
Abstract
Background and Objective
Trastuzumab emtansine (T-DM1) is an antibody–drug conjugate recently approved by the US Food and Drug Administration for the treatment of human epidermal growth factor receptor 2 (HER2)–positive metastatic breast cancer previously treated with trastuzumab and taxane chemotherapy. It comprises the microtubule inhibitory cytotoxic agent DM1 conjugated to the HER2-targeted humanized monoclonal antibody trastuzumab via a stable linker. To characterize the pharmacokinetics of T-DM1 in patients with metastatic breast cancer, concentrations of multiple analytes were quantified, including serum concentrations of T-DM1 conjugate and total trastuzumab (the sum of conjugated and unconjugated trastuzumab), as well as plasma concentrations of DM1. The clearance of T-DM1 conjugate is approximately 2 to 3 times faster than its parent antibody, trastuzumab. However, the clearance pathways accounting for this faster clearance rate are unclear. An integrated population pharmacokinetic model that simultaneously fits the pharmacokinetics of T-DM1 conjugate and total trastuzumab can help to elucidate the clearance pathways of T-DM1. The model can also be used to predict total trastuzumab pharmacokinetic profiles based on T-DM1 conjugate pharmacokinetic data and sparse total trastuzumab pharmacokinetic data, thereby reducing the frequency of pharmacokinetic sampling.
Methods
T-DM1 conjugate and total trastuzumab serum concentration data, including baseline trastuzumab concentrations prior to T-DM1 treatment, from phase I and II studies were used to develop this integrated population pharmacokinetic model. Based on a hypothetical T-DM1 catabolism scheme, two-compartment models for T-DM1 conjugate and trastuzumab were integrated by assuming a one-step deconjugation clearance from T-DM1 conjugate to trastuzumab. The ability of the model to predict the total trastuzumab pharmacokinetic profile based on T-DM1 conjugate pharmacokinetics and various sampling schemes of total trastuzumab pharmacokinetics was assessed to evaluate total trastuzumab sampling schemes.
Results
The final model reflects a simplified catabolism scheme of T-DM1, suggesting that T-DM1 clearance pathways include both deconjugation and proteolytic degradation. The model fits T-DM1 conjugate and total trastuzumab pharmacokinetic data simultaneously. The deconjugation clearance of T-DM1 was estimated to be ~0.4 L/day. Proteolytic degradation clearances for T-DM1 and trastuzumab were similar (~0.3 L/day). This model accurately predicts total trastuzumab pharmacokinetic profiles based on T-DM1 conjugate pharmacokinetic data and sparse total trastuzumab pharmacokinetic data sampled at preinfusion and end of infusion in cycle 1, and in one additional steady state cycle.
Conclusions
This semi-mechanistic integrated model links T-DM1 conjugate and total trastuzumab pharmacokinetic data, and supports the inclusion of both proteolytic degradation and deconjugation as clearance pathways in the hypothetical T-DM1 catabolism scheme. The model attributes a faster T-DM1 conjugate clearance versus that of trastuzumab to the presence of a deconjugation process and suggests a similar proteolytic clearance of T-DM1 and trastuzumab. Based on the model and T-DM1 conjugate pharmacokinetic data, a sparse pharmacokinetic sampling scheme for total trastuzumab provides an entire pharmacokinetic profile with similar predictive accuracy to that of a dense pharmacokinetic sampling scheme.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Background
Breast cancer accounts for approximately 28 % of all new cases of cancer in women in the United States [1]. Gene amplification of the human epidermal growth factor receptor 2 (HER2) tyrosine kinase occurs in approximately 15 %–25 % of breast cancers [2–4] and is associated with poor prognosis [5–7].
Antibody–drug conjugates (ADCs) are a novel type of biologic therapy that aims to reduce systemic exposure to cytotoxic chemotherapy via linkage to a targeting monoclonal antibody. Trastuzumab emtansine (T-DM1) is a HER2-directed ADC composed of the humanized monoclonal antibody trastuzumab conjugated to the microtubule-inhibitory cytotoxic agent DM1 via a stable thioether linker [8]. T-DM1 incorporates the antitumor activities of trastuzumab (mediation of antibody-dependent cellular cytotoxicity, inhibition of HER2-mediated intracellular signaling, and inhibition of HER2 shedding) with the targeted delivery of the cytotoxic agent DM1 [9, 10]. T-DM1 has demonstrated clinical efficacy as a single agent in several phase II studies [11–13] and in a phase III study [14] in patients with HER2-positive breast cancer, and was recently approved by the US Food and Drug Administration for the treatment of HER2-positive metastatic breast cancer (MBC) previously treated with trastuzumab and taxane chemotherapy.
The pharmacokinetics of multiple analytes (T-DM1 conjugate, total trastuzumab and DM1) have been characterized in several clinical trials to understand T-DM1 disposition. Based on the mechanism of action of T-DM1, total trastuzumab, in addition to T-DM1, is considered a clinically relevant analyte that correlates with efficacy in HER2-positive breast cancer patients. The T-DM1 conjugate administered is a mixture of species with 1–8 DM1 molecules conjugated to each molecule of trastuzumab (average drug-to-antibody ratio [DAR] = 3.5). The bioanalytical method used to measure serum T-DM1 conjugate quantifies the total concentration of this heterogeneous mixture (Fig. 1a); for serum total trastuzumab, the total concentrations of both unconjugated trastuzumab and T-DM1 conjugate are measured (Fig. 1b). In vivo, T-DM1 catabolic clearance causes the concentrations of individual components to change with time, resulting in a gradual decrease of the average DAR over time, which was observed in cynomolgus monkeys [15]. Exposures of T-DM1 and total trastuzumab are highly correlated due to their overlapping species; it is important to quantify the relationship between T-DM1 conjugate and total trastuzumab to provide insight into the catabolic clearance pathways of T-DM1.

Serum T-DM1 conjugate: trastuzumab with 1–8 DM1 conjugated (a); serum total trastuzumab: both unconjugated trastuzumab and T-DM1 conjugate (b); hypothetical schematic diagram of T-DM1 catabolism by deconjugation and proteolytic degradation (c). Proteolytic clearance includes non-specific and target-mediated degradation. DAR = drug-to-antibody ratio; T-DM1 = trastuzumab emtansine
Based on T-DM1 and total trastuzumab pharmacokinetic parameters determined by a non-compartmental analysis, T-DM1 conjugate clearance is approximately 2- to 3-fold faster than that of total trastuzumab [16]. Similarly, population pharmacokinetic models of T-DM1 conjugate [17] and trastuzumab [18] have estimated faster clearance rates for T-DM1 (~0.7 L/day) [17] than for trastuzumab (~0.23 L/day) [18]. The structure and pharmacokinetic properties of T-DM1 suggest that it may undergo multiple catabolic clearance pathways, resulting in unconjugated trastuzumab and DM1-containing catabolites (Fig. 1c). T-DM1 may undergo proteolytic degradation similar to typical monoclonal antibody clearance pathways that include both non-specific proteolytic degradation and target-mediated disposition [19]. In addition, T-DM1 may undergo a chemical or enzymatic process that deconjugates DM1 from the antibody component, thereby converting high DAR species to either low DAR species or to unconjugated trastuzumab. It is hypothesized that the faster clearance of T-DM1 compared with trastuzumab could be largely due to this deconjugation clearance pathway.
The integrated population pharmacokinetic model reported here reflects the proposed, simplified catabolic mechanisms of T-DM1 (see Fig. 1c) and simultaneously fits the pharmacokinetic data of T-DM1 conjugate and total trastuzumab. Total trastuzumab exposure can also be influenced by baseline trastuzumab concentrations resulting from prior trastuzumab treatment; this was included as an initial condition in the model. The model quantifies the relationship between T-DM1 conjugate and total trastuzumab by estimating the deconjugation rate at which T-DM1 conjugate is converted to trastuzumab, and confirms that T-DM1 catabolic clearance is composed of proteolytic degradation and deconjugation pathways. The model further enables pharmacokinetic sampling reduction of total trastuzumab for future clinical trials. The reduced pharmacokinetic sampling design enables the evaluation of total trastuzumab exposure for further exposure–response analysis while greatly reducing the clinical sample numbers.
The pharmacokinetics of DM1 were not integrated into this model because the majority of values postdose were below the limit of quantification [16].
2 Methods
2.1 Studies and Population Pharmacokinetic Dataset
The integrated population pharmacokinetic model was developed using a model-building dataset containing 5333 T-DM1 conjugate and 5370 total trastuzumab concentration-time records from 273 patients with HER2-positive MBC enrolled in three studies: phase I study TDM3569g [20], and phase II studies TDM4258g [11] and TDM4374g [12] (Table 1). The external dataset for model validation contains 498 T-DM1 and 495 total trastuzumab concentration-time records from 51 patients in the phase II study TDM4688g [21]. Pharmacokinetic sampling schemes have been described previously [16, 17]. The serum pharmacokinetic sampling scheme was the same for both T-DM1 and total trastuzumab in all clinical studies to date. Relatively intensive pharmacokinetic samples were obtained in cycles 1 and 4 for TDM3569g, TDM4258g and TDM4374g, and in cycles 1 and 3 for TDM4688g. Pharmacokinetic sampling was limited to pre-T-DM1 and post-T-DM1 infusion in other cycles (see Table 1).
All patients provided written informed consent. Each study was reviewed and approved by the institutional review board at each site according to local clinical guidelines. All studies were performed in accordance with assurances filed with and approved by the US Department of Health and Human Services.
Summary statistics of patient covariates for the model-building dataset were reported previously [17]. Pharmacokinetic covariate values were comparable for patients in both the model-building and external model-validation datasets. Baseline trastuzumab concentrations in these patients (resulting from previous trastuzumab treatment) were included as initial conditions in the model (see the Electronic Supplementary Material). Forty-nine percent of patients (of 273) in the model-building dataset and 61 % of patients (of 51) in the model-validation dataset had measurable baseline trastuzumab concentrations ranging from 0.044 to 122 μg/mL and 0.040 to 148 μg/mL, respectively.
2.2 Bioanalytical Methods
T-DM1 conjugate and total trastuzumab concentrations in serum samples were analyzed using validated indirect sandwich enzyme-linked immunosorbent assay (ELISA) methods [20]. The T-DM1 ELISA using anti-DM1 monoclonal antibody as the coat reagent measures T-DM1 conjugate that contains 1–8 covalently bound DM1 molecules but excludes measurement of unconjugated trastuzumab. The total trastuzumab ELISA using recombinant HER2 extracellular domain as capture reagent measures both T-DM1 conjugate and fully unconjugated trastuzumab. The minimum quantifiable concentration (MQC) in serum for both assays is 40 ng/mL. Assay methods are as previously described [20]. The total trastuzumab concentration is considered a sum of T-DM1 concentration and fully unconjugated trastuzumab concentration.
2.3 Integrated Population Pharmacokinetic Model
A model based on simplification of the hypothetical catabolic T-DM1 scheme (see Fig. 1c) was proposed (Fig. 2). Several assumptions were made for this base model. First, two-compartment models with linear elimination from the central compartment were assumed for both T-DM1 and trastuzumab based on historical population pharmacokinetic models for both drugs [17, 18]. Second, T-DM1 is cleared by both deconjugation into trastuzumab and proteolytic degradation; trastuzumab is cleared only by proteolytic degradation. In Fig. 2, parameters CL1, CL2 and CL3 represent deconjugation clearance of T-DM1, proteolytic degradation clearance of T-DM1 and proteolytic degradation clearance of trastuzumab, respectively. While mechanistically these clearance processes could also occur in the peripheral compartment, it was assumed that they only occur in the central compartment. Alternative models with deconjugation in the peripheral compartment were tested. Third, since trastuzumab was considered to be a metabolite of T-DM1, and to ensure that the model parameter would be identifiable [22], the central volume of distribution (Vc) for trastuzumab was assumed to be the same as that for T-DM1. This is based on the similar Vc values (~3 L) for both T-DM1 and trastuzumab from historical population pharmacokinetic models [17, 18]. Fourth, based on the hypothesis of gradual conversion from high to low DAR species (see Fig. 1c), a simplified scheme of a one-step first-order kinetic process to quantify the deconjugation rate was used for this model. Due to the lack of concentration data for each DAR species, this simplified scheme was sufficient to optimally fit the data. Fifth, to set the baseline trastuzumab levels as initial model conditions, baseline trastuzumab concentrations after prior trastuzumab treatment were assumed to be in the terminal elimination phase (i.e. equilibrium had been reached between central and peripheral compartments).

Base model structure of the integrated pharmacokinetic model. For differential equations, model compartment set-up in NONMEM and initial conditions of each compartment, see Electronic Supplementary Material. CL1 = T-DM1 deconjugation clearance; CL2 = T-DM1 proteolytic degradation clearance; CL3 = trastuzumab proteolytic degradation clearance; QT-DM1 = distributional clearance of T-DM1; Qtrastuzumab = distributional clearance of trastuzumab; T-DM1 = trastuzumab emtansine; Vc = central volume of distribution of T-DM1 or trastuzumab; Vp, T-DM1 = peripheral volume of distribution of T-DM1; Vp, trastuzumab = peripheral volume of distribution of trastuzumab
Non-linear mixed-effect modeling was performed by NONMEM VII (ICON Development Solutions, Ellicott City, MD, USA), and first-order conditional estimation with interaction was used in all runs to estimate population pharmacokinetic parameters [23]. Additive error models were used for T-DM1 and total trastuzumab.
The differential equations, model compartment set-up in NONMEM and initial conditions of each compartment are included in the Electronic Supplementary Material. Baseline trastuzumab concentrations resulting from prior trastuzumab treatment were included as initial conditions for the central and peripheral compartments of trastuzumab by the following method: (1) In the NONMEM dataset, the doses at time zero for compartments 2 and 4 were set to the baseline trastuzumab concentration for each individual patient, if the values were above the MQC. (2) In the NONMEM control stream, compartment 2 bioavailability was set to Vc. The initial amount of trastuzumab in the peripheral compartment was computed based on measured concentrations in the central compartment. Compartment 4 bioavailability was set to the peripheral volume of trastuzumab at the terminal phase, which equaled the terminal volume of distribution (Vβ) minus Vc (see supplementary materials). This is based on the assumption that baseline trastuzumab concentrations resulting from prior treatment were entering the terminal phase (i.e. distribution equilibrium between central and peripheral compartments was achieved, thus the concentrations in the central and peripheral compartments were identical). For a two-compartment model after intravenous administration, Vβ was calculated as shown in supplementary materials [24].
Multiple base models (Base A to G; Table 2) were tested to optimize model structure and parameterization. Model Base A had the highest number of parameters with the lowest objective function value (OFV). Base models with simplified model parameterization (Base B to E) were tested. If an alternative model with a reduced number of model parameters had acceptable numerical stability and a difference of OFV <6.63 (p > 0.01, log-likelihood ratio test, degree of freedom = 1) compared with Base A, it was considered to be a simpler and acceptable base model. Two additional base models with deconjugation from both central and peripheral compartments of T-DM1 (Base F) or deconjugation from the peripheral compartment (Base G) were also tested. The numerical stability of the various models was assessed to assist in model selection.
The intent of this integrated population pharmacokinetic model is to understand T-DM1 catabolism and evaluate pharmacokinetic sampling. As this purpose can be fulfilled using the base model, a comprehensive covariate model to inform potential dose adjustment was not built. Covariates for T-DM1 conjugate and trastuzumab pharmacokinetics were separately identified in historical models [17, 18]; hence, no covariate analysis was performed for the integrated population pharmacokinetic model described here.
2.4 Model Validation
The model was validated using both the model-building and external datasets. Standard model diagnosis plots, including goodness-of-fit and individual fitting plots, were examined. Conditional weighted residual (CWRES) was used for model diagnosis [25]. To assess the predictive performance of the model, visual predictive checks (VPC) and numerical predictive checks (NPC) [26] were also performed.
2.5 Total Trastuzumab Pharmacokinetic Profile Prediction to Evaluate Reduced Pharmacokinetic Sampling Schedule
To evaluate a reduced sampling scheme of total trastuzumab in future clinical trials, the model was applied to predict total trastuzumab pharmacokinetic profiles for each patient based on intensive T-DM1 pharmacokinetic data and sparse total trastuzumab pharmacokinetic data. The model-building and model-validation datasets contained dense pharmacokinetic data for both T-DM1 and total trastuzumab, which included >25 points per patient, with relatively intensive pharmacokinetic samples in cycle 1 and one cycle at steady state. To test reduced sampling schemes of total trastuzumab based on the existing datasets, all T-DM1 observations were retained and total trastuzumab observations were reduced according to various sampling schemes (see “Results”). Using the Bayesian feedback method [23], the newly constructed dataset was used to predict the total trastuzumab pharmacokinetic profiles with intensive sampling for individual patients based on the integrated model. Predicted total trastuzumab concentrations were compared with observed concentrations from the original dataset to assess predictive accuracy. To create a benchmark of predictive accuracy, all observed total trastuzumab pharmacokinetic data in the original dataset were also used to make predictions and were compared with the observed data. A value of median absolute prediction error (MEAPE) [27, 28] was calculated for each patient to quantify the predictive accuracy of multiple pharmacokinetic observations for each patient (Eq. 1). To compare the predictive accuracy of the entire population for various sampling schemes of total trastuzumab, the cumulative probability density (CPD) distribution curves of MEAPE for all patients from the model-building dataset or from the external dataset were obtained, and the 90th-percentile MEAPE value for each sampling scheme was derived. A smaller 90th-percentile MEAPE value indicates better predictive accuracy for the population. S-PLUS statistical software (Version 8.2, Seattle, Washington) was used for these analyses.
where DV is the observed value, IPRED is the individual prediction value and MEAPE is the median absolute prediction error.
3 Results
3.1 Integrated Population Pharmacokinetic Model: Final Model Structure and Parameter Values
Model Base B was chosen as the best base model owing to the OFVs and model numerical stability of the various base models (see Table 2). Model Base B reduced one parameter, compared with model Base A, by assuming the same distributional clearance of T-DM1 and trastuzumab (QT-DM1 = Qtrastuzumab) to their peripheral compartments. Further model parameter simplification was not supported by model comparison. Model Base D, which assumes the same peripheral volume of distribution for T-DM1 and trastuzumab (Vp,T-DM1 = Vp, trastuzumab), resulted in a much higher OFV (p < 0.001) compared with model Base A and was therefore not recommended. The censored-data likelihood method (M3 method in NONMEM) was not used in the current analysis since <5 % of T-DM1 or total trastuzumab pharmacokinetic data were below the MQC.
Interindividual variability (IIV) for all parameters was estimated; however, IIV for distributional clearance (Q) was not estimated with good precision and was therefore not included in the final model. In addition, using the method proposed by Savic and Karlsson [29], the percentages of the shrinkage for IIV in CL1, CL2, CL3, Vc, Vp, T-DM1 and Vp, trastuzumab were estimated to be 46.1 %, 10.1 %, 30.7 %, 9.13 %, 25.7 % and 24.1 %, respectively. These results suggest that there was relatively abundant information in the dataset to estimate IIV for most of the parameters except for CL1, for which a relatively large shrinkage of IIV was observed. A high shrinkage value might impact a reliable covariate analysis for that parameter. However, a covariate analysis was not performed in this analysis and thus is not a concern. The off-diagonal elements in IIV were not estimated based on the lack of apparent correlations among IIVs estimated from the base model (data not shown).
Pharmacokinetic parameter estimates and the IIV for the best base model (model Base B) are listed in Table 3. The T-DM1 deconjugation clearance (CL1) was estimated to be ~0.4 L/day, and Vc was ~3.4 L, indicating a deconjugation half-life of ~6 days in patients with HER2-positive MBC. The model estimated similar proteolytic degradation clearance values (0.3 L/day) for T-DM1 (CL2) and trastuzumab (CL3). A smaller peripheral volume of distribution was estimated for T-DM1 (~0.9 L) compared with trastuzumab (~5 L; see Table 3). Median post hoc pharmacokinetic parameter estimates for T-DM1 and trastuzumab from the integrated population pharmacokinetic model were similar to those from the population pharmacokinetic single-agent models of T-DM1 [17] and trastuzumab [18, 30] (Table 4), implying reasonable values of parameter estimation by the integrated model. The difference in Q between this model and the historical trastuzumab model might have been because the IIV of Q was not estimated in both models due to insufficient data for a precise estimation.
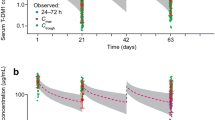
There was agreement between the observed and predicted concentrations for T-DM1 and total trastuzumab for the model-building dataset (Figs. 3 and 4) and the external validation dataset (Fig. 5). VPC plots showed that observed pharmacokinetic concentrations for T-DM1 and total trastuzumab were largely within the 90 % prediction intervals of the model (Figs. 6 and 7). Using the NPC method, it was calculated that in the model-building dataset, observed pharmacokinetic concentrations for 95.3 % of T-DM1 data and 91.5 % of total trastuzumab data were within the 90 % prediction interval of the model; in the external dataset, observed pharmacokinetic concentrations for 96.0 % of T-DM1 data and for 96.4 % of total trastuzumab data were within the 90 % prediction intervals of the model. These results indicated acceptable performance of the final model in predicting the central tendency and the variability of serum concentrations.

Standard basic model diagnosis plots for T-DM1 (a) and total trastuzumab (b) for the model-building dataset. Blue solid lines denote locally weighted scatterplot smoothing lines. CWRES = conditional weighted residuals; WRES = weighted residuals

Typical individual fitting plots using the model-building dataset. Black circles denote T-DM1 observations; gray circles denote total trastuzumab observations; black solid line denotes IPRED for T-DM1; gray solid line denotes IPRED for total trastuzumab; black dotted line denotes PRED for T-DM1; and gray dotted line denoted PRED for total trastuzumab. The fitting for four individual patients’ pharmacokinetic profiles are shown here as examples. IPRED = individual-predicted concentrations; PRED = population-predicted concentrations; T-DM1 = trastuzumab emtansine

Goodness-of-fit plot using the external dataset (circles) for T-DM1 (a) and total trastuzumab (b). Dotted lines denote locally weighted scatter-plot smoothing lines. T-DM1 = trastuzumab emtansine

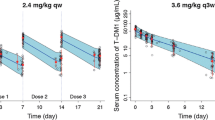
Visual predictive check plots for T-DM1 (a) and total trastuzumab (b) using the patients in the model-building dataset who received T-DM1 3.6 mg/kg every 3 weeks (n = 236). Circles denote observations; solid line denotes median of observed data; dashed lines denote 2.5th and 97.5th percentile values of observed data; gray bands denote 5th to 95th percentile intervals based on 500 repeated trials for the simulated median value, 2.5th and 97.5th percentile values of pharmacokinetic data from each trial containing 236 patients. T-DM1 = trastuzumab emtansine

Visual predictive check plots for T-DM1 (a) and total trastuzumab (b) using the patients in the external dataset who received T-DM1 3.6 mg/kg every 3 weeks (n = 51). Circles denote observations; solid line denotes median of observed data; dashed lines denote 2.5th and 97.5th percentile values of observed data; gray bands denote 5th to 95th percentile intervals based on 500 repeated trials for the simulated median value, 2.5th and 97.5th percentile values of pharmacokinetic data from each trial containing 51 patients. T-DM1 = trastuzumab emtansine
3.2 Total Trastuzumab Pharmacokinetic Profile Prediction for Reduced Pharmacokinetic Sampling Scheme Evaluation
The predictive accuracy for total trastuzumab pharmacokinetic profiles was dependent on the pharmacokinetic sampling scheme of total trastuzumab (sparse or intensive) used for prediction. The accuracy comparisons (as assessed by CPD distribution of MEAPE values of the population for the model-building dataset or the model-validation dataset) are shown in Table 5 and Fig. 8.

Cumulative probability density distribution of MEAPE for predicting total trastuzumab pharmacokinetic profiles by the Bayesian feedback method based on all observed T-DM1 pharmacokinetic data and inclusion of various scenarios of observed total trastuzumab pharmacokinetic data using (a) the model-building dataset (n = 202 patients) and (b) the external dataset (n = 48 patients). Note that only 202 out of 273 patients in the model-building dataset had observed steady state (i.e. cycle 4) T-DM1 and total trastuzumab pharmacokinetic data and were used for this analysis, and that only 48 out of 51 patients in the external model-validation dataset had observed steady state (i.e. cycle 3) T-DM1 and total trastuzumab pharmacokinetic data and were used for this analysis. MEAPE = median absolute performance error; T-DM1 = trastuzumab emtansine
For the model-building dataset, inclusion of the observed sparse total trastuzumab pharmacokinetic data (predose and end of infusion) at cycle 1 and steady state (cycle 4), which included only three pharmacokinetic sampling points of total trastuzumab post-T-DM1 dose, resulted in an MEAPE <24.8 % for >90 % of patients (see Table 5). This was considerably better than the inclusion of the observed intensive total trastuzumab pharmacokinetic data from cycle 1 (MEAPE <35.2 % for >90 % of patients) or the exclusion of any observed total trastuzumab pharmacokinetic data (MEAPE <33.3 % for >90 % of patients). The value of MEAPE <24.8 % was also close to the benchmark scenario of the predictive accuracy (MEAPE <20.7 % for >90 % of patients) when all available total trastuzumab pharmacokinetic data were used for model prediction (>25 time points). This result clearly suggests that steady state sparse total trastuzumab pharmacokinetic data are more informative for improving the predictive accuracy than cycle 1 intensive total trastuzumab pharmacokinetic data.
A similar analysis using the external model-validation dataset from TDM4688g confirmed the recommended sampling scheme for total trastuzumab. Predictions based on T-DM1 pharmacokinetic data and observed total trastuzumab pharmacokinetic data at the time points of preinfusion and end of infusion of cycle 1 and steady state (cycle 3) resulted in MEAPE <19.2 % for >90 % of patients (see Table 5). This predictive accuracy was similar to that based on all observed intensive total trastuzumab pharmacokinetic data (MEAPE <19.5 % for >90 % of patients). There is also a relatively large improvement upon the inclusion of observed intensive total trastuzumab pharmacokinetic data from cycle 1 or the exclusion of any observed total trastuzumab pharmacokinetic data. Therefore, the sparse steady state (cycle 3+) total trastuzumab pharmacokinetic data are confirmed to be critical for an accurate prediction of the entire profile using the integrated model.
These results suggest the most effective and patient-friendly sampling scenario for total trastuzumab is taking sparse pharmacokinetic samples at predose and end of infusion during cycle 1 and one steady state cycle (i.e. one predose and three postdose total trastuzumab pharmacokinetic samples).
4 Discussion
A novel integrated population pharmacokinetic model was developed to reflect the hypothetical catabolic mechanisms of T-DM1 and to simultaneously fit both T-DM1 and total trastuzumab pharmacokinetic data from patients with previously treated HER2-positive MBC after T-DM1 treatment [11, 12, 20, 21]. The acceptable model-fitting performance suggests the plausibility of the model scheme to reflect two essential T-DM1 catabolic pathways: proteolytic degradation and deconjugation.
The proteolytic degradation process is most likely related to target-mediated and non-specific proteolysis of T-DM1, a process consistent with the disposition of monoclonal antibodies. Non-specific degradation, which involves FcRn-mediated recycling, predominantly occurs in endothelial cells and mononuclear phagocytes; target-mediated degradation, which involves both receptor-mediated internalization and intracellular lysosomal degradation, primarily occurs in target-expressing cells [31]. The proteolytic degradation clearances of T-DM1 and unconjugated trastuzumab were found to be similar (~0.3 L/day) in this model. This result is in alignment with data from in vitro studies showing that T-DM1 binds to HER2 and Fc gamma receptors with similar affinity as trastuzumab [9, 10]. The specific and non-specific binding properties of the trastuzumab component appeared to be unchanged when conjugated with DM1.
The deconjugation process is most likely related to linker chemistry. For T-DM1, the thioether linker conjugating DM1 to trastuzumab is considered non-cleavable [32]. However, some deconjugation appears to occur because total trastuzumab concentrations were higher than T-DM1 conjugate concentrations at each pharmacokinetic time point, with an increasing difference at later pharmacokinetic time points. This suggests a gradual formation of unconjugated trastuzumab [16]. The mechanisms of thioether linker deconjugation for T-DM1 are unclear. Based on the observation of maleimide exchange-resulted deconjugation in thiomabs-containing maleimide-type linkers [33], one hypothetical deconjugation mechanism for T-DM1 may involve a similar maleimide exchange with reactive thiols in albumin, free cysteine, or glutathione, resulting in the loss of DM1 from the antibody. It is not clear whether deconjugation occurs in plasma or the intracellular space of tissues. This model suggests that the faster clearance of T-DM1 compared with trastuzumab is likely explained by the elimination of T-DM1 by deconjugation, in addition to its proteolytic degradation; however, trastuzumab only undergoes proteolytic degradation. The deconjugation clearance of ~0.4 L/day estimated by the model suggests comparable deconjugation and proteolytic degradation rates. Similar results were found in a pharmacokinetic study of the ADC cantuzumab mertansine. Cantuzumab mertansine conjugate had a faster clearance than that of its parent antibody; this was thought to be due to deconjugation of the cytotoxic drug from the conjugate via linker cleavage, in addition to conjugate degradation [34].
A linear clearance model was used for both T-DM1 conjugate and trastuzumab compartments in this model, which is consistent with historical models of T-DM1 and trastuzumab [17, 18]. A non-linear pharmacokinetic model was not used, since only five patients receiving lower doses of T-DM1 (0.3, 0.6 and 1.2 mg/kg once every 3 weeks) had faster clearance values than the remaining 268 patients receiving higher doses, and the model parameters associated with non-linear elimination were not estimated with good precision [17]. This observation also suggests that target-mediated disposition is saturated at clinically relevant doses of T-DM1 (2.4–3.6 mg/kg), and the deconjugation clearance does not change with dose.
The trastuzumab peripheral volume of distribution (~3–5 L) was consistent with other immunoglobulin G1 monoclonal antibodies, such as bevacizumab [35], pertuzumab [36] and rituximab [37], yet a much smaller peripheral volume of distribution was estimated for T-DM1 (~0.9 L). It is not apparent why T-DM1 would have a smaller peripheral volume of distribution than trastuzumab, since T-DM1 and trastuzumab were found to distribute to similar peripheral organs in rats [33]. One possible explanation could be the existence of substantial T-DM1 elimination from the peripheral compartment. This would be a deviation from a basic assumption for mammillary compartmental models, which assume the site of drug elimination is in rapid equilibrium with serum (i.e. elimination is only from the “central” compartment). This is assumed because the elimination rate from the peripheral compartment is unidentifiable due to the lack of tissue pharmacokinetic data. However, this assumption may not be valid for antibodies that are internalized and catabolized to a non-negligible extent in the peripheral tissue. When the rate of drug catabolism in peripheral tissue is much faster than the rate of drug distribution from tissue to blood, the estimated peripheral volume of distribution may be inappropriately small [38]. T-DM1 may have relatively more rapid elimination from peripheral tissue compared with trastuzumab because of both deconjugation and proteolytic degradation, resulting in an apparently “smaller” peripheral volume of distribution for T-DM1. In addition, it has been suggested that a three-compartment model may provide a more physiologically accurate estimate of volume of distribution for antibodies by adding a slow-distribution compartment [39]. However, the pharmacokinetic data are only available for most patients up to 3 weeks post-T-DM1 dose, which may not be long enough to characterize the third slow-distribution compartment of T-DM1 conjugate and trastuzumab.
A historical semi-mechanistic multi-compartment model was used to simultaneously fit the pharmacokinetic data of each individual DAR species (DAR = 0–8) in cynomolgus monkeys after T-DM1 dosing, by considering sequential deconjugation from high to low DAR species [15]. This model was further translated to a semi-mechanistic population pharmacokinetic model with multiple transit compartments to characterize T-DM1 and total trastuzumab pharmacokinetics in patients [40]. However, since pharmacokinetic data of each DAR species of T-DM1 are not measured in patients, the further simplified model reported here, with a one-step deconjugation process of T-DM1 to convert to unconjugated trastuzumab, is considered optimal to describe the data and maintain predictive performance of the model. Furthermore, the addition of multiple transit compartments to describe a sequential conversion from high to low DAR species did not improve the model fit significantly (data not shown).
The integrated model was able to predict total trastuzumab pharmacokinetic profiles using intensive T-DM1 pharmacokinetic data and sparse total trastuzumab pharmacokinetic data at cycle 1 and steady state, with predictive accuracy close to that obtained with intensive pharmacokinetic data for both analytes. The steady state pharmacokinetic data were more informative than cycle 1 pharmacokinetic data for making the predictions. This may be because total trastuzumab has a relatively long half-life of approximately 8–10 days (taking at least three cycles to reach steady state), thus the cycle 1 pharmacokinetic data may not provide sufficient information to accurately estimate trastuzumab clearance. The cycle 1 end-of-infusion sample provided limited value for improving prediction accuracy (data not shown). However, considering not every patient can be sampled at steady state, this cycle 1 sample is still included in the final recommendation.
5 Conclusions
In summary, the integrated pharmacokinetic model reported here supports the hypothesis that both deconjugation and proteolytic degradation are important clearance pathways of T-DM1. This model also provides basis for the reduced total trastuzumab pharmacokinetic sampling evaluation in future clinical studies.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012; 62(1):10–29.
Sjögren S, Inganäs M, Lindgren A, et al. Prognostic and predictive value of c-erbB-2 overexpression in primary breast cancer, alone and in combination with other prognostic markers. J Clin Oncol. 1998;16(2):462–9.
Ross JS, Slodkowska EA, Symmans WF, et al. The HER-2 receptor and breast cancer: ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist. 2009;14(4):320–68.
Dawood S, Broglio K, Buzdar AU, et al. Prognosis of women with metastatic breast cancer by HER2 status and trastuzumab treatment: an institutional-based review. J Clin Oncol. 2010;28(1):92–8.
Slamon DJ, Clark GM, Wong SG, et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–82.
Seshadri R, Firgaira FA, Horsfall DJ, et al. Clinical significance of HER-2/neu oncogene amplification in primary breast cancer. The South Australian Breast Cancer Study Group. J Clin Oncol. 1993;11(10):1936–42.
Gabos Z, Sinha R, Hanson J, et al. Prognostic significance of human epidermal growth factor receptor positivity for the development of brain metastases after newly diagnosed breast cancer. J Clin Oncol. 2006;24(36):5658–63.
Lewis Phillips GD, Li G, Dugger DL, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68(22):9280–90.
Junttila TT, Li G, Parsons K, et al. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res Treat. 2011;128(2):347–56.
Barok M, Tanner M, Köninki K, et al. Trastuzumab-DM1 is highly effective in preclinical models of HER2-positive gastric cancer. Cancer Lett. 2011;306(2):171–9.
Burris HA 3rd, Rugo HS, Vukelja SJ, et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J Clin Oncol. 2011;29(4):398–405.
Krop I, LoRusso P, Miller KD, et al. A phase II study of trastuzumab emtansine in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer who were previously treated with trastuzumab, lapatinib, an anthracycline, a taxane, and capecitabine. J Clin Oncol. 2012;30(26):3234–41.
Hurvitz SA, Dirix L, Kocsis J, et al. A phase 2 randomized study of trastuzumab emtansine (T-DM1) versus trastuzumab + docetaxel in patients with HER2-positive metastatic breast cancer. J Clin Oncol. 2013;31(9):1157–63.
Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367(19):1783–91.
Leipold D, Bender B, Xu K, et al. (2009). Understanding the de-conjugation of Trastuzumab-MCC-DM1 through application of a multi-compartmental model of individual drug:antibody species in cynomolgus monkey [abstract no. 2914]. American Association for Cancer Research (AACR) Meeting; 2009 Apr 18; Denver (CO).
Girish S, Gupta M, Wang B, et al. Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother Pharmacol. 2012;69(5):1229–40.
Gupta M, LoRusso PM, Wang B, et al. Clinical implications of pathophysiological and demographic covariates on the population pharmacokinetics of trastuzumab-emtansine, a HER2-targeted antibody–drug conjugate, in patients with HER2-positive metastatic breast cancer. J Clin Pharmacol. 2012;52(5):691–703.
Bruno R, Washington CB, Lu JF, et al. Population pharmacokinetics of trastuzumab in patients with HER2 + metastatic breast cancer. Cancer Chemother Pharmacol. 2005;56(4):361–9.
Kuester K, Kloft C. Pharmacokinetics of monoclonal antibodies. In: Meibohm B, editor. Pharmacokinetics and pharmacodynamics of biotech drugs. 1st ed. Weinheim (Germany): Wiley-VCH GmbH & Co. KGaA; 2006. p. 45–91.
Krop IE, Beeram M, Modi S, et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clin Oncol. 2010;28(16):2698–704.
Gupta M, Wang B, Carrothers TJ, et al. Effects of trastuzumab emtansine (T-DM1) on QT interval and safety of pertuzumab plus T-DM1 in patients with previously treated human epidermal growth factor receptor 2–positive metastatic breast cancer. Clin Pharmacol Drug Devel. 2013 Feb 26 (E-pub)
Bonate PL. Pharmacokinetic-pharmacodynamic modeling and simulation. 2nd ed. New York: Springer; 2005.
Boeckman AJ, Beal SL, Sheiner LB, Beal. NONMEM User’s Guide—Part IV (online). San Francisco (CA): NONMEM Project Group, University of California at San Francisco, 1992. Available from URL: ftp://nonmem.iconplc.com/Public/nonmem720/guides/iv.pdf. Accessed 2012 Jul 18.
Rowland M, Tozer TN. Clinical pharmacokinetics: concepts and applications. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 1995.
Hooker AC, Staatz CE, Karlsson MO. Conditional weighted residuals (CWRES): a model diagnostic for the FOCE method. Pharm Res. 2007;24(12):2187–97.
Karlsson MO, Savic RM. Diagnosing model diagnostics. Clin Pharmacol Ther. 2007;82(1):17–20.
Raemer DB, Buschman A, Varvel JR, et al. The prospective use of population pharmacokinetics in a computer-driven infusion system for alfentanil. Anesthesiology. 1990;73(1):66–72.
Varvel JR, Donoho DL, Shafer SL. Measuring the predictive performance of computer-controlled infusion pumps. J Pharmacokinet Biopharm. 1992;20(1):63–94.
Savic RM, Karlsson MO. Importance of shrinkage in empirical bayes estimates for diagnostics: problems and solutions. AAPS J. 2009;11(3):558–69.
Fukushima Y, Charoin J-E. Report No. 1034069. Population pharmacokinetic analysis of combined Phase II/III studies: BO15899, BO15935, WO16229, M77004 and MO16982. Basel: Hoffman-La Roche, Inc.;2009. Data on file.
Meibohm B, editor. Pharmacokinetics and pharmacodynamics of biotech drugs: Principles and case studies in drug development. San Francisco: Wiley-VCH GmbH & Co.;2006.
Erickson HK, Widdison WC, Mayo MF, et al. Tumor delivery and in vivo processing of disulfide-linked and thioether-linked antibody-maytansinoid conjugates. Bioconjug Chem. 2010;21(1):84–92.
Shen BQ, Xu K, Liu L, et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol. 2012;30(2):184–9.
Xie H, Audette C, Hoffee M, et al. Pharmacokinetics and biodistribution of the antitumor immunoconjugate, cantuzumab mertansine (huC242-DM1), and its two components in mice. J Pharmacol Exp Ther. 2004;308(3):1073–82.
Lu JF, Bruno R, Eppler S, et al. Clinical pharmacokinetics of bevacizumab in patients with solid tumors. Cancer Chemother Pharmacol. 2008;62(5):779–86.
Ng CM, Lum BL, Gimenez V, et al. Rationale for fixed dosing of pertuzumab in cancer patients based on population pharmacokinetic analysis. Pharm Res. 2006;23(6):1275–84.
Li J, Zhi J, Wenger M, et al. Population pharmacokinetics of rituximab in patients with chronic lymphocytic leukemia. J Clin Pharmacol. 2012;52(12):1918–26.
Lobo ED, Hansen RJ, Blathasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93(11):2645–68.
Garzone PD, Atkinson AJ Jr. In search of physiologically based distribution volume estimates for macromolecules. Clin Pharmacol Ther. 2012;92(4):419–21.
Chudasama VL, Schaedeli Stark F, Harrold JM, et al. Semi-mechanistic population pharmacokinetic model of multivalent trastuzumab emtansine in patients with metastatic breast cancer. Clin Pharmacol Ther. 2012;92(4):520–7.
Acknowledgments
The authors acknowledge the investigators, patients and their families who participated in these clinical trials. The authors acknowledge Jin Yan Jin, PhD, for scientific input and helpful discussions during the preparation of this report. Support for third-party editorial assistance for this manuscript, furnished by Denise Chun, Ph.D. (CodonMedical), was provided by Genentech, Inc. The clinical studies reported here were sponsored by Genentech, Inc. Dan Lu, Amita Joshi, Bei Wang, Steve Olsen, Joo-Hee Yi and Sandhya Girish are employees of Genentech, Inc., and have Roche stock/stock options. Ian E. Krop receives clinical trial support from Genentech, Inc. Howard A. Burris has no conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Lu, D., Joshi, A., Wang, B. et al. An Integrated Multiple-Analyte Pharmacokinetic Model to Characterize Trastuzumab Emtansine (T-DM1) Clearance Pathways and to Evaluate Reduced Pharmacokinetic Sampling in Patients with HER2-Positive Metastatic Breast Cancer. Clin Pharmacokinet 52, 657–672 (2013). https://doi.org/10.1007/s40262-013-0060-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-013-0060-y