
Abstract
Background
Biologicals are important treatment options for various chronic diseases. After the introduction of the first biosimilars, animated debate arose in the scientific community about the actual benefit–risk profile of these drugs. In this context, a comparative safety evaluation of biologicals and biosimilars in clinical practice is warranted.
Methods
We identified all suspected adverse drug reactions (ADRs) concerning biological/biosimilars (excluding vaccines, toxins, blood derivatives, and radio-pharmaceuticals), and further classified them into mechanistic classes. We described the frequency of biological/biosimilar class- and compound-specific ADRs by system organ class (SOC) and type of reporter. We also separately explored the traceability of biologicals and biosimilar-related ADR reports.
Results
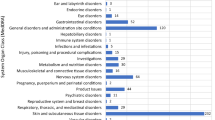
Overall 171,201 ADR reports were collected during the observation period; 9,601 (5.6 %) of these concerned biologicals. Biological-related reports were mainly issued by hospital-based physicians (78.7 %). Most of these reports involved monoclonal antibodies and fusion proteins (66.3 %). Reported ADRs were mainly ‘skin and subcutaneous tissue disorders’ (21 %), ‘general and administration site disorders’ (17 %), and ‘gastrointestinal disorders’ (13.6 %). In terms of traceability, 94.8 % of biological-related reports included an identifiable product name, whilst only 8.6 % indicated the corresponding batch number. Regarding biosimilars, 298 reports were identified, with a low proportion indicating drug ineffectiveness (10.1 %).
Conclusions
Most ADRs attributed to biologicals are ‘skin and subcutaneous tissue disorders’. Anticancer monoclonal antibodies are most frequently associated with ADRs. A low proportion of ADR reports concern biosimilars.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Biological drugs have substantial differences in the safety profile as compared with non-biological drugs. |
Traceability of biological drugs, including biosimilars, requires further improvement in the spontaneous reporting databases with respect to information on batch number. |
Spontaneous reporting databases represent a useful source to evaluate the safety profile and detect emerging safety signals for different classes of biological drugs. |
1 Introduction
A biological medicinal product is a product in which the active material is a biological substance. A biological substance is one that is produced by or extracted from a biological source, such as micro-organisms, organs and tissues of either plant or animal origin, cells or fluids (including blood or plasma) of human or animal origin, or biotechnological cell constructs (cell substrates, whether they are recombinant or not, including primary cells) and for which a combination of physico-chemical-biological testing and the production process and its control is needed for its characterization and the determination of its quality [1].
These drugs have dramatically changed the treatment of important chronic and severe diseases, such as rheumatoid arthritis and some types of cancer.
Biologicals are larger, more complex molecules than traditional chemically synthesized small molecules. Only living organisms are able to produce such complex molecules. Their complexity, as well as the way in which they are synthesized, may result in a degree of variability in molecules of the same active substance, particularly across different batches of the medicine [2].
Most commercially available biologicals are derived from recombinant DNA and hybridoma technologies, which enable the large-scale production of biologicals. Several safety issues, such as the risk of infection, malignancy, or administration reactions, may arise during therapy with many biological medicines.
Biologicals can also trigger immune responses/immunogenicity through the production of anti-drug antibodies. Other particular adverse effects are specifically associated with individual biological agents as a result of their mechanism of action involving selected targets [3, 4].
Although randomized, controlled trials provide preliminary clinically relevant information about major risks of newly marketed therapeutic agents, particularly biologicals, post-marketing data are necessary to gain a better insight into the drug safety profile in routine clinical care. Consequently, it is important to study the safety profile of biological products, including in comparison with chemical entities, by exploring national spontaneous reporting databases.
For suspected adverse reactions relating to biological medicinal products, the definite identification of the product concerned with regard to its manufacturing is of particular importance. As a consequence, and to support pharmacovigilance monitoring, the specific biological medicinal product given to the patient should be clearly identified in the adverse drug reaction (ADR) reports. According to new European legislation, all appropriate measures should be taken to clearly identify the name of the biological product and the batch number [5].
A few years after the expiry of the patents for some biologicals, biosimilars are introduced into the market. Biosimilars are highly similar, but not identical, to the reference product, without any clinically meaningful differences in terms of the quality, safety, and potency of the product. However, clinicians have expressed major concerns about comparative benefit–risk profiles as well as interchangeability of biosimilars and reference products. By 2020, the patents for most of the ‘blockbuster’ biologicals will have expired. Widespread use of biosimilars may substantially decrease expenditure on biologicals, thus contributing to the sustainability of the national health systems [6].
In this context, comparative safety evaluation of biologicals and biosimilars in real-world clinical practice is needed. To date, published data from national/international spontaneous reporting systems (SRSs) about the safety of biological and biosimilars are limited, and none of the available studies were carried out in Italy [7, 8].
The aim of this study is to provide an overview of the safety regarding biologicals and biosimilars using the data from the Italian SRS.
2 Methods
2.1 Data Source
The national SRS database is managed by the Italian Medicines Agency (AIFA) and contains all spontaneous reports of suspected ADRs that have been sent by consumers and all healthcare professionals practicing in Italy since January 2001. Drugs implicated in the ADRs are categorized according to the anatomical therapeutic chemical (ATC) classification. Suspected ADRs are coded according to the Medical Dictionary for Regulatory Activities (MedDRA®).
In the Italian SRS, a procedure has been put in place to automatically detect duplicate cases during data entry. An individual case is identified as a duplicate of another individual case previously submitted on the basis of patient’s initials, age, sex, suspected medicinal products, adverse reactions, and date of onset.
For this study, we selected and analyzed ADR reports that included at least one biological/biosimilar drug during the period 1 January 2001–30 June 2013. We included in the study ADR reports with a certain, probable, or possible causality assessment, according to the Naranjo algorithm.
2.2 Biological Medicines
We compiled a list of biological products based on a previous study [8]. Biological products were further classified into the following mechanistic classes: (1) monoclonal antibodies and fusion proteins; (2) cytokines and antagonists; (3) enzymes and coagulation factors; and (4) recombinant hormones. Vaccines, toxins, blood derivatives, and radio-pharmaceuticals were excluded from the analysis.
From this list, we extracted for our analysis the following biological drugs that are currently marketed in Italy:
-
1.
Recombinant hormones: Insulins and analogs for injection (ATC: A10A), liraglutide (ATC: A10BX07), gonadotropins (ATC: G03A), somatropin (ATC: H01AC01), mecasermin (ATC: H01AC03), somatostatin and analogs (ATC: H01CB), pegvisomant (ATC: H01AX01), glucagon (ATC: H04AA01), teriparatide (ATC: H05AA02), thyrotropin alfa (ATC: H01AB01).
-
2.
Enzymes and coagulation factors: alimentary tract and metabolism enzymes (ATC: A16AB), alteplase (ATC: B01AD02), reteplase (ATC: B01AD07), tenecteplase (ATC: B01AD11), lepirudin (ATC: B01AE02), blood coagulation factors (ATC: B02BD), romiplostim (ATC: B02BX04), pegloticase (ATC: M04AX02), bone morphogenetic proteins (ATC: M05BC), collagenase clostridium histolyticum (ATC: M09AB02), dornase alfa (ATC: R05CB13), rasburicase (ATC: V03AF07), palifermin (ATC: V03AF08).
-
3.
Citokines and antagonists: erythropoietins (ATC: B03XA), colony-stimulating factors (ATC: L03AA), interferons (ATC: L03AB), aldesleukin (ATC: L03AC01).
-
4.
Monoclonal antibodies and chimeras with recombinant immunoglobulin (Ig) parts: abciximab (ATC: B01AC13), antineoplastic monoclonal antibodies (ATC: L01XC), tasonermin (ATC: L03AX11), natalizumab (ATC: L04AA23), abatacept (ATC: L04AA24), eculizumab (ATC: L04AA25), belimumab (ATC: L04AA26), tumor necrosis factor (TNF) alpha inhibitors (ATC: L04AB), interleukin inhibitors (ATC: L04AC), denosumab (ATC: M05BX04), omalizumab (ATC: R03DX05), ranibizumab (ATC: S01LA04).
Biosimilars are currently available in Italy for only three biological products: epoetin alfa, filgrastim, and somatropin. The biosimilars are distinguished by the corresponding reference products looking at the unique Italian national drug code (AIC).
All ADR reports concerning drugs other than biologicals/biosimilars were used as reference group.
2.3 Data Analysis
Frequency analyses were conducted to separately explore the main characteristics of the ADR reports concerning biologicals and biosimilars versus non-biological drugs. ADR reports for biologicals/biosimilars were analyzed as a whole as well as by mechanistic class and individual compounds. In particular, the frequency of type of reporter, ADR seriousness (i.e. fatal, leading to hospitalization, life-threatening), the age and sex distribution of patients affected by the ADR, and the temporal trend in ADR reporting were analyzed.
A comparison of suspected ADRs reported for each biological product of the five mechanistic classes was calculated both overall and by system organ class (SOC). The chi-squared test was used to perform a statistical comparison of the distribution of ADR reports by SOC between biologicals and non-biologicals; differences with p values <0.05 were considered statistically significant.
We further examined the traceability of biologicals and biosimilars by exploring the frequency of availability of the product name and batch numbers in the ADR reports.
3 Results
Overall, 171,201 ADR reports were collected in the Italian SRS during the period January 2001–June 2013. Of these, 9,601 (5.6 %) were related to biologicals and 161,600 were reported for all other drugs (the reference group). Vaccines, toxins, blood derivatives, and radio-pharmaceuticals were excluded from the analysis. When the Naranjo algorithm was applied, the majority of biological-related reports had a possible or probable causal relationship, whilst 20 reports had a score >9 (certain causality). The biological-related reports with an ‘unlikely’ causal relationship (n = 39) were not included in the analysis.
The mean age of patients with a biological-related ADR was higher than those with non-biological ADR reports (57.0 ± 16.3 vs. 49.5 ± 26.0 years) (see Table 1). Two-thirds of ADR reports for biologicals were collected during the period 1 January 2010–30 June 2013. Serious ADRs accounted for 37.8 % (n = 3,629; 155 [4.3 %] fatal cases) of total biological-related reports and 29.5 % (n = 47,736) of non-biological-related reports.
ADRs for biologicals were more frequently reported by hospital-based physicians (78.7 %), while the contribution of these clinicians for non-biological-related ADR reporting was lower (49.0 %), even though this increased when serious ADR reports were specifically analyzed (68.4 %). On the other hand, general practitioners accounted for 16.9 % of non-biological-related ADR reports and much less for those concerning biologicals (2.0 %) (see Fig. 1).

Distribution of adverse drug reaction (%) reports of biologicals by type of reporter. Others = lawyers, pharmaceutical companies; other healthcare professionals: nurses, dentistry, and family pediatricians
Overall, the 9,601 biological-related reports included 13,611 single ADRs (1.4 ADRs per report), whilst the reports concerning the reference group contained 195,256 suspected ADRs (1.2 ADRs per report).
A higher, statistically significant rate of biological-related reports compared with those for non-biologicals was mostly found for ‘blood and lymphatic system disorders’, ‘infections and infestations’, ‘cardiac disorders’, ‘endocrine disorders’, ‘general disorders and administration site conditions’, ‘metabolism and nutrition disorders’, ‘neoplasms benign, malignant and unspecified (including cysts and polyps)’, ‘respiratory, thoracic and mediastinal disorders’, and ‘vascular disorders’ (p < 0.001). Interestingly, significantly higher reporting in favor of biologicals was also observed for the SOC ‘pregnancy, puerperium and perinatal conditions’ (p < 0.05) (see Table 2).
The safety profile of biologicals was studied in more detail by stratifying by mechanistic classes (see Table 3): 6,362 reports (66.3 %) were associated with monoclonal antibodies and fusion proteins, 1,650 (17.2 %) with cytokines and antagonists, 1,365 (14.2 %) with recombinant hormones, 231 (2.4 %) with enzymes and coagulation factors. Antibodies and fusion proteins were mainly associated with cutaneous reactions; cytokines and antagonists with blood and lymphatic system disorders; enzymes and coagulation factors with skin reactions; and hormones with metabolism and nutrition disorders. Neoplasms, infections, and immune system disorders were more frequently reported with monoclonal antibodies.
Concerning therapeutic classes, two-thirds of all ADR reports involved anti-cancer monoclonal antibodies (n = 4,127; 43 %), TNF-alpha inhibitors (n = 1,175; 12.2 %), and interferons (n = 1,150; 12 %). The individual biological agents most frequently implicated in ADRs were bevacizumab (n = 1,295; 13.5 %), cetuximab (n = 1,190; 12.4 %), and rituximab (n = 985; 10.3 %).
Looking at individual compounds and SOCs, ‘neoplasms’ (n = 219) were mainly reported for etanercept (n = 76; 34.7 %), adalimumab (n = 63; 28,8 %) and natalizumab (n = 24; 11 %); among 888 reports of ‘infections’, 20.0 % were attributed to cetuximab (n = 178), 13.7 % to etanercept (n = 122), and 10.6 % to adalimumab (n = 94); ‘metabolic disorders’ (n = 827) (e.g. hypoglycemia) were mainly reported for insulin glargine (n = 551; 66.6 %); and ‘vascular disorders’ (n = 714) for bevacizumab (n = 301, 42.2 %). Most cases of ‘endocrine disorders’ (n = 52) were related to thyroid disorders and were attributed to interferons (n = 48; 92.3 %). ‘Pregnancy, puerperium and perinatal conditions’ included 12 cases of spontaneous abortion, most of which were related to natalizumab and interferons.
3.1 Biosimilars
Concerning biologicals with an expired patent, 298 reports (3.1 % of total biological-related reports) were identified. Among these, 124 (41.6 %) concerned filgrastim, 69 (23.2 %) somatropin, and 105 (35.2 %) epoetin alfa. Specifically, 135 reports were related to biosimilars (45.3 %), and 160 reports (53.7 %) concerned originator drugs; three reports were not assessable because the trade name of the drug substance was not specified.
Stratifying data for these drugs by type of reporter, most of the ADR reports related to innovators and biosimilars were sent by hospital-based physicians (71.9 % for innovators, 79.3 % for biosimilars), in line with general results. Pharmaceutical companies had reported some cases related to biosimilars (8.1 % of the total 135 reports), but not for innovators, whilst specialists’ reporting was slightly higher for innovators (14.4 %) than biosimilars (8.9 %) (data not shown).
A comparison of the nature of the suspected ADRs stratified by SOCs reported for biosimilar products and innovators of the three biologicals with an expired patent (filgrastim, epoetin alfa, somatropin) showed no relevant differences between the suspected ADRs reported in the two groups (see Table 4). Higher, statistically significant rates were found for ‘blood and lymphatic system disorders’, ‘skin and subcutaneous tissue disorders’, and ‘neoplasms’ for innovator drugs, and ‘musculoskeletal and connective tissue disorders’ for biosimilar products. However, the small number of reports does not allow us to draw firm conclusions.
Out of the total 298 reports, 30 (10.1 %) concerned drug ineffectiveness (epoetin alfa 16 and filgrastim 14). By independently applying the Naranjo algorithm through case-by-case revision of therapeutic failures of epoetin alfa, 12 were assessed as possible and four as probable. For filgrastim, ten reports were assessed as possible and four as probable. However, most of the therapeutic failure reports did not contain enough clinical information for an accurate evaluation of causality assessment (e.g. complete laboratory tests, doses, and concomitant drugs).
3.2 Traceability of Biological Products
Traceability of biologicals was evaluated on the basis of the presence of the batch number and the brand name of the suspect drugs in the reports (see Fig. 2). Overall, an identifiable brand name was indicated in 94.8 % of biological-related reports, whilst batch number was present in only 8.6 % of the reports. A higher level of completeness was available for those biologicals with expired patent (brand name of the product present in 98.7 % of reports; batch number in 13.4 %).

Traceability of biologicals in the Italian spontaneous reporting database, measured by the availability of brand name and batch number
4 Discussion
To our knowledge, the present study was the first Italian analysis aimed at exploring the safety of biologicals using the national SRS database. Our data showed that approximately 6 % of all ADR reports collected by the Italian SRS concerned biologicals, with a growing trend during recent years. More than two-thirds of biological-related ADR reports were collected in the period 2010–2013. In addition, as a result of the Directive 2010/84/EC promoting intensive monitoring of biological products, which came into force in Italy in July 2012, the number of biological-related ADR reports is expected to substantially increase in the next few years. This is in line with the 25.8 % increase in the total number of biological-related reports collected in the period July 2012–June 2013 compared with the previous year.
Patients treated with biologicals who experienced a reported ADR were older than patients with ADRs related to non-biological drugs. This result differs from those of another study published in 2010 concerning pharmacovigilance of biologicals in Vigibase [7], in which patients treated with biologicals were younger than patients with ADRs related to the reference group of non-biological drugs.
The percentage of serious ADRs is higher for biologicals than for the reference group (37.8 vs. 29.5 %). This finding may be because most of the biologicals are used for the treatment of severe diseases or disabling conditions, thus influencing the severity of the reported ADRs. In addition, our study showed that 78.7 % of the total biological-related reports were submitted by hospital-based physicians. Specialists play a key role in the pharmacovigilance of these products, as most biologicals are prescribed and monitored in the hospital setting. On the other hand, our data showed that general practitioners (2.0 %) and pharmacists (6.5 %) played a minor role in collecting ADR reports of biologicals. Strategies to incentivize the active involvement of consumers as well as other healthcare professionals (e.g. pharmacists and general practitioners) in ADR reporting concerning biologicals/biosimilars should be implemented in routine care.
In the present study, about two-thirds of all ADR reports involved anti-cancer monoclonal antibodies, TNF-alpha inhibitors, and interferons. The most frequently implicated biological agents were bevacizumab, cetuximab, and rituximab. Even this result differs from the analysis of Vigibase reported by Giezen et al. [7], in which the most reported active substances until 2008 were etanercept, infliximab, and adalimumab. This difference may suggest that the pharmacovigilance of biologicals is dynamic, continuously adapting to new emerging safety issues that are also influenced by drug consumption trends. In addition, as early as 2006, the Italian Medicines Agency implemented registry-based monitoring mainly for anti-cancer biologicals and later, also for other innovative biotechnological drugs used in the therapy of diseases such as psoriasis and rheumatoid arthritis. This may have substantially facilitated ADR reporting in recent years for these drug classes.
Our analysis confirmed that the safety profile for biologicals differs from that for non-biological products and, additionally, showed that substantial differences exist across various mechanistic classes of biologicals.
Most of the ADRs attributed to biologicals are ‘skin and subcutaneous tissue disorders’, and, of these, antibodies and fusion proteins were more frequently reported than other mechanistic classes. According to previous pharmacovigilance studies [3, 7], adverse systemic disorders and administration site reactions, infections, and tumors were significantly associated with the use of biologicals. In our analysis, reporting of ‘general disorders and administration site conditions’, ‘infections’ and ‘neoplasms’ was more likely with biologicals than with non-biologicals. ‘Neoplasms’ were particularly related with anti-TNF-alpha agents, etanercept, and adalimumab.
Biologicals are mainly administered intravenously, which explains why these drugs are frequently associated with infusion-related reactions.
It is well known that serious infections are the most important risks associated with anti-TNF-alpha therapy. In several clinical studies [9, 10], a significant increase in serious infections in patients treated with these drugs has been reported. In addition, post-marketing literature data highlight the risk of developing lymphomas, leukemia, or other malignancies in users of anti-TNF-alpha agents [11]. Suspected ADRs related to the SOC ‘pregnancy, puerperium and perinatal conditions’ were significantly higher for biologicals than for non-biological drugs. These reports included 12 cases of spontaneous abortion, most of which were related to natalizumab and interferons. The published literature includes some reports of spontaneous abortion, mainly due to exposure to interferons [12, 13], thus highlighting that the effect of biological use in pregnant women requires further investigation. Few data are available regarding natalizumab use in pregnancy. A case series of 277 exposed pregnancies to mothers with multiple sclerosis found 31 spontaneous abortions and 23 congenital anomalies [14].
As regards biosimilars, a low number of ADR reports were received in the Italian SRS, in line with a low penetration of these drugs in the market.
In Italy, health policy interventions regarding the management of biologicals/biosimilars interchangeability have been defined more or less recently on a regional basis and in line with the position paper on biosimilars that was issued by the Italian Medicines Agency in 2013 [15]. In particular, the Agency recommends that biosimilars should be preferentially prescribed for treatment-naïve patients. On the other hand, interchangeability is not automatically recommended if a patient has already been treated successfully, irrespective of whether originators or biosimilars were used. In any case, the decision on the drug to be prescribed should be left to the physician.
The late implementation of health policy interventions promoting the use of biosimilars in several regions, as well as clinicians’ skepticism about the actual comparability of the benefit–risk profile between the biological originator and corresponding biosimilars, may have partly contributed to slowing the market penetration of biosimilars in Italy in the early post-authorization phases.
In a general population of Southern Italy, only around 2 % of epoetin users received a biosimilar during the years 2010–2011 [16]. The Italian national report on medicine use showed that epoetin biosimilars accounted only for around 10 % of total epoetin consumption (measured as defined daily dose per 1,000 inhabitants per day) in 2012; this proportion was twice that of the previous year [17]. More recently, a progressively increasing use of biosimilars has been documented in Italy, as reported in the 2013 Italian national report on drug use [ 18 ]. A larger number of reports for biosimilars are thus expected in future years due to healthcare policies promoting widespread use of these less costly drugs, which may contribute to the sustainability of the healthcare system.
Interestingly, 10 % of ADR reports for biosimilars indicate drug ineffectiveness. This finding, in our opinion, should be interpreted cautiously in light of the limited clinical information available in the reports to substantiate the association between drugs and ineffectiveness.
In general, SRSs can hardly be used for assessing the effectiveness of a product because of the interference of several uncontrolled variables, including the clinical characteristics of the drug or the individual patient’s response to the effects of the drug. Furthermore, patterns of suspected stimulated reporting could be identified in spontaneous reporting databases, related for instance to business competition, prescriber’s skepticism, or healthcare policies focused on implementing biosimilar prescriptions [19]. This makes the evaluation of therapeutic failure reports difficult. On the other hand, spontaneous reports may help in identifying cases of therapeutic ineffectiveness due to pharmaceutical defects or drug interactions.
In general, the quality of biological products depends strictly on their manufacturing process. Therefore, careful monitoring of the safety of biologicals for each specific batch and brand name is needed in clinical practice. Consequently, to ensure traceability and robust post-marketing assessment of biological products, European regulatory authorities encourage clinicians to report ADRs concerning biological products by specifying the exact brand name and batch number [20].
In the Italian SRS, almost all biological-related reports indicated the brand name of the product, while only a few also contained the batch number. A previous study found that the batch number was present in a higher percentage (21.1 % reports) of biopharmaceutical-ADR reports from EudraVigilance [8]. In our study, ADR reports for biologicals whose patent had expired were more complete. A similar pattern was seen in EudraVigilance [8].
To enable the identification of safety problems at an early stage, traceability of the biologicals implicated in the ADRs should be promoted, stimulating ADR reporters to also register data on the batch numbers [3]. Accurate batch traceability is pivotal for the identification of any batch-related ADR problems with biologicals, e.g. pathogen-contaminated batches [21] or other host cell impurities [22].
This study adds important information to the general knowledge about the safety profile of biologicals. However, several potential limitations warrant warnings. Under-reporting of suspected ADRs is a well recognized problem when using data from SRSs. The absence of drug exposure data does not allow any estimation of the incidence rate of ADRs. Another limitation is that the reporting rate can depend on the length of time the drug was on the market and by the frequently selective reporting of biologicals involved in intensive ADR-monitoring projects and ad hoc prescription registries (see also http://monitoraggio-farmaci.agenziafarmaco.it/) or implicated in health policy reimbursement procedures that require ADR assessment.
As regards biological medicinal products, causality assessment of spontaneous reports are potentially difficult because these drugs are often indicated to treat severe and/or life-threatening diseases (confounding by indication might have influenced the results) and because of the lack of important clinical information (i.e. missing data) for a proper evaluation of causality. In particular, the evaluation of causality assessment for therapeutic failure reports is limited by the lack of relevant clinical information as well as details about the manufacturing process. Nevertheless, in light of the inherent limitations of the pre-marketing randomized clinical trials in identifying all the possible ADRs occurring during treatment with newly marketed drugs, spontaneous reporting remains an important tool for early detection of signals.
5 Conclusions
The study results highlight the importance of national spontaneous reporting databases to explore the safety profile of biologicals compared with chemical entities. We documented substantial differences between biologicals and non-biologicals concerning the reporting of some serious ADRs such as infections and tumors.
In the Italian SRS, almost all biological-related reports indicated the brand name of the product. However, the traceability of biologicals (both originators and biosimilars) through to the batch number is currently limited. As the number of biological medicines on the market as well as their use in clinical practice is expected to significantly increase in the future, strategies to further increase the traceability of biological drugs at batch level are needed.
References
Coordination Group for Mutual Recognition and Decentralised Procedures. Questions & answers biologicals. CMDh/269/2012, October 2012. http://www.hma.eu. Accessed 30 July 2014.
European Medicinal Agency. Questions and answers on biosimilar medicines (similar biological medicinal products) 2012. EMA/837805/2011. http://www.ema.europa.eu. Accessed 30 July 2014.
Giezen TJ, Mantel-Teeuwisse AK, Leufkens HGM. Pharmacovigilance of biopharmaceuticals. Drug Saf. 2009;32(10):811–7.
Giezen TJ, Mantel-Teeuwisse AK, Straus SMJM, Schellekens H, Leufkens HG, Egberts AC. Safety-related regulatory actions for biologicals approved in the United States and the European Union. JAMA. 2008;300(16):1887–96.
European Medicines Agency, Heads of Medicines Agencies. Guideline on good pharmacovigilance practices (GVP)—module VI 2012. EMA/873138/2011. http://www.ema.europa.eu. Accessed 30 July 2014.
Simoens S. Biosimilar medicines and cost-effectiveness. ClinicoEcon Outcomes Res. 2011;3:29–36.
Giezen TJ, Mantel-Teeuwisse AK, Meyboom RHB, Straus SMJM, Leufkens HGM, Egberts TCG. Mapping the safety profile of biologicals. A disproportionality analysis using the WHO Adverse Drug Reaction Database, VigiBase. Drug Saf. 2010;33:865–78.
Vermeer NS, Straus SMJM, Mantel-Teeuwisse AK, Domergue F, Egberts TCG, Leufkens HGM, De Bruin ML. Traceability of biopharmaceuticals in spontaneous reporting systems: a cross-sectional study in the FDA Adverse Event Reporting System (FAERS) and EudraVigilance Databases. Drug Saf. 2013;36:617–25.
Strangfeld A, Listing J, Herzer P, Liebhaber A, Rockwitz K, Richter C, Zink A. Risk of herpes zoster in patients with rheumatoid arthritis treated with anti TNF-alpha agents. JAMA. 2009;301(7):737–44.
Listing J, Strangfeld A, Kary S, Rau R, Von Hinueber U, Stoyanova-Scholz M, Gromnica-Ihle E, Antoni C, Herzer P, Kekow J, Schneider M, Zink A. Infections in patients with rheumatoid arthritis treated with biologic agents. Arthritis Rheum. 2005;52(11):3403–12.
Meyboom RH, Star K, Bate J, Savage R, Edwards IR. TNF-a inhibitors and leukaemia: international pharmacovigilance reports. Drug Saf. 2008;31(5):445–7.
Sandberg-Wollheim M, Alteri E, Stam Moraga M, Kornmann G. Pregnancy outcomes in multiple sclerosis following subcutaneous interferon beta-1a therapy. Mult Scler J. 2011;17(4):423–30.
Lu E, Wang BW, Guimond C, Synnes A, Sadovnick D, Tremlett H. Disease-modifying drugs for multiple sclerosis in pregnancy: a systematic review. Neurology. 2012;79(11):1130–5.
Cristiano LM, Bozic C, Bloomgren G. Preliminary evaluation of pregnancy outcomes from the Tysabri (natalizumab) pregnancy exposure registry. Mult Scler. 2011;17(suppl 10):A457 (Abstract).
Italian Medines Agency. AIFA Position Paper. I farmaci biosimilari. 2013. http://www.agenziafarmaco.gov.it/sites/default/files/AIFA_POSITION_PAPER_FARMACI_BIOSIMILARI.pdf. Accessed 30 July 2014.
Loiacono C, Sgroi C, Coppolino S, Cannata A, Ferrara R, Arcoraci V, Cananzi P, Savica V, Schuemie M, Caputi AP, Trifirò G. How much are biosimilars used in Southern Italy? A retrospective analysis of epoetin utilization in the Local Health Unit of Messina in the Years 2010–2011.
Agenzia Italiana del Farmaco. L’uso dei farmaci in Italia. Rapporto Nazionale Anno. 2012.http://www.agenziafarmaco.gov.it/it/content/osservatorio-sull%E2%80%99impiego-dei-medicinali-osmed. Accessed 5 February 2014.
Agenzia Italiana del Farmaco. L’uso dei farmaci in Italia. Rapporto Nazionale Anno. 2013. http://www.agenziafarmaco.gov.it/it/content/osservatorio-sull%E2%80%99impiego-dei-medicinali-osmed. Accessed 28 July 2014.
Gagne JJ, Bykov K. On analyzing therapeutic ineffectiveness reports. Pharmacoepidemiol Drug Saf. 2013;22:207–8.
Calvo B, Zuñiga L. EU’s New Pharmacovigilance Legislation: considerations for biosimilars. Drug Saf. 2014;37:9–18.
Vonberg RP, Gastmeier P. Hospital-acquired infections related to contaminated substances. J Hosp Infect. 2007;65(1):15–23.
Thorpe SJ, Fox BJ, Dolman CD, Lawrence J, Thorpe R. Batches of intravenous immunoglobulin associated with adverse reactions in recipients contain atypically high anti-Rh D activity. Vox Sang. 2003;85(2):80–4.
Funding
The study was conducted in the context of the project “Assessment of short and long term risk-benefit profile of biologics through healthcare database network in Italy”, code: RF-2010-2320172, which was funded by the Italian Health Ministry.
Conflict of interest
Professor Achille P. Caputi has received consulting fees that are not related to the content of this study from the following pharmaceutical companies: Bristol-Myers-Squibb, Otsuka, Novartis, Teva and Novo Nordisk. Paola M. Cutroneo, Valentina Isgrò, Alessandra Russo, Valentina Ientile, Laura Sottosanti, Giuseppe Pimpinella, Anita Conforti, Ugo Moretti, and Gianluca Trifirò have no conflicts of interest that are directly relevant to the content of this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Cutroneo, P.M., Isgrò, V., Russo, A. et al. Safety Profile of Biological Medicines as Compared with Non-Biologicals: An Analysis of the Italian Spontaneous Reporting System Database. Drug Saf 37, 961–970 (2014). https://doi.org/10.1007/s40264-014-0224-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-014-0224-1