
Abstract
Background
Adverse drug reactions (ADRs) of biopharmaceuticals can be batch or product specific, resulting from small differences in the manufacturing process. Detailed exposure information should be readily available in systems for postmarketing safety surveillance of biopharmaceuticals, including spontaneous reporting systems (SRSs), in which reports of ADRs are collected.
Objective
The aim of this study was to explore the current status of traceability of biopharmaceuticals in the US and the EU up to patient level in SRSs.
Design and Setting
A cross-sectional study was conducted over the period 2004–2010, including ADR reports from two major SRSs: the FDA Adverse Event Reporting System (FAERS) in the US and EudraVigilance (EV) in the EU.
Main Outcome Measures
The availability of batch numbers was determined for biopharmaceuticals, and compared with small molecule drugs. For biopharmaceuticals for which a biosimilar has been approved for marketing in the EU, the identifiability of the product (i.e. the possibility of distinguishing the biosimilar from the reference biopharmaceutical) was determined.
Results
A total of 2,028,600 unique ADR reports were identified in the FAERS, reporting a total of 591,380 biopharmaceuticals (of which 487,065 were suspected). In EV there were 2,108,742 unique ADR reports, reporting a total of 439,971 biopharmaceuticals (356,293 suspected). Overall, for 24.0 % of the suspected biopharmaceuticals in the FAERS and 7.4 % of the suspected small molecule drugs (p < 0.001) batch numbers were available. A similar pattern was seen in EV: for 21.1 % of the suspected biopharmaceuticals batch numbers were available, compared with only 3.6 % of the small molecule drugs (p < 0.001). In both SRSs, consumers were most likely to report a batch number for suspected biologicals (36.3 % in the FAERS and 40.7 % in EV). A total of 13,790 biopharmaceuticals (9,759 suspected) for which a biosimilar has been approved in the EU were identified in EV. For 90.4 % of these biopharmaceuticals and 96.2 % of the suspected biopharmaceuticals the product was clearly identifiable.
Conclusion
This study underlines the need for improving traceability of biopharmaceuticals, in particular with respect to individual batches, allowing better identification and monitoring of postmarketing safety issues related to biopharmaceuticals.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Background
Biopharmaceuticals provide innovative and effective therapies for often severe and life-threatening diseases. Because of their specific characteristics, biopharmaceuticals have been associated with specific safety concerns, and challenges in pharmacovigilance and risk management [1, 2]. One of the distinctive properties of biopharmaceuticals is that the safety profile may change over time, resulting from changes in the manufacturing or formulating process. The unexpected increased occurrence of pure red cell aplasia in patients treated with recombinant human erythropoietin outside the US is an example of a postmarketing safety issue associated with such a specific characteristic of biopharmaceuticals [3]. More than 200 cases of this rare and severe haematological disorder have been attributed to the altered immunogenicity of an epoetin alfa for which formulation changes had recently been issued [4].
Biopharmaceuticals are subject to frequent manufacturing changes once a marketing authorization has been granted. Although these changes are adopted to benefit public health, e.g. by improving product properties or product yield, alterations in the production process may adversely impact the product quality attributes [5, 6]. Consequently, the clinical efficacy and safety, in particular immunogenicity, may be affected. Several examples, apart from epoetin alfa, are known whereby immunogenicity with potentially serious clinical consequences was associated with manufacturing and/or formulation changes, including thrombocytopenia with thrombopoietin and neutralizing antibodies with human growth hormone [6]. To ensure patient safety, regulatory authorities in the US and EU have adopted extensive guidance for evaluating comparability of biopharmaceuticals pre- and postmanufacturing changes [7, 8]. This comparability exercise represents a challenging task for manufacturers and regulatory authorities as there is no set of analytical techniques that can fully describe the structural properties of the biopharmaceutical [9]. Moreover, when substantial alterations in the structure products are found, regulatory authorities have the difficult task of deciding whether the identified changes are acceptable [10], i.e. that the changes don’t affect the clinical performance of the biopharmaceutical.
Determining therapeutic equivalence is an even more difficult task when the whole manufacturing process of a biopharmaceutical is redeveloped by a second manufacturer, which is the case for biosimilars [11]. Manufacturers of biosimilars do not have access to the manufacturing process of the reference product since this is proprietary knowledge [12]. Consequently, the independent development of a new manufacturing process is likely to result in structural differences between biosimilars and their reference products, possibly affecting the product’s immunogenicity [13]. Recently, biosimilar development has been receiving increasing attention due to expiring patent protection of top-selling biopharmaceuticals [14] and the need for cutting healthcare spending in the Western world [15]. It is already estimated that in 2015 biosimilars will represent approximately 40 % of the total worldwide biopharmaceutical market [16].
Ensuring the traceability of biopharmaceuticals up to batch and product level is essential in view of the risk of batch- or product-specific adverse drug reactions (ADRs). Detailed exposure information should be readily available in systems for postmarketing safety surveillance of biopharmaceuticals, including spontaneous reporting systems (SRSs), in which reports of ADRs are collected. It is known that SRS have played a pivotal role in detecting postmarketing safety issues for small molecule drugs in the past. These systems could also play an essential role in detecting and monitoring any future batch- or product-specific safety issues of biopharmaceuticals, provided that this information is captured. The current study therefore aims to explore the current status of traceability of biopharmaceuticals in the US and the EU up to patient level in SRSs.
2 Methods
The traceability of biopharmaceuticals was studied in a cross-sectional study over the period 2004–2010, including spontaneous ADR reports from two major SRS: the FDA Adverse Event Reporting System (FAERS) in the US and EudraVigilance (EV) in the EU.
2.1 FDA Adverse Event Reporting System Data Content and Structure
The FAERS was established in 1969 to support the postmarketing safety surveillance of the US FDA. The FAERS database encompasses individual case safety reports (ICSRs) for the majority of FDA-approved medicinal products. An ICSR is defined as the information provided by a primary source to describe suspected ADRs related to the administration of one or more medicinal products to an individual patient at a particular point of time [17]. The FDA receives ICSRs directly from consumers or healthcare professionals, or indirectly through manufacturers, when the ADR is initially brought to their attention. Manufacturers have an obligation to periodically report both serious and non-serious ADRs that occurred in domestic (US) clinical practice, and serious unexpected ADRs occurring in the US or a third country [18, 19]. FAERS data is freely accessible under the Freedom of Information Act, and FAERS data from 2004 onwards are directly available from the website of the FDA (http://www.fda.gov/Drugs/InformationOnDrugs/ucm135151.htm).
The FDA may receive multiple ICSRs, e.g. follow-up reports from the previous or a second reporter, referring to the same occurrence of ADRs in the same patient on the same time. All initial and follow-up ICSRs appear in the FAERS. If correctly linked, follow-up reports on an initial ICSR are identifiable in the FAERS upon identical ‘case numbers’.
2.2 EudraVigilance Data Content and Structure
EV was established in 2001 to collect ICSRs of (serious) ADRs to medicines licensed within the EU. ICSRs are received indirectly through EU national competent authorities and pharmaceutical companies. Pharmaceutical companies have a legal obligation to report all serious unexpected ADRs and any suspected transmission via a medicinal product of any infectious agents occurring outside the EU which are brought to their attention. National competent authorities are required to report any serious ADR occurring within the EU [20]. EV data was obtained through a request for access to data according to the EV Access Policy, as data from EV has only recently become accessible for research purposes. Recently, the European Medicines Agency (EMA) has begun publishing suspected ADR reports on their website to foster transparency (http://www.adrreports.eu).
An ICSR in EV reflects the most recent and comprehensive information on an event of an ADR. Follow-up ICSRs by the same reporter will automatically replace the previous ICSR. Duplicate ICSRs from multiple reporters are merged into ‘master cases’, containing the most comprehensive information from the individual ICSRs [17].
2.3 Data Extraction and Handling
All spontaneous ICSRs over the period 2004–2010 were selected from FAERS and EV. ICSRs originating from literature and clinical studies were not of interest as the current study aims to describe batch and product traceability in clinical practice. All literature and study ICSRs were therefore omitted from the FAERS (n = 156,776). Only spontaneous ICSRs were requested from EV.
All drug information from ICSRs in the FAERS referring to the same case number were merged into a single cumulative ICSR, similar to the EV approach mentioned above. One cumulative ICSR contains all unique drugs reported over time within these ICSRs. Duplicate drugs within the same ICSR were merged in EV. These steps were undertaken to avoid duplication of data in our final unit of analysis: one medicinal product, subject of a suspected ADR report, administered to an individual patient at a particular point of time. Figure 1 illustrates how EV and FAERS data were processed to our unit of analysis.

Processing original FAERS and EV ICSRs to cumulative ICSRs. EV EudraVigilance, FAERS FDA Adverse Event Reporting System, ICSR Individual Case Safety Report
The following data was subsequently extracted from all cumulative ICSRs from EV and the FAERS: information on name of the drug and/or active substances, batch number, name of marketing authorization holder (EV only), role code of the drug (see Sect. 2.4), type of reporter and reporting date. In line with the International Conference on Harmonisation (ICH) E2B guideline, the following reporters are distinguished: physician, pharmacist, other health professional, lawyer and consumer. Since the reporter on a cumulative ICSR may be non-unique (due to duplicate reporting) a separate category was assigned for multiple reporters.
2.4 Classification of Medicinal Products
Biopharmaceuticals were defined as protein- or nucleic-based pharmaceutical products used for therapeutic or in vivo diagnostic purposes [21]. Medicinal products were classified into two groups: (i) biopharmaceuticals; and (ii) small molecule drugs using the WHO Anatomical Therapeutic Chemical (ATC) classification system (http://www.whocc.no/atc_ddd_index/; see Table S1 [Online Resource 1]). Two pharmacists confirmed the classification of the data. Medicinal products not classifiable to either group (e.g. verbatim data entered as ‘unspecified drug’ or ‘radiation therapy’) were excluded. Moreover, vaccines (ATC class J07) and whole blood or components of whole blood (ATC class B05A) were excluded as they are subject to different reporting requirements. The role code of the medicinal product was recoded into suspected (classified as ‘primary suspect’ or ‘secondary suspect’ in the FAERS or ‘suspect’ in EV) and non-suspected (classified as ‘interacting’ or ‘concomitant’ in both SRSs).
2.5 Classification of Traceability
Verbatim data entered in the designated field for batch number was recoded to a dichotomous variable describing the availability of the batch number. Any verbatim data entered into the designated field for batch number was considered to be a batch number. To validate whether the verbatim data did not contain any information referring to the unavailability of a batch number (e.g. ‘unknown’ or ‘discarded package’), data were aggregated and carefully reviewed. A second reviewer reviewed the determination of the availability of batch numbers.
For biopharmaceuticals for which a biosimilar has been approved for marketing in the EU (see Table S2 [Online Resource 1]), the identifiability of the product in the cumulative ICSRs in EV was determined. For this analysis we included only ICSRs reported from the month following the approval of the first biosimilar within that product class: epoetin alfa, filgrastim and somatropin. Product names were considered identifiable when the brand name or the international nonproprietary name (INN) plus the name of the marketing authorization holder were available. Products for which only the INN was available were considered non-identifiable, except for epoetin zeta, for which product the INN differs from the innovator (epoetin alfa).
2.6 Data Analysis
Descriptive statistics were calculated as proportions to describe the traceability of batch numbers for biopharmaceuticals and small molecule drugs in the FAERS and EV. Results were further stratified by the role code of drug and the type of reporter to identify whether proportions differed over the different variables. For the top eight most frequently reported ATC classes (see Table S1 [Online Resource 1]) the availability of batch numbers was calculated overall, and further stratified by type of reporter (medical doctor, pharmacist or consumer). The product identifiability, and subsequent batch traceability, for biopharmaceuticals for which a biosimilar has been approved in the EU were also calculated as proportions. Significance was tested using Chi-square statistics. All analyses were performed using SPSS statistical software version 18 (IBM Software, Chicago, IL, USA).
3 Results
A total of 2,028,600 and 2,108,742 cumulative ICSRs were reported in the FAERS and EV, respectively. Within these cumulative ICSRs, a respective total of 591,380 (from the 6,603,489) and 439,971 (from the 6,431,175) medicinal products concerned biopharmaceuticals (see Fig. 2).

Number of ICSRs and medicinal products in original dataset, and dataset for analysis. EV EudraVigilance, FAERS FDA Adverse Event Reporting System, ICSR Individual Case Safety Report
Overall, for 24.0 % of the suspected biopharmaceuticals in the FAERS and 7.4 % of the suspected small molecule drugs (p < 0.001), batch numbers were available. A similar pattern was seen in EV: for 21.1 % of the suspected biopharmaceuticals, batch numbers were available, compared with only 3.6 % of the small molecule drugs (p < 0.001). The traceability of individual batches for the overall group of drugs in the FAERS and EV, including concomitant and interacting drugs, was less well guaranteed (see Table 1).
For the 487,065 suspected biopharmaceuticals in the FAERS and 356,293 suspected biopharmaceuticals in EV, the availability of batch numbers was calculated for different reporter types. Biopharmaceuticals reported by consumers most frequently contained a batch number (36.3 % in the FAERS and 40.7 % in EV). Batch traceability of biopharmaceuticals reported by physicians in the FAERS (13.3 %) and EV (7.0 %) was substantially lower (both p < 0.001). Biopharmaceuticals reported by pharmacists or other healthcare professionals were also less likely to contain a batch number (see Table 2).
This pattern differed, however, between the eight most frequently reported pharmacological and therapeutic subgroups of biopharmaceuticals. Overall, batch numbers were most frequently available for parathyroid hormone (H05AA) in the FAERS (43.1 %) and immunoglobulins (J06) in EV (42.0 %). Most notably, the overall availability of batch numbers for parathyroid hormone in the FAERS was higher than in all three separate reporter categories (see Fig. 3), representing the high availability of batch numbers in reports of which the reporter was not known (62.5 %, not shown in Fig. 3). Respectively, 43.1 % and 44.2 % of the tumour necrosis factor (TNF)-α inhibitors, the most frequently reported ATC class, reported by consumers contained a batch number in the FAERS and EV. Batch traceability of TNF-α inhibitors reported by physicians and pharmacists was lower in the FAERS (16.2 % and 13.5 %, respectively), and particularly in EV (3.8 % and 10.5 %, respectively). Pharmacists did remarkably well in reporting batch numbers for immunoglobulins in the FAERS (59.4 %) and EV (55.2 %). The traceability of antineoplastic monoclonal antibodies (ATC class L01XC) was overall poorly maintained (see Fig. 3).

Availability of batch numbers (%1) for eight groups of suspected biopharmaceuticals in FAERS (a) and EV (b), stratified by type of reporter. 1Calculated as number of biopharmaceuticals containing batch number/number of biopharmaceuticals reported by reporter within group. EV EudraVigilance, FAERS FDA Adverse Event Reporting System
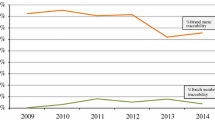
There was variability in the traceability of suspected biopharmaceuticals over time for EV and the FAERS (see Fig. 4). Batch traceability of biopharmaceuticals in EV showed a sharp increase between 2007 (10.7 % of reported biopharmaceuticals contained a batch number) and 2008 (22.8 % contained a batch number). Batch traceability of biopharmaceuticals in the FAERS showed a peak in 2007, with 35.8 % of the reported biopharmaceuticals containing a batch number, but declined thereafter.

Availability of batch number for suspected biopharmaceuticals from 2004 until 2010 in FAERS and EV. EV EudraVigilance, FAERS FDA Adverse Event Reporting System
A total of 13,790 biopharmaceuticals (of which 9,759 were suspected) for which a biosimilar has been approved in the EU were extracted from cumulative ICSRs in EV. For 90.4 % of these biopharmaceuticals, and 96.2 % of the suspected biopharmaceuticals, the product was clearly identifiable. The batch traceability of biosimilars was, interestingly, poorly maintained (see Table 3).
4 Discussion
The present study showed that for 24.0 % of the suspected biopharmaceuticals in the FAERS and 21.1 % of the suspected biopharmaceuticals in EV a batch number was available. In addition, the current study showed that for 96.2 % of the suspected biopharmaceuticals for which a biosimilar was available in the EU the product name was clearly identifiable in EV. Accurate traceability of biopharmaceuticals up to batch and product level in these SRSs is essential for identifying and monitoring any batch- or product-specific safety issues, resulting from differences in the manufacturing process. Batch traceability may, in addition, help to distinguish between, and assess the safety profile over, different pharmaceutical forms and dosage strengths of biopharmaceuticals. Furthermore, accurate batch traceability is pivotal for relating any batch-related problems of biopharmaceuticals, e.g. pathogen-contaminated batches [22] or other host cell impurities [23], to reported ADRs. Biopharmaceuticals might be at increased risk of batch-related problems as the production process, which involves living expression systems, gives rise to a large number of host cell-, process- and product-related impurities.
The proportions of biopharmaceuticals containing a batch number in the FAERS and EV were much higher than we found for suspected small molecule drugs (7.4 % and 3.6 %, respectively), but lower than elsewhere reported for vaccines (54.4 %) [24]. The lack of information on batch numbers for approximately three in four biopharmaceuticals in the FAERS (approximately four in five in EV) could either be the result of incomplete recording of exposure information at dispensing or incomplete reporting of the available information to regulatory authorities and/or manufacturers. The reported differences in batch traceability between different pharmacological/therapeutic groups of biopharmaceuticals suggest, in particular, a role for incomplete recording of exposure information in clinical practice. Whereas consumers reported a batch number for 36.2–46.8 % of frequently home-administered insulins, they reported a batch number for only 1.3–7.5 % of antineoplastic monoclonal antibodies, which are primarily administered in hospitals. For consumers it is relatively easy to obtain batch numbers if the medicine is applied at home, as is the case for insulins. As in the case of antineoplastic agents, the preparation, administration and reporting might very well be by different persons, and the patient or physician confronted with the ADR might not have access to the batch information. This indicates that once the batch numbers are readily available consumers are likely to report this information.
Another finding from the current study was that batch traceability was, overall, well maintained for immunoglobulins, particularly when reported by pharmacists. Overall, batch numbers were available for 36.7–42.0 % of the reported immunoglobulins, and 55.2–59.4 % of the immunoglobulins reported by pharmacists. This might be explained by the fact that immunoglobulins were historically plasma-derived medicinal products, for which separate regulations are in place. The safety of blood-derived products has been under close scrutiny, especially in Europe, following the HIV-infected blood-products scandal that occurred in France in the 1990s [25], and concerns for transmission of Creutzfeldt-Jakob Disease via whole blood and plasma-derived products in Europe [26], although the latter has only been a theoretical risk until 2009 [27].
The current study showed that patients play an important role in ensuring traceability of biopharmaceuticals. In 36.3 % and 40.7 % of the consumer reports on biopharmaceuticals a batch number was available in the FAERS and EV, respectively. These results underpin the importance of patient reporting of adverse events. Patients have been able to report adverse events to the FAERS since its establishment in 1969. In most European countries, patient reporting schemes have only recently been established and in some countries patients are still not able to report adverse events directly to the competent authorities [28]. On the other hand, we showed that physicians played only a minor role in ensuring traceability of biopharmaceuticals. Despite being a major contributor in the absolute number of reports, only in 13.3 % and 7.0 % of the reports on biopharmaceuticals in the FAERS and EV, respectively, was a batch number available.
The present study showed that product identifiability of biopharmaceuticals for which a biosimilars has been approved in the EU is reasonably well ensured in Europe, especially for epoetin alfa. This is an important finding, taking into account that biosimilars are frequently given the same INN as the reference innovator. Of the six currently approved biosimilars in Europe (sold under 12 different trade names), five contain the same INN as the innovator (see Table S2 [Online Resource 1]). It has therefore been recognized that the INN system, although playing an important role in global pharmacovigilance, cannot be relied upon for product identification of biosimilars [29]. As the number of biosimilars on the market is expected to increase in a vast pace, and a road for biosimilar registration is currently been paved in the US [30], traceability of biosimilars will only become increasingly important.
The need to ensure traceability is not unique to biopharmaceuticals, but is also a well-known aspect in numerous other industries [31]. In particular, the traceability of medical devices is receiving increased scrutiny following recent concerns in Europe of a possible association between frequently used PIP (Poly Implant Prothèse) breast implants and cancer [32]. The European Commission has already announced plans to enhance traceability of medical devices [33]. Similarly, several initiatives are currently ongoing to further promote the traceability of biopharmaceuticals. These initiatives are not only fuelled by increased interest in drug safety, but also by the need for improving supply chain efficacy and the need for taking measures against counterfeit medicines [31]. Two-dimensional barcodes that could include detailed product information such as batch numbers is one of the presented solutions for promoting traceability of biopharmaceuticals [34]. When such information is automatically recorded in clinical practice, it is essential that patients and healthcare professionals are aware that this accurate exposure information is necessary to link the adverse event properly to the specific product. Regulatory authorities in the product information of biopharmaceuticals may encourage patients and healthcare professionals to report such information. This may be one of the increased efforts taken by the EU and its Member States to ensure traceability of biopharmaceuticals, demanded by recently adopted new European pharmacovigilance legislation [35].
It should be noted that SRSs may not be the sole point in the community where detailed exposure information on biopharmaceuticals is captured. In the current study we did not assess whether population-based databases or disease registries contain the necessary exposure information to monitor and ascertain the safety of biopharmaceuticals over different batches or products. A second limitation that should be addressed is that the databases we have used might contain a large number of duplicate reports. In particular, extensive duplication of reports has been reported in the FAERS [36]. To limit any influence of duplicate information on our study results, efforts were undertaken to reduce data duplication. As we were, however, not interested in the frequency of certain adverse events but only in the reporting quality of the submitted reports, we feel any residual duplication might have little influence on our results.
5 Conclusion
The present study was, to our knowledge, the first study to explore the current status of traceability of biopharmaceuticals in major SRSs of ADRs in the US and the EU. We have reported that the current system insufficiently ensures the traceability of individual batches of biopharmaceuticals, although the identifiability of biosimilars is reasonably well ensured. Stakeholders in pharmacovigilance should undertake efforts to improve the traceability of biopharmaceuticals.
References
Giezen TJ, Mantel-Teeuwisse AK, Leufkens HG. Pharmacovigilance of biopharmaceuticals: challenges remain. Drug Saf. 2009;32(10):811–7.
Declerck PJ. Biotherapeutics in the era of biosimilars: what really matters is patient safety. Drug Saf. 2007;30(12):1087–92.
Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian JJ, Martin-Dupont P, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346(7):469–75.
Ebbers HC, Mantel-Teeuwisse AK, Moors EH, Schellekens H, Leufkens HG. Today’s challenges in pharmacovigilance: what can we learn from epoetins? Drug Saf. 2011;34(4):273–87.
Chirino AJ, Mire-Sluis A. Characterizing biological products and assessing comparability following manufacturing changes. Nat Biotechnol. 2004;22(11):1383–91.
Sharma B. Immunogenicity of therapeutic proteins. Part 3: impact of manufacturing changes. Biotechnol Adv. 2007;25(3):325–31.
European Medicines Agency. Note for guidance on biotechnological/biological products subject to changes in their manufacturing process (CPMP/ICH/5271/03). June 2005. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002805.pdf. Accessed 7 May 2013.
US FDA. Demonstration of comparability of human biological products, including therapeutic biotechnology-derived products. April 1996. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm122879.htm. Accessed 7 May 2013.
Schellekens H. When biotech proteins go off-patent. Trends Biotechnol. 2004;22(8):406–10.
Schiestl M, Stangler T, Torella C, Cepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29(4):310–2.
Buttel IC, Chamberlain P, Chowers Y, Ehmann F, Greinacher A, Jefferis R, et al. Taking immunogenicity assessment of therapeutic proteins to the next level. Biologicals. 2011;39(2):100–9.
Mellstedt H, Niederwieser D, Ludwig H. The challenge of biosimilars. Ann Oncol. 2008;19(3):411–9.
Schellekens H. Bioequivalence and the immunogenicity of biopharmaceuticals. Nat Rev Drug Discov. 2002;1(6):457–62.
Walsh G. Biopharmaceutical benchmarks 2010. Nat Biotechnol. 2010;28(9):917–24.
Wadman M. Bills target biosimilar drugs. Nature. 2009;458:394–5.
McCamish M, Woollett G. Worldwide experience with biosimilar development. MAbs. 2011;3(2):209–17.
European Medicines Agency. Note for guidance—EudraVigilance human—processing of safety messages and individual case safety reports (ICSRs). 15 October 2010. http://eudravigilance.ema.europa.eu/human/docs/guid%C2%AFP%C2%AFTechnical%20Documentation%C2%AFEMEA-H-20665-04-en-Final_Revision_2.pdf. Accessed 7 May 2013.
US FDA. Postmarketing reporting of adverse experiences. Code of Federal Regulations Title 21 (21 CFR 600.80). Silver Spring: US FDA; 2012.
US FDA. Postmarketing reporting of adverse drug experiences, Code of Federal Regulation Title 21, (21 CFR 314.80). Silver Spring: US FDA; 2012.
Regulation (EC) No 726/2004 of The European Parliament and of the Council of 31 March 2004, laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency. http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2004:136:0001:0033:en:PDF. Accessed 7 May 2013.
Crommelin DJA, Storm G, Verrijk R, de Leede L, Jiskoot W, Hennink WE. Shifting paradigms: biopharmaceuticals versus low molecular weight drugs. Int J Pharm. 2003;266(1–2):3–16.
Vonberg RP, Gastmeier P. Hospital-acquired infections related to contaminated substances. J Hosp Infect. 2007;65(1):15–23.
Thorpe SJ, Fox BJ, Dolman CD, Lawrence J, Thorpe R. Batches of intravenous immunoglobulin associated with adverse reactions in recipients contain atypically high anti-Rh D activity. Vox Sang. 2003;85(2):80–4.
Kurz X, Domergue F, Slattery J, Segec A, Szmigiel A, Hidalgo-Simon A. Safety monitoring of Influenza A/H1N1 pandemic vaccines in EudraVigilance. Vaccine. 2011;29(26):4378–87.
de Bousingen DD. Former French ministers on trial for infected blood-products scandal. Lancet. 1999;353(9153):653.
Esmonde TF, Will RG, Slattery JM, Knight R, Harries-Jones R, de Silva R, et al. Creutzfeldt-Jakob disease and blood transfusion. Lancet. 1993;341(8839):205–7.
Eaton L. Haemophilia patient had variant CJD agent in spleen. BMJ. 2009;18(338):b705.
van Hunsel F, Harmark L, Pal S, Olsson S, van Grootheest K. Experiences with adverse drug reaction reporting by patients: an 11-country survey. Drug Saf. 2012;35(1):45–60.
Zuñiga L, Calvo B. Biosimilars: pharmacovigilance and risk management. Pharmacoepidemiol Drug Saf. 2010;19(7):661–9.
Fox JL. Debate over details of US biosimilar pathway continues to rage. Nat Biotechnol. 2012;30(7):577.
Lovis C. Traceability in healthcare: crossing boundaries. Yearb Med Inform. 2008;47(Suppl 1):105–13.
O’Dowd A. UK launches inquiry into safety of PIP breast implants. BMJ. 2012;3(344):e11.
Petitjean S. Commission to enhance traceability of medical devices. http://www.europolitics.info/commission-to-enhance-traceability-of-medical-devices-art324118.html. Accessed 22 Mar 2012.
European Federation of Pharmaceutical Industries and Associations. EFPIA product verification project. April 2011. http://www.efpia.eu. Accessed 16 Jan 2011.
Directive 2010/84/EU of The European Parliament and of the Council of 15 December 2010, amending, as regards pharmacovigilance, Directive 2001/83/EC on the Community code relating to medicinal products for human use. http://eudravigilance.ema.europa.eu/human/docs/Directives/dir_2010_84_en.pdf. Accessed 7 May 2013.
Hauben M, Reich L, DeMicco J, Kim K. ‘Extreme duplication’ in the US FDA Adverse Events Reporting System database. Drug Saf. 2007;30(6):551–4.
Acknowledgments
The Department of Pharmacoepidemiology and Clinical Pharmacology, Utrecht Institute for Pharmaceutical Sciences, has received unrestricted research funding from the Netherlands Organisation for Health Research and Development (ZonMW), the Dutch Health Care Insurance Board (CVZ), the Royal Dutch Pharmacists Association (KNMP), the private-public funded Top Institute Pharma (www.tipharma.nl, includes co-funding from universities, government and industry), the EU Innovative Medicines Initiative (IMI), EU 7th Framework Program (FP7), the Dutch Medicines Evaluation Board and the Dutch Ministry of Health and Industry (including GlaxoSmithKline, Pfizer and others). The views expressed in this article are the personal views of the author(s) and may not be understood or quoted as being made on behalf of or reflecting the position of the EMA or one of its committees or working parties.
Conflicts of interest
Niels S. Vermeer, Sabine M.J.M. Straus, Aukje K. Mantel-Teeuwisse, Francois Domergue, Toine C.G. Egberts, Hubert G.M. Leufkens and Marie L. De Bruin have no conflicts of interest that are directly relevant to the content of this review.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below are the links to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Vermeer, N.S., Straus, S.M.J.M., Mantel-Teeuwisse, A.K. et al. Traceability of Biopharmaceuticals in Spontaneous Reporting Systems: A Cross-Sectional Study in the FDA Adverse Event Reporting System (FAERS) and EudraVigilance Databases. Drug Saf 36, 617–625 (2013). https://doi.org/10.1007/s40264-013-0073-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-013-0073-3