res. 2017;23(20):6012–20. https://doi.org/10.1158/1078-0432.ccr-17-1012.
Zoete V, Irving M, Ferber M, Cuendet MA, Michielin O. Structure-based, rational design of T cell receptors. Front Immunol. 2013;4:268. https://doi.org/10.3389/fimmu.2013.00268.
Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol. 2005;23(3):349–54. https://doi.org/10.1038/nbt1070.
Raman MC, Rizkallah PJ, Simmons R, Donnellan Z, Dukes J, Bossi G, et al. Direct molecular mimicry enables off-target cardiovascular toxicity by an enhanced affinity TCR designed for cancer immunotherapy. Sci Rep. 2016;6:18851. https://doi.org/10.1038/srep18851.
van Loenen MM, de Boer R, Amir AL, Hagedoorn RS, Volbeda GL, Willemze R, et al. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc Natl Acad Sci USA. 2010;107(24):10972–7. https://doi.org/10.1073/pnas.1005802107.
Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med. 2010;16(5):565–70, 1p following 70. https://doi.org/10.1038/nm.2128.
Spear TT, Foley KC, Garrett-Mayer E, Nishimura MI. TCR modifications that enhance chain pairing in gene-modified T cells can augment cross-reactivity and alleviate CD8 dependence. J Leukoc Biol. 2018;103(5):973–83. https://doi.org/10.1002/JLB.5A0817-314R.
Chodon T, Comin-Anduix B, Chmielowski B, Koya RC, Wu Z, Auerbach M, et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin Cancer Res. 2014;20(9):2457–65. https://doi.org/10.1158/1078-0432.CCR-13-3017.
Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114(3):535–46. https://doi.org/10.1182/blood-2009-03-211714.
Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122(6):863–71. https://doi.org/10.1182/blood-2013-03-490565.
Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19(3):620–6. https://doi.org/10.1038/mt.2010.272.
Lu YC, Parker LL, Lu T, Zheng Z, Toomey MA, White DE, et al. Treatment of patients with metastatic cancer using a major histocompatibility complex class II-restricted T-cell receptor targeting the cancer germline antigen MAGE-A3. J Clin Oncol. 2017;35(29):3322–9. https://doi.org/10.1200/JCO.2017.74.5463.
Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36(2):133–51. https://doi.org/10.1097/CJI.0b013e3182829903.
Davis JL, Theoret MR, Zheng Z, Lamers CH, Rosenberg SA, Morgan RA. Development of human anti-murine T-cell receptor antibodies in both responding and nonresponding patients enrolled in TCR gene therapy trials. Clin Cancer Res. 2010;16(23):5852–61. https://doi.org/10.1158/1078-0432.CCR-10-1280.
Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med. 2015;21(8):914–21. https://doi.org/10.1038/nm.3910.
Tawara I, Kageyama S, Miyahara Y, Fujiwara H, Nishida T, Akatsuka Y, et al. Safety and persistence of WT1-specific T-cell receptor gene-transduced lymphocytes in patients with AML and MDS. Blood. 2017;130(18):1985–94. https://doi.org/10.1182/blood-2017-06-791202.
Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–24. https://doi.org/10.1200/JCO.2010.32.2537.
Yang JC. Toxicities associated with adoptive T-cell transfer for cancer. Cancer J. 2015;21(6):506–9. https://doi.org/10.1097/PPO.0000000000000157.
Janeway CATP, Walport M, Shlomchik MJ. Immunobiology: the immune system in health and disease. Immunobiology, 5th edition. New York: Garland Science; 2001.
Debets R, Donnadieu E, Chouaib S, Coukos G. TCR-engineered T cells to treat tumors: seeing but not touching? Semin Immunol. 2016;28(1):10–21. https://doi.org/10.1016/j.smim.2016.03.002.
Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. 2013;5(197):197ra03. https://doi.org/10.1126/scitranslmed.3006034.
Hurwitz AA, Cuss SM, Stagliano KE, Zhu Z. T cell avidity and tumor immunity: problems and solutions. Cancer Microenviron. 2014;7(1–2):1–9. https://doi.org/10.1007/s12307-013-0143-1.
Heemskerk MH, de Paus RA, Lurvink EG, Koning F, Mulder A, Willemze R, et al. Dual HLA class I and class II restricted recognition of alloreactive T lymphocytes mediated by a single T cell receptor complex. Proc Natl Acad Sci USA. 2001;98(12):6806–11. https://doi.org/10.1073/pnas.111162298.
Obenaus M, Leitao C, Leisegang M, Chen X, Gavvovidis I, van der Bruggen P, et al. Identification of human T-cell receptors with optimal affinity to cancer antigens using antigen-negative humanized mice. Nat Biotechnol. 2015;33(4):402–7. https://doi.org/10.1038/nbt.3147.
Bijen HM, van der Steen DM, Hagedoorn RS, Wouters AK, Wooldridge L, Falkenburg JHF, et al. Preclinical strategies to identify off-target toxicity of high-affinity TCRs. Mol Ther. 2018;26(5):1206–14. https://doi.org/10.1016/j.ymthe.2018.02.017.
Govers C, Sebestyen Z, Coccoris M, Willemsen RA, Debets R. T cell receptor gene therapy: strategies for optimizing transgenic TCR pairing. Trends Mol Med. 2010;16(2):77–87. https://doi.org/10.1016/j.molmed.2009.12.004.
Okamoto S, Mineno J, Ikeda H, Fujiwara H, Yasukawa M, Shiku H, et al. Improved expression and reactivity of transduced tumor-specific TCRs in human lymphocytes by specific silencing of endogenous TCR. Cancer Res. 2009;69(23):9003–11. https://doi.org/10.1158/0008-5472.CAN-09-1450.
Lu YC, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res. 2014;20(13):3401–10. https://doi.org/10.1158/1078-0432.CCR-14-0433.
Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350(6266):1387–90. https://doi.org/10.1126/science.aad1253.
Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014;24(5):743–50. https://doi.org/10.1101/gr.165985.113.
van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013;31(32):e439–42. https://doi.org/10.1200/JCO.2012.47.7521.
Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med. 2016;22(1):26–36. https://doi.org/10.1038/nm.4015.
Gilham DE, Maher J. ‘Atypical’ CAR T cells: NKG2D and Erb-B as examples of natural receptor/ligands to target recalcitrant solid tumors. Immunotherapy. 2017;9(9):723–33. https://doi.org/10.2217/imt-2017-0045.
Cavalieri S, Cazzaniga S, Geuna M, Magnani Z, Bordignon C, Naldini L, et al. Human T lymphocytes transduced by lentiviral vectors in the absence of TCR activation maintain an intact immune competence. Blood. 2003;102(2):497–505. https://doi.org/10.1182/blood-2003-01-0297.
Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3(1):35–45. https://doi.org/10.1038/nrc971.
Thomas S, Hart DP, Xue SA, Cesco-Gaspere M, Stauss HJ. T-cell receptor gene therapy for cancer: the progress to date and future objectives. Expert Opin Biol Ther. 2007;7(8):1207–18. https://doi.org/10.1517/14712598.7.8.1207.
Zhou X, Cui Y, Huang X, Yu Z, Thomas AM, Ye Z, et al. Lentivirus-mediated gene transfer and expression in established human tumor antigen-specific cytotoxic T cells and primary unstimulated T cells. Hum Gene Ther. 2003;14(11):1089–105. https://doi.org/10.1089/104303403322124800.
Zhang Z, Qiu S, Zhang X, Chen W. Optimized DNA electroporation for primary human T cell engineering. BMC Biotechnol. 2018;18(1):4. https://doi.org/10.1186/s12896-018-0419-0.
Singh H, Manuri PR, Olivares S, Dara N, Dawson MJ, Huls H, et al. Redirecting specificity of T-cell populations for CD19 using the sleeping beauty system. Cancer Res. 2008;68(8):2961–71. https://doi.org/10.1158/0008-5472.CAN-07-5600.
Tipanee J, Chai YC, VandenDriessche T, Chuah MK. Preclinical and clinical advances in transposon-based gene therapy. Biosci Rep. 2017;37(6). doi:https://doi.org/10.1042/BSR20160614
Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3(11):999–1005. https://doi.org/10.1038/ni1102-999.
Casucci M, Hawkins RE, Dotti G, Bondanza A. Overcoming the toxicity hurdles of genetically targeted T cells. Cancer Immunol Immunother. 2015;64(1):123–30. https://doi.org/10.1007/s00262-014-1641-9.
Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra38. https://doi.org/10.1126/scitranslmed.3005930.
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25. https://doi.org/10.1126/scitranslmed.3008226.
Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. https://doi.org/10.1126/scitranslmed.3002842.
Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–20. https://doi.org/10.1182/blood-2011-10-384388.
Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540–9. https://doi.org/10.1200/JCO.2014.56.2025.
Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116(20):4099–102. https://doi.org/10.1182/blood-2010-04-281931.
Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8 + and CD4 + CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016;8(355):355ra116. https://doi.org/10.1126/scitranslmed.aaf8621.
Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20(2):119–22. https://doi.org/10.1097/PPO.0000000000000035.
Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy—assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15(1):47–62. https://doi.org/10.1038/nrclinonc.2017.148.
Wang Z, Han W. Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy. Biomark Res. 2018;6:4. https://doi.org/10.1186/s40364-018-0116-0.
Mahadeo KM, Khazal SJ, Abdel-Azim H, Fitzgerald JC, Taraseviciute A, Bollard CM, et al. Management guidelines for paediatric patients receiving chimeric antigen receptor T cell therapy. Nat Rev Clin Oncol. 2018;22:22. https://doi.org/10.1038/s41571-018-0075-2.
Teachey DT, Bishop MR, Maloney DG, Grupp SA. Toxicity management after chimeric antigen receptor T cell therapy: one size does not fit ‘ALL’. Nat Rev Clin Oncol. 2018;15(4):218. https://doi.org/10.1038/nrclinonc.2018.19.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–44. https://doi.org/10.1056/NEJMoa1707447.
Schuster SJ, Bishop MR, Tam C, Waller EK, Borchmann P, Mcguirk J et al. Global pivotal phase 2 trial of the CD19-targeted therapy CTL019 in adult patients with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL)—an interim analysis. In: Oncology H, editor. 14th International conference on malignant lymphoma; 07.06.2017; Lugano (Switzerland): Hematological Oncology; 2017.
Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–95. https://doi.org/10.1182/blood-2014-05-552729.
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–18. https://doi.org/10.1056/NEJMoa1215134.
Klaver Y, van Steenbergen SC, Sleijfer S, Debets R, Lamers CH. T cell maturation stage prior to and during GMP processing informs on CAR T cell expansion in patients. Front Immunol. 2016;7:648. https://doi.org/10.3389/fimmu.2016.00648.
Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018;24(6):731–8. https://doi.org/10.1038/s41591-018-0041-7.
Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739–48. https://doi.org/10.1038/s41591-018-0036-4.
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–28. https://doi.org/10.1016/S0140-6736(14)61403-3.
Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. 2017;7(12):1404–19. https://doi.org/10.1158/2159-8290.CD-17-0698.
Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21(4):904–12. https://doi.org/10.1038/mt.2013.17.
Irving M, de Silly RV, Scholten K, Dilek N, Coukos G. Engineering chimeric antigen receptor T-cells for racing in solid tumors: don’t forget the fuel. Front Immunol. 2017;8:267. https://doi.org/10.3389/fimmu.2017.00267.
Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31(1):71–5. https://doi.org/10.1038/nbt.2459.
Wiseman AC. Immunosuppressive Medications. Clin J Am Soc Nephrol. 2016;11(2):332–43. https://doi.org/10.2215/CJN.08570814.
Jones BS, Lamb LS, Goldman F, Di Stasi A. Improving the safety of cell therapy products by suicide gene transfer. Front Pharmacol. 2014;5:254. https://doi.org/10.3389/fphar.2014.00254.
Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276(5319):1719–24.
Ciceri F, Bonini C, Stanghellini MT, Bondanza A, Traversari C, Salomoni M, et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. Lancet Oncol. 2009;10(5):489–500. https://doi.org/10.1016/S1470-2045(09)70074-9.
Oliveira G, Greco R, Lupo-Stanghellini MT, Vago L, Bonini C. Use of TK-cells in haploidentical hematopoietic stem cell transplantation. Curr Opin Hematol. 2012;19(6):427–33. https://doi.org/10.1097/MOH.0b013e32835822f5.
Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365(18):1673–83. https://doi.org/10.1056/NEJMoa1106152.
Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24(6):1160–70. https://doi.org/10.1038/leu.2010.75.
Straathof KC, Pule MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105(11):4247–54. https://doi.org/10.1182/blood-2004-11-4564.
Introna M, Barbui AM, Bambacioni F, Casati C, Gaipa G, Borleri G, et al. Genetic modification of human T cells with CD20: a strategy to purify and lyse transduced cells with anti-CD20 antibodies. Hum Gene Ther. 2000;11(4):611–20. https://doi.org/10.1089/10430340050015798.
Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. 2014;124(8):1277–87. https://doi.org/10.1182/blood-2014-01-545020.
Kieback E, Charo J, Sommermeyer D, Blankenstein T, Uckert W. A safeguard eliminates T cell receptor gene-modified autoreactive T cells after adoptive transfer. Proc Natl Acad Sci USA. 2008;105(2):623–8. https://doi.org/10.1073/pnas.0710198105.
Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118(5):1255–63. https://doi.org/10.1182/blood-2011-02-337360.
Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. 2016;3:16011. https://doi.org/10.1038/mto.2016.11.
Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115(5):925–35. https://doi.org/10.1182/blood-2009-08-239186.
Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012;4(132):132ra53. https://doi.org/10.1126/scitranslmed.3003761.
Karpanen T, Olweus J. T-cell receptor gene therapy—ready to go viral? Mol Oncol. 2015;9(10):2019–42. https://doi.org/10.1016/j.molonc.2015.10.006.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Institutional Review Board approval was not required because this is a review of the literature.
Conflict of interest
The authors of this manuscript declare relationships with the following companies: Prof. George Coukos, Dr Lionel Trueb, Dr Stefan Zimmermann, Dr Benita Wolf, Dr Melita Irving and Prof. Carline Arber are co-investigators in Iovance and Kite Pharma sponsored clinical studies at CHUV, Lausanne. Prof. Caroline Arber receives royalties from Cell Medica. Prof. Carline Arber has one patent issued in the field of engineered T-cell therapies licensed to Cell Medica in 2016. Prof. Caroline Arber has two provisional patent applications filed in the field of engineered T-cell therapies. Prof. Caroline Arber received consulting fees from Kite/Gilead.
Funding
The authors state that this work has not received any funding.
Additional information
Part of a theme issue on “Safety of Novel Anticancer Therapies: Future Perspectives”. Guest Editors: Rashmi R Shah, Giuseppe Curigliano.
Rights and permissions
About this article

Cite this article
Wolf, B., Zimmermann, S., Arber, C. et al. Safety and Tolerability of Adoptive Cell Therapy in Cancer. Drug Saf 42, 315–334 (2019). https://doi.org/10.1007/s40264-018-0779-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-018-0779-3