
Abstract
Objective
To characterize the population pharmacokinetics of bevacizumab and the influence of demographic factors, disease severity, and concomitantly used chemotherapy agents on it’s pharmacokinetic behavior.
Patients and methods
Data from eight clinical trials with bevacizumab administered by intravenous infusion were included. A total of 4,629 bevacizumab concentrations from 491 patients with solid tumors, who received bevacizumab doses ranging from 1 to 20 mg/kg at a dosing frequency ranging from weekly to every 3 weeks, were analyzed using a nonlinear mixed-effects modeling approach (NONMEM).
Results
The best structural model was a two-compartment model with first-order elimination. In the final model, estimated clearance (CL) and central compartment volume of distribution (V c) were 0.207 L/day and 2.39 L for a typical female. The terminal half-life estimate was ∼20 days for both men and women. Body weight and gender were the most significant covariates to explain interpatient variability for CL and V c. Clearance was 26% faster in men than in women. Patients with low serum albumin and high serum alkaline phosphatase had 19 and 23% faster CL, respectively, than a typical patient. Consistent with the long elimination half life, simulations showed that similar steady-state exposures can be maintained when the weekly mg/kg dose rate is maintained, therefore allowing administration of bevacizumab to coincide with the frequency of administration of the cytotoxic agents.
Conclusion
The PK parameters were consistent with those of other IgG molecules. The results support dosing bevacizumab on a once every 2 weeks or once every 3 weeks dosing schedule on a mg/kg basis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Bevacizumab (AVASTIN™, Genentech, Inc., South San Francisco, CA, USA), also known as rhuMAb VEGF, is a recombinant humanized IgG1 antibody that binds and inhibits effects induced by vascular endothelial growth factor (VEGF) in vitro and in vivo. Vascular endothelial growth factor, a diffusible glycoprotein produced by normal and neoplastic cells, is an important regulator of physiologic and pathologic angiogenesis [6]. A large, randomized, controlled Phase III clinical trial has demonstrated that the addition of bevacizumab (5 mg/kg every 2 weeks) to standard chemotherapy with the bolus-IFL [irinotecan/5-fluorouracil (5-FU)/leucovorin] regimen improves survival in patients with previously untreated metastatic colorectal cancer [10]. Bevacizumab is the first biologic agent clinically proven to extend survival in metastatic colorectal cancer and is approved in the US as a first-line treatment for metastatic carcinoma of the colon or rectum in combination with intravenous 5-FU-based chemotherapy [2]. Recently, bevacizumab plus paclitaxel has also been demonstrated to provide a progression-free survival benefit over paclitaxel alone in first-line metastatic breast cancer [17] and to prolong survival in combination with chemotherapy (paclitaxel and carboplatin) relative to chemotherapy alone in patients with advanced nonsquamous non-small-cell lung cancer (NSCLC) [5].
The pharmacokinetic (PK) and pharmacodynamic (PD) information were collected during Phase I to Phase III trials in the development program. In a preliminary analysis of a dataset consisting of full profile data from 52 individual patients in Phase I and II trials, the PK of bevacizumab was best described by an open two-compartment model [9].
The primary objective of the present analysis was to utilize data pooled from eight clinical trials investigating several different bevacizumab dosing regimens, patient populations, and concomitant antineoplastic regimens to determine the population PK of bevacizumab and its relationship to demographics, pathophysiological covariates, disease severity, and combination therapy with various chemotherapy regimens.
Methods
Patient population and data collection
The concentration data analyzed were obtained from eight bevacizumab clinical studies: three trials in which the drug was used as a single agent and five in which the drug was administered concomitantly with five different cytotoxic chemotherapy regimens. All patients gave written informed consent and the study protocols were approved by the medical ethics committee in all study centers. A summary of studies included in the analysis is given in Table 1.
Phase I trials enrolled patients with various types of solid tumors [8, 14]. Phase II and Phase III trials were conducted in patients with colorectal carcinoma [10, 12], hormone refractory prostate carcinoma [16], metastatic breast carcinoma [15] and NSCLC [11]. In these studies, bevacizumab doses ranged from 0.1 to 20 mg/kg, and dosing frequency ranged from weekly to every 3 weeks. Bevacizumab was administered initially as a 90-min infusion. If this first infusion was well tolerated, the infusion duration could be decreased in 30-min decrements to a minimum duration of 30 min. Intensive PK samples were taken for the two Phase I trials and multiple peaks and troughs were obtained for Phase II and III trials. Data from the ten patients, who received doses of 0.1 and 0.3 mg/kg were not included in this population PK analysis because bevacizumab clearance at both of these doses was approximately two to three times faster than at doses ≥1 mg/kg and these lower doses were not investigated further in any clinical trial.
Covariates and patient characteristics considered in the analyses are shown in Table 2. A total of 15 pathophysiologic covariates were tested on bevacizumab clearance and volume of distribution. The effect of chemotherapies on bevacizumab clearance was also assessed.
Assay method
For all studies, serum bevacizumab concentrations were determined at Genentech, Inc. using an enzyme-linked immunosorbent assay (ELISA) that used recombinant human VEGF (rhVEGF) for capture and a goat antibody to human IgG conjugated to horseradish peroxidase for detection [1]. The lower limit of sensitivity of this assay was 78 ng/mL. Concentrations of rhVEGF up to 500 ng/mL, which are 5–10,000 times higher than systemically circulating VEGF, have been shown to not interfere with the determination of bevacizumab concentrations. It is therefore unlikely that a patient’s circulating VEGF would interfere with the determination of bevacizumab concentrations under these conditions.
Population PK analyses
The PK data were analyzed using the nonlinear mixed-effects modeling (NONMEM) program (Version V, level 1.0, GloboMax LLC, Hanover, MD, USA) [3] together with the S-PLUS (Insightful Corp., Seattle, WA, USA) programs for goodness-of-fit assessment and covariate model building. The first-order estimation method (FO) in NONMEM was used for model development. First-order conditional estimation method (FOCE) was used for the final model.
The following strategy was used to develop the final model. First, the structural PK model was developed (e.g., a one- or a two-compartment model). Significant pathophysiological covariates were then identified and incorporated into the model, followed by assessment of the influence of combination chemotherapy on bevacizumab disposition to yield the final model. The final model was then evaluated with bootstrap for its robustness.
Interpatient variability in the PK parameters was assumed log-normally distributed (exponential model). For example, CL:
where η j,CL denotes the proportional difference between the “true” parameter (CL j ) of individual patient j and the typical value \( {{\rm C}}{\hat{{\rm L}}}_{j} \) in the population, adjusted for values of covariates equal to those of the individual patient. The ηs represented random effects with mean zero and variance ϖ 2.
Residual error was modeled as a combination of proportional and additive components.
where Cp ij and \( {\rm C}\hat{{\rm p}}_{{ij}} \) are the ith measured and model predicted (true) concentrations, respectively, for patient j, and ε ij,prop and ε ij,add denote the proportional and additive residual intraindividual random errors, distributed with zero means and variances δ 2 prop and δ 2 add.
Covariate model development
The covariate effect analysis was limited to CL and V c. Following identification of the basic structural model, individual patient random effects and PK parameter estimates were generated using post hoc Bayesian estimation. Graphs of random effects (e.g., η j,CL) versus covariates were produced with a smoothing of the data (SUPSMU function in S-plus) to allow visual inspection of the influence of covariates on the PK parameters.
The construction of the regression model for CL and V c of bevacizumab using NONMEM was based on the likelihood ratio test. The objective function value (OFV) is proportional to the −2 times the log-likelihood of the data, given the model; therefore, the difference between two hierarchical models in OFV (ΔOFV) is approximately χ2-distributed. When comparing two models, where the more complex model included one more parameter than the simpler one, the more complex model was selected only when the ΔOFV was greater than 7.88 (df = 1, P < 0.005). Use of this rather strict criterion was motivated by the multiple testing involved in the model development, as well as the fact that the ΔOFV is only approximately χ2-distributed [7, 20].
The regression model for CL and V c was constructed in three steps: (a) screening of covariate effects on CL and V c separately, by adding covariates one by one to the base model; (b) construction of the full model by incorporating the significant covariates from step (a); (c) elaboration of the reduced model, by testing the full model against restricted models by removing each covariate one at a time. The reduced models obtained separately for CL and V c at (c) were then combined in a new full model, and the final model for pathophysiological covariates was elaborated by testing the full model against restricted models by removing each covariate one at a time.
A multiplicative covariate regression model was implemented as follows [e.g., weight (WT) and gender (GDR) effect on CL]:
where θs are the regression coefficients to be estimated for continuous (e.g., WT) or dichotomous (e.g., GDR) covariates. Continuous covariates were centered around their median [med(WT)] values, thus allowing θ 1 to represent the clearance estimate for the typical patient with median covariates. Dichotomous covariates were coded 0 or 1 (e.g., 0 = female; 1 = male).
The effect of chemotherapy on CL was tested last. Five different combination chemotherapy regimens were used in different trials (see Table 1). The regimen used in the pivotal study (i.e., the bolus-IFL regimen) was used as the reference and was compared with either bevacizumab as a single agent or bevacizumab when combined with all other chemotherapy regimens. The chemotherapy effect was implemented as shown below:
Simulation of temporal dosing patterns
Based on the final model, a total of 500 patients were randomly drawn from the pivotal Phase III study with replacement. The steady-state PK profiles in 500 patients following administration of 2.5 mg/kg per week (i.e., 5 mg/kg every 2 weeks and 7.5 mg/kg every 3 weeks) were simulated with parameters drawn from the population distribution of the final model. The difference in steady-state exposure for the two dosing regimens was evaluated and compared with the observed concentrations.
Results
Population PK analysis
The final dataset for analysis contained 4,629 concentrations from 491 patients, with treatment dose ranging from 1 to 20 mg/kg. On average, approximately 9.4 samples were available per patient (range 1–36), with collection time up to 1 year (97.5 percentile of 382 days). The important demographic and other baseline characteristics are summarized in Table 2. The patient population was predominantly Caucasian (85%) and female (56%), with a median age of 59 years and median weight of 74 kg. There were several types of carcinomas; the majority of patients (79%) received bevacizumab in combination with various chemotherapy regimens.
A two-compartment model was found to describe the data better than a one-compartment model, with a decrease in objective function (OFV) of 1,173, and therefore was used as the base model. Sixteen pathophysiological covariates were evaluated for bevacizumab CL and V c. Body weight (WT), gender, and albumin had the largest effect on CL. In addition, alkaline phosphatase and SGOT had significant effects on CL, and body weight, gender, and albumin had significant effects on V c.
The effect of chemotherapy on CL was tested last, and was significant (ΔOFV > 62.7, df = 2, P < .005). However, as indicated by overlap in the parameter estimates, single-agent CL was not different from CL with the reference bolus-IFL regimen, while the CL with the bolus-IFL regimen was slower than that with all other chemotherapy regimens. Combined chemotherapy effect (CHEMEF) was incorporated to obtain the final model for CL and V as follows:
where GDR is gender, WT is body weight, ALBU is albumin, ALK is alkaline phosphatase, SGOT is serum glutamic oxalacetic transaminase, and CHEMEF is the effect of concomitantly administered chemotherapies on bevacizumab.
The final model using FOCE provided very similar parameter estimates when compared with FO. Parameter estimates from the final model and the 95% confidence interval from the bootstrap are shown in Table 3. The covariate effects in the final model explained about 40% of interpatient variance for V c and 60% of interpatient variance for CL. Body weight and gender were the most significant covariates to explain interpatient variability for both CL and V c. Bevacizumab CL was 26% faster in males relative to females. In the final model, estimated CL and V were 0.207 L/day and 2.39 L for a typical female, and 0.262 L/day and 3.29L for a typical male. The median terminal half-life estimate, based on the final model individual predicted values, was 19.9 days and similar for both genders (males 19.5 days, females 20.6 days).
Subjects with low serum albumin (a value <29 g/L) had a 19% faster CL, while patients with higher ALK (>483 IU/L) had a 23% faster CL than a typical patient.
Although there was significant effect of chemotherapy, single-agent CL was not different from the reference bolus-IFL regimen (95% CI of parameter estimate includes 0), while the CL in the bolus IFL regimen was 17% slower compared to all other chemotherapy regimens combined.
Evaluation of the final model
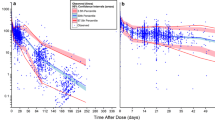
Figure 1 shows the goodness-of-fit for the final model. The IWRES versus IPRED plots suggested no significant deviations of IWRES from the line of identity, with the exception of a slight overproduction at low concentrations (also seen in the plot of IPRED vs. DV). However, no such trend was observed in the plot of WRES versus PRED, suggesting that the model can adequately predict low and high concentrations. The IWRES is calculated based on the following equation:

Model diagnostic plots. a Observed versus predicted bevacizumab concentrations. The solid line is the line of unity. b Weighted residuals versus predicted bevacizumab concentrations. The dashed line is a LOESS smooth of data
Simulation results
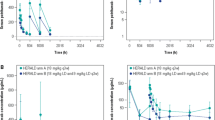
While the steady-state peak concentrations were approximately 20% higher with dosing of 7.5 mg/kg every 3 weeks compared with dosing of 5 mg/kg every 2 weeks (270 vs. 215 μg/mL), the steady-state average and trough concentrations were similar for the two regimens (73 vs. 85 μg/mL).
Discussion
The population PK analysis combined data representing both single- and multiple-dose administration and rich and sparse data, and took advantage of drug accumulation information to estimate the long half-life of bevacizumab. In addition, the effect of pathophysiological covariates and concomitant therapy on PK parameters in the target patient population was evaluated.
Among all covariates tested, body weight and gender were the most important for explaining interindividual variability for CL. The CL of bevacizumab for extreme body weight (from 49 to 114 kg) was up to 30% around the typical value. After adjusting for weight, gender remained a significant effect, with a 26% faster CL in males than in females. The gender difference in bevacizumab clearance may be related to greater muscular mass in males than females.
Patients with low serum albumin concentrations (≤29 g/L) or a high concentration of serum alkaline phosphatase (≥483 U/L) had approximately a 20% increase in CL compared with a typical patient. Low albumin (a sign of inflammation) and high alkaline phosphatase might be related to disease severity, and were correlated with tumor burden in this dataset. Tumor burden could not be assessed as a covariate for CL in this analysis since data were not available in all studies. Another measure of patients’ disease severity, ECOG performance status, was tested and was not significant. It should be noted that only nine patients had an ECOG status >2, which may explain the apparent lack of relationship for this measure of disease severity. The effect of SGOT was of limited magnitude, with a 10% decrease in CL in patients with elevated SGOT (114 U/L). It is presently unknown if impaired liver function significantly alters the CL of bevacizumab, but is not considered likely, as bevacizumab complexes are cleared by the Fc receptor within the endothelial system and not the hepatocyte or biliary system per se [18]. Our preclinical data indicated that hepatic dysfunction resulting from bile duct ligation did not have a significant effect on bevacizumab PK in a rodent model [13]. Although different combination therapies were given to patients with different tumor types, the effect of chemotherapy on bevacizumab CL was minimal. Clearance of bevacizumab following repeated administration of the bevacizumab/bolus-IFL regimen combination in colorectal cancer patients was similar to that following treatment with bevacizumab as a single agent.
Despite these covariate effects, however, from a PD point of view these effects would have minimal effect on the level of unbound VEGF. Although concentrations of unbound (i.e., free) circulating VEGF could not be measured in clinical trials after bevacizumab treatment, based on the proposed clinical dose rates of 2.5 mg/kg per week in colorectal cancer and 5 mg/kg per week in breast cancer and NCSLC and the apparent IC50 of bevacizumab for VEGF, the circulating VEGF is predicted to be, on average, >98% bound with bevacizumab [1]. These data suggest that a 20–30% potential change in CL would not have a significant impact on the free fraction of VEGF and therefore the impact of these covariates on efficacy would be minimal. The definition of population PK model and covariates impacting bevacizumab disposition provides an important tool for the simulation of alternative dose schedules for future study.
In summary, the accumulation of bevacizumab to steady state concentration after long-term treatment can be predicted by a linear two-compartment model. The population PK parameters for bevacizumab are similar to those for other IgG antibodies [4, 19]. Weight and gender were the covariates with the greatest influence on bevacizumab CL and V c, which support body weight based dosing of bevacizumab. Since bevacizumab display linear PK, similar exposure can be achieved with flexible dosage regimens administered on a mg/kg basis, such as Q2, or Q3 weekly dosing (Fig. 2).

Comparison of model predicted steady-state concentrations following 5 mg/kg every 2 weeks (left) and 7.5 mg/kg every 3 weeks (right) dosing of bevacizumab
References
Avastin Biological License Application (BLA) (2003) Data on file Genentech, Inc., South San Francisco
AVASTIN (package insert) (2004) Genentech, Inc., South San Francisco, December
Beal SL, Sheiner LB (1992) NONMEM User’s Guide. NONMEM Project Group. University of California, San Francisco
Bruno R, Washington CB, Lu JF, Lieberman G, Banken L, Klein P (2005) Population pharmacokinetics of trastuzumab in patients with HER2+ metastatic breast cancer. Cancer Chemother Pharmacol 56:361–369
Giantonio BJ, Catalano PJ, Meropol NJ et al (2005) High-dose bevacizumab improves survival when combined with FOLFOX4 in previously treated advanced colorectal cancer: results from the Eastern Cooperative Oncology Group (ECOG) study E3200. J Clin Oncol [ASCO Annual Meeting Proceedings. 23 (no. 16S, Part I of II, June 1 Supplement), 2005:2]
Ferrara N, Gerber HP, LeCouter J (2003) The biology of VEGF and its receptors. Nat Med 9:669076
Gobburu JVS, Lawrence J (2002) Application of resampling techniques to estimate exact significance levels for covariate selection during nonlinear mixed effects model building: some inferences. Pharmacol Res 19:9–28
Gordon MS, Margolin K, Talpaz M, Sledge GW Jr, Holmgren E, Benjamin R et al (2002) Phase I safety and pharmacokinetic study of recombinant human anti-vascular endothelial growth factor in patients with advanced cancer. J Clin Oncol 19:843–850
Hsei VC, Novotny WF, Margolin K et al (2001) Population pharmacokinetic (PK) analysis of bevacizumab (BV) in cancer subjects. Proc Am Soc Clin Oncol 20 (abstract 272)
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W et al (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350:2335–2342
Johnson DH, Fehrenbacher L, Novotny WF et al (2004) Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol 22:2184–2191
Kabbinavar F, Hurwitz HI, Fehrenbacher L et al (2003) Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J Clin Oncol 21:60–65
Lobo ED, Hansen RJ, Balthasar JP (2004) Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci 93:2645–2668
Margolin K, Gordon MS, Holmgren E, Gaudreault J, Novotny W, Fyfe G et al (2001) Phase Ib trial of intravenous recombinant humanized monoclonal antibody to vascular endothelial growth factor in combination with chemotherapy in patients with advanced cancer: pharmacologic and long-term safety data. J Clin Oncol 19:851–856
Miller KD, Chap LI, Holmes FA et al (2005) Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J Clin Oncol 23:792–799
Reese D, Frohlich D, Bok R, Corry M, Novotny W, Homgren E, Small E (1999) A Phase II trial of humanized monoclonal antivascular endothelial growth factor antibody (rhuMAb VEGF) in hormone refractory prostate cancer (HRPC). Proc Am Soc Clin Oncol 18:1355a
Sandler AB, Gray R, Brahmer J et al (2005) Randomized phase II/III trial of paclitaxel (P) plus carboplatin (C) with or without bevacizumab (NSC #704865) in patients with advanced non-squamous non-small cell lung cancer (NSCLC): An Eastern Cooperative Oncology Group (ECOG) Trial-E4599. J Clin Oncol [ASCO Annual Meeting Proceedings, vol 23, no 16S, Part I of II (June 1 Supplement), 2005:4]
Shen BQ et al (2006) Hepatic dysfunction resulting from common bile duct ligation did not affect the pharmacokinetics of bevacizumab (Avastin) in Sprague–Dawley rats. In: Proceedings AACR, vol 47, p 731
Trang JM (1992) Pharmacokinetics and metabolism of therapeutic and diagnostic antibodies. In: Ferraiolo BL, Mohler MA, Gloff CA (eds) Protein pharmacokinetics and metabolism. Pharmaceutical biotechnology, vol 1. Plenum Press, New York, pp 223–270
Wahlby U, Jonsson EN, Karlsson MO (2001) Assessment of actual significance levels for covariate effects in NONMEM. J Pharmacokinet Pharmacodyn 28:231–252
Acknowledgments
The authors thank Dr. Julie Hambleton (Genentech, Inc.) for her review and insightful comments and to the investigators, the clinical research support teams, and to the patients participating in these trials.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lu, JF., Bruno, R., Eppler, S. et al. Clinical pharmacokinetics of bevacizumab in patients with solid tumors. Cancer Chemother Pharmacol 62, 779–786 (2008). https://doi.org/10.1007/s00280-007-0664-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-007-0664-8