
Abstract
Background and Objective
Organic anion-transporting polypeptide (OATP)-mediated drug–drug interactions (DDIs) are among the most important classes of clinically relevant DDIs. Accurate prediction of the OATP-mediated DDIs is not successful due to the sequential disposition pathways of OATP substrates in humans. Intestinal and hepatic uptake transporters, efflux transporters, and cytochrome P450 (CYP) enzymes are often involved in the sequential disposition pathways of typical OATP substrates. The aim of this proof-of-concept study is to develop and validate a novel approach which can be used to predict OATP-mediated DDIs with significantly increased accuracy and decreased false-negatives.
Methods
The feasibility of using a disposition pathway-dependent prediction (DPDP) approach to predict the ratios of the area under the plasma concentration–time curve (AUCR) in the presence and absence of the inhibitor was investigated. A total of 62 clinical DDI studies were included in this feasibility study. The disposition pathways governing the outcome of DDIs were first identified for each substrate using the information within learning sets, and then substrate-specific algorithms were used to predict the DDI risks of the external validation set (51 DDIs).
Results
The method predicted AUCR within 50–200 % for 50 studies (98 %), and the false-negative rate was 9.8 %. The DPDP approach showed significant improvement over an existing approach and was used to forecast the magnitude of 198 DDIs that have not been studied.
Conclusion
This approach can be used to avoid unnecessary clinical DDI studies during new drug development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The pharmacokinetic interactions between an investigational new drug and other drugs should be defined during drug development as part of an adequate assessment of the drug’s safety and effectiveness [1]. Predicting clinical drug–drug interactions (DDIs) during drug development is essential to the design of clinical DDI studies and/or the elimination of unnecessary clinical DDI studies.
Organic anion-transporting polypeptide (OATP) 1B1 (OATP1B1) has, along with OATP1B3, been shortlisted as a transporter of considerable importance for drug disposition [2]. In fact, OATP-mediated DDIs are among the most important classes of clinically relevant DDIs [2]. In order to avoid unnecessary clinical DDI studies in the drug development process, transporter-mediated clinical DDIs should be estimated from the results of in vitro experiments [1].
The first static model for predicting OATP-mediated DDIs was recently established and validated by being applied to a large number of DDI studies [3]. This impressive model was developed based on in vitro inhibition potencies and several sets of assumptions. By assuming maximal increase in intestinal availability, maximum unbound concentration at the inlet to the liver, and inhibition of only the hepatic uptake, the prediction accuracy of this model was within 50–200 % for 44 studies (76 %); however, the false-negative prediction was not satisfactory (28 %). The minimal false-negative predictions were obtained (19 %) when the inhibitory effects on both hepatic uptake and efflux/metabolism were included, but then the overall prediction accuracy became worse as compared to the former assumption. In light of these results, the author hypothesized that the inhibitory effects on both hepatic uptake and efflux/metabolism should be considered together for some substrates, while only the hepatic uptake should be considered for others.
The disposition of OATP substrates in humans often involves multiple transporters [OATP1, P-glycoprotein, multidrug resistance-associated protein 2 (MRP2), or breast cancer resistance protein (BCRP)] and drug-metabolizing enzymes [cytochrome P450 (CYP) 34A, CYP2C8, CYP2C9, or UDP-glucuronosyltransferase]. In addition, the role of uptake transporters, efflux transporters, and drug-metabolizing enzymes involved in the clearance of OATP substrates are sequential. The prediction for the DDIs involving substrates with parallel pathways of clearance (such as typical CYP substrates) has already been dealt with, and this has been quite successful [4, 5]. However, the prediction for the DDIs involving the OATP substrates which have sequential clearance processes still requires more effort. The major challenge for the prediction of OATP-mediated DDIs is how to identify the specific disposition pathway (or the rate-determining process) of an OATP substrate that controls the magnitude of a DDI.
In order to overcome this difficulty, I have used the information of typical clinical DDIs (a so-called learning set) to determine the disposition pathways of a substrate which contribute to the observed DDIs. This idea was inspired by a recent publication by our group which showed that an in vivo information-guided prediction approach is more robust than a purely in vitro data-based approach [4]. Here, I report the results of a proof-of-concept study, the primary aim of which was to develop and validate a disposition pathway-dependent prediction (DPDP) approach which can predict OATP-mediated DDIs with significantly increased accuracy and decreased false-negatives. The second objective of this study was to forecast with confidence the magnitude of a large number of drug interactions that have not been studied.
2 Methods
2.1 Data Source
The relevant data of clinical DDIs associated with OATP substrates were mainly retrieved from a single recent report which is based on studies extracted from the PubMed database [3]. Whenever available, additional data from the literature were included. The clinical DDIs that involved intravenously administered inhibitors were excluded. The following information was abstracted from the literature: doses of the inhibitors, inhibition constant (K i) of reversible inhibitors, concentration of inhibitor required to achieve half-maximal inactivation (K I) and k inact (maximal rate constant of enzyme inactivation) of mechanism-based inhibitors, intestinal availability (F g) of the substrates, the ratios (AUCR) of the area under the plasma concentration–time curve (AUC) in the presence and absence of the inhibitor, transporters and metabolic enzymes involved in the disposition of substrates, and estimated maximum unbound concentrations of the inhibitors at the inlet to the liver [I]. An algebraic mean of the AUC increase was used in the calculation when multiple studies were available for a single combination of substrate and inhibitor (if the same dose of inhibitor was used in these studies).
2.2 Assumptions for the Disposition Pathway-Dependent Prediction (DPDP) Approach
Although disposition sites other than liver and intestine can play roles in the clearance pathway of substrates, only hepatic and intestinal transporters or enzymes were assumed to contribute to the observed DDIs. The fraction of the substrate transported by way of the specific uptake transporter (OATP1B1 or 1B3), efflux transporter [multidrug resistance protein 1 (MDR1), MRP2, or BCRP], the fraction absorbed (F a) of the substrates and inhibitors, and the blood-to-plasma concentration ratio value of inhibitors were assumed to be 1.0. Only one primary uptake or efflux transporter was considered in the prediction of each DDI.
2.3 Initial Prediction Models
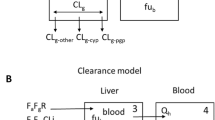
Thirteen OATP substrates were divided into six classes based on the transporters and metabolic enzymes involved in their disposition in humans [3]. Initial prediction models for each class of substrates were derived (Table 1) based on the reported static prediction models for OATP- and CYP-mediated DDIs [3, 5, 6], and the disposition information from the literature [3]. The generic algorithm used to calculate AUCR is listed below (Eq. 1). All of the potential contributing factors (CYP enzymes, uptake, and efflux transporters in the liver and intestine) are included in this algorithm, and the consecutive pathways of disposition are addressed by it. The first part of Eq. 1 indicates the contribution of hepatic OATP transporters; the second part, the contribution of hepatic efflux transporters; the third part, the contribution of hepatic CYP enzymes; the last part, the contribution of the intestinal CYP3A enzyme and efflux transporters which determine the intestinal availability (F g).
where cypK i is the inhibition constant of the CYP enzyme, [I g] is the concentration if the inhibitor in the gut and oatK i is the inhibition constant for OATP1B1 or 1B3. For the contribution of a particular hepatic CYP enzyme to an observed DDI, a novel parameter was introduced (named as CRCYP) in the prediction models (Table 1). CRCYP reflects the apparent contribution ratio of the specific hepatic CYP enzyme to an observed DDI. CRCYP is not necessarily equal to f mCYP, which is the contribution of the specific hepatic CYP to the total clearance of a substrate. In theoretical considerations, CRCYP (0 ≤ CRCYP ≤ f mCYP) is equal to f mCYP when the specific hepatic CYP contributes ‘completely’ to the DDI. CRCYP is less than f mCYP if the specific hepatic CYP contributes partially to the DDI, and it is assumed to be 0 when the inhibition of CYP by the inhibitor has no impact on the DDI involving an OATP substrate. With regard to the calculation method, f mCYP was often estimated from the in vitro phenotyping and mass balance studies; in contrast, CRCYP was estimated from the in vivo information (a reported clinical DDI). A similar parameter for the hepatic efflux transporter was also introduced (CRE). However, for the sake of simplicity, only two possible values were set for this parameter (0 and 1). The value of CRE was tentatively set to 1.0 for the initial prediction models.
A recent study revealed an equal contribution from CYP2C8 and CYP3A to the metabolism of repaglinide [7] and, therefore, both enzymes were taken into account for repaglinide. The contribution of gut metabolism and efflux to a DDI was only considered for atorvastatin, pravastatin, rosuvastatin, and bosentan because the F g of these substrates is relatively low (0.39, 0.50, 0.43, and 0.47, respectively) [3, 8]. Although the F g of fimasartan was not reported, the contribution of gut metabolism to the DDI was also considered for fimasartan because CYP3A is responsible for the metabolism of this drug. Consequently, the F g value of fimasartan was estimated in the development of the final prediction models. All four of these substrates are CYP3A, MDR1, MRP2, and/or BCRP substrates. Both reversible and irreversible CYP inhibitors were included in the analysis. Only the equations for reversible inhibitions are shown in Table 1. For the irreversible inhibitions, Eq. 2 was used to replace the corresponding part (Eq. 3) of the equations in Table 1. k deg is the enzyme degradation rate constant (0.000481 min−1 for intestinal CYP3A and 0.00097 min−1 for CYP2C8) [5, 9].
2.4 Final Prediction Models
The final prediction models were developed based on the key disposition information and selected learning sets of these OATP substrates (Table 2). For atorvastatin and bosentan, hepatic uptake via OATP is the rate-determining process for drug clearance [10, 11]. Therefore, inhibition of the hepatic efflux/metabolic processes was not considered in the final prediction model for atorvastatin and bosentan (CRCYP = 0). The contribution of CYP2C9 to the observed DDI of glibenclamide cannot be determined due to the lack of relevant data. In order to enable avoidance of false-negative predictions to a great extent and rationally exclude unnecessary clinical DDI studies, CYP2C9 was included in the final prediction model for glibenclamide. The CRCYP2C9 value was assumed to be the same as f mCYP2C9 (0.60) [12], which was estimated using the pharmacogenetic method [4]. For the other OATP substrates, a reported DDI (learning set) was selected for each substrate (Table 2). CRCYP, CRE, and F g values were estimated based on the data of the learning set in combination with the initial prediction model (Table 1). In order to best estimate the CRCYP, CRE, or F g value of a OATP substrate, the criteria of selection of the learning set was defined as follows: (1) the AUCR of a DDI was ≥1.25 and (2) the inhibitor of a DDI shows relatively potent inhibition on a CYP enzyme and/or a efflux transporter.
The maximum unbound concentrations of the inhibitors at the inlet to the liver [I] is estimated by Eq. 3, where k a is the absorption rate constant (0.03 min−1, an assumed average value [13]), D is the dose of the inhibitor, Q h is the liver blood flow (1,240 mL/min) [3], and f u is the unbound fraction of drug in plasma. [I] of inhibitors were directly obtained from the literature [3] (calculated based on Eq. 4) except for sildenafil and ketoconazole, whose [I] were calculated in house using Eq. 3.
The concentration in the gut, [I]g, was defined by Eq. 5, where Q g is the enterocytic blood flow (248 mL/min) [6].
2.5 Assessment of the Predictive Performance
To evaluate the performance of the prediction models, the prediction accuracy was defined as the proportion of DDI studies in which the predicted AUCR was within a twofold range of the observed AUCR. If the observed AUCR of a DDI was ≥1.25 and the predicted AUCR was <1.25, then it was defined as a false-negative prediction. This threshold was selected in accordance with U.S. Food and Drug Administration standards for ‘weak’ inhibition [1]. The prediction bias of each assumption was calculated as an average deviation (AD) of the predicted versus observed AUC ratios (Eq. 6). The precision of each assumption was calculated as the root-mean-square error (RMSE) (Eq. 7).
3 Results
3.1 Collection of Clinical Drug-Drug Interaction (DDI) Studies
A total of 62 studies involving 13 OATP substrates and 20 inhibitors were included in this study. Among these, 24 studies showed a greater than threefold increase in the AUC of substrate drugs, and 14 studies showed a greater than fivefold increase. A total of 11 studies were selected as the learning set, and the remaining 51 studies were included in the validation set.
3.2 Prediction of the Ratio of the Area Under the Plasma Concentration-Time Curve Using the DPDP Approach
The details of the data of 62 clinical studies are provided in the Electronic Supplementary Material. The method successfully predicted AUCR within 66–150 % of the observed AUCR for 38 studies (75 %, 38/51 studies) and within 50–200 % for 50 studies (98 %, 50/51 studies) (Fig. 1). The number of false-negative predictions was only five (9.8 %, 5/51 studies). The predictive error (AD) and precision (RMSE) were −0.09 and 2.58, respectively. The predictive performance of the DPDP approach shows significant improvement over the existing approach. For example, the prediction accuracy of the published model was within 50–200 % for 44 studies (76 %), whereas the false-negative prediction was 28 % [3]. The predictive precisions ranged from 3.46 to 15.6 [3].

Predicted versus observed AUCR values in the external validation set. Solid, dashed, dotted lines Line of unity, the 66–150 % range, and the 50–200 % range of the observed AUCR, respectively. Closed diamond atorvastatin, closed square cerivastatin, fluvastatin, and pitavastatin, closed triangle pravastatin, closed circle rosuvastatin, open circle glibenclamide, glimepiride, nateglinide, and repaglinide, open diamond, bosentan, fimasartan, and irbesartan. AUC R Ratio of the area under the plasma concentration–time curve in the presence and absence of the inhibitor
3.3 Forecasting the Magnitude of DDIs That Have Not Yet Been Studied
The AUCR values of 260 possible interactions between the 13 substrates and the 20 inhibitors were estimated (Fig. 2a). Only a small proportion (24 %) of all possible combinations between substrates and inhibitors have as yet been studied in humans. Gemfibrozil, clarithromycin, protease inhibitors, rifampicin, and cyclosporin are the most potent inhibitors. According to the sums of AUCR values (the ‘drug interaction number’—given in following text after each drug in parenthesis), atorvastatin (78), bosentan (66), fimasartan (63), and repaglinide (57) are the OATP substrates most sensitive to inhibition (Fig. 2b), and pitavastatin (42), rosuvastatin (42), and glimepiride (39) are the least sensitive substrates. In general, there was an association between the drug interaction number and the disposition pathway of OATP substrates (Fig. 2b). OATP1B1 and CYP3A double substrates were found to be the most easily inhibited by inhibiting drugs, with OATP1B1 and CYP2C8/CYP2C9/MRP2 double substrates moderately inhibited. OATP1B1 single substrates were also moderately influenced, but to a lesser extent. In addition to OATP1B1, CYP3A appears to be the most important factor influencing the outcome of a DDI involving OATP substrates.

a Predicted AUCR values of organic anion-transporting polypeptide (OATP) substrates in the presence of various inhibitors. b Drug interaction number for OATP substrates. Drug interaction number is the sum of AUCR values for each substrate. The unit for dose is milligrams. ATO Atorvastatin, 1B1 OATP1B1, 1B3 OATP1B3, 2C8 CYP2C8, 2C9 CYP2C9, 3A CYP3A, CER cerivastatin, CYP cytochrome P450, FLU fluvastatin, PIT pitavastatin, PRA pravastatin, ROS rosuvastatin, GLB glibenclamide, GLM glimepiride, NAT nateglinide, REP repaglinide, BOS bosentan, FIM fimasartan, IRB irbesartan, OATP organic anion-transporting polypeptide, P-gp P-glycoprotein
4 Discussion
The primary finding of this study is that the DPDP approach can accurately predict the DDI risks involving OATP substrates. The predictive performance of the DPDP approach shows significant improvement over an existing approach. Taking advantage of this novel approach, the magnitude of a large number of drug interactions that have not been studied were forecasted with confidence.
To my knowledge, this is the first proof-of-concept study demonstrating that the DPDP approach is a useful tool for the prediction of drug interaction risks associated with OATP substrates. Successful prediction of an OATP-mediated DDI is difficult because the disposition of OATP substrates in humans often involves multiple intestinal and hepatic uptake and efflux transporters, as well as CYP enzymes. What is more, the disposition pathway often varies from one substrate to another substrate. In order to overcome these difficulties, in the DPDP approach, the key disposition pathways governing the outcome of DDIs were identified for each substrate using the information from a newly developed learning set, as well as relevant pharmacokinetic data. Thereafter, a substrate-specific prediction model was developed for each OATP substrate.
In order to improve decision-making in drug development and discovery, the DPDP approach can be used during the course of new drug development and discovery. Utilization of a DPDP approach can reasonably preclude unnecessary clinical DDI studies. If a new drug is a potential victim drug and the disposition pathway of this drug in human is already known, the initial prediction model for clinical DDIs involving this drug can be developed. One clinical DDI study for this victim drug (co-administered with a strong inhibitor) is needed to investigate the potential DDI risk [1]. Subsequently, the values of CRCYP, CRE, and/or F g can be estimated based on this clinical study. The final prediction model can be derived based on the initial model, in combination with the values of CRCYP, CRE, and/or F g, and this final prediction model enables the DDI risks of this victim drug to be predicted if co-administered with other inhibitors. According to the new Federal Drug Administration draft guidance, if a strong inhibitor alters the kinetics of this victim drug, subsequent clinical DDI studies are advised to define interactions with other less potent inhibitors. However, if no significant DDI is predicted between this victim drug and other weaker inhibitors based on DPDP approach, a secondary clinical study may not be necessary. If a new drug is a potential perpetrator, a pilot clinical DDI study is not needed. Most of the DDIs involving this drug can be readily predicted using the final prediction model of various OATP substrates (shown in Table 2).
It is noteworthy that the impact of SLCO (encoding OATP) polymorphisms was not considered in the prediction models reported here. It has been well documented that the magnitude of OATP1B1-mediated DDIs is influenced by the SLCO1B1 genotype. For example, the effect of cyclosporin on repaglinide AUC was reported to be 42 % lower in subjects with the SLCO1B1 521TC genotype than in subjects with the 521TT (reference) genotype [14]. This finding may be explained by the reduced activity of OATP1B1 in carriers of the variant SLCO1B1 c.521C allele. Therefore, the fractional activity (FA) of OATP1B1 resulting from the combination of mutated alleles relative to the activity of the reference genotype could be incorporated into the current prediction model. Further studies are required to test the predictive performance of the modified model involving SLCO polymorphisms.
The limitations of this study need to be considered. The DPDP approach may not applicable to DDIs involving a new OATP substrate whose full disposition characteristics in humans are not clear. In this situation, the DDI can only be reasonably predicted if the perpetrator is a pure OATP inhibitor (without any other effect on transporters or drug-metabolizing enzymes). The DPDP approach relies on the data of a learning set (or key pharmacokinetic information) to establish the final prediction model. Successful application of the DPDP approach is therefore not feasible without these relevant data. However, in this situation, the initial prediction model with some assumptions (assuming CRCYP = f mCYP, CRE = 1, F g = 1) may be used for predicting OATP-mediated DDIs. While this approach may overestimate DDI risks, it enables avoidance of false-negative predictions to a great extent and rationally excludes unnecessary clinical DDI studies. Indeed, a recent report on the prediction of OATP-mediated DDIs showed that the overall prediction accuracy became worse (although the false-negative prediction decreased) when the inhibitory effects on both hepatic uptake and efflux/metabolism were considered [3].
5 Conclusions
A novel approach for the prediction of OATP transporters-mediated clinical DDIs is proposed. The results suggest that AUCR can be predicted with more accuracy using this algorithm than with an existing method. In addition, predictive distributions for 260 possible DDIs were obtained, giving detailed information on some drugs or inhibitors that have been poorly studied to date. The DPDP approach can be used in the course of new drug development to reasonably avoid unnecessary clinical DDI studies.
References
U.S. Food and Drug Administration (FDA). Draft guidance for industry. Drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations. 2012. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf. Accessed 16 Sept 2012.
Giacomini KM, Huang SM, Tweedie DJ, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–36.
Yoshida K, Maeda K, Sugiyama Y. Transporter-mediated drug–drug interactions involving OATP substrates: predictions based on in vitro inhibition studies. Clin Pharmacol Ther. 2012;91:1053–64.
Hu ZY, Parker RB, Laizure SC. In vivo information-guided prediction approach for assessing the risks of drug–drug interactions associated with circulating inhibitory metabolites. Drug Metab Dispos. 2012;40:1487–94.
Shardlow CE, Generaux GT, MacLauchlin CC, et al. Utilizing drug–drug interaction prediction tools during drug development: enhanced decision making based on clinical risk. Drug Metab Dispos. 2011;39:2076–84.
Hinton LK, Galetin A, Houston JB. Multiple inhibition mechanisms and prediction of drug–drug interactions: status of metabolism and transporter models as exemplified by gemfibrozil–drug interactions. Pharm Res. 2008;25:1063–74.
Sall C, Houston JB, Galetin A. A comprehensive assessment of repaglinide metabolic pathways: impact of choice of in vitro system and relative enzyme contribution to in vitro clearance. Drug Metab Dispos. 2012;40:1279–89.
Shitara Y. Clinical importance of OATP1B1 and OATP1B3 in drug–drug interactions. Drug Metab Pharmacokinet. 2011;26:220–7.
Obach RS, Walsky RL, Venkatakrishnan K. Mechanism-based inactivation of human cytochrome p450 enzymes and the prediction of drug–drug interactions. Drug Metab Dispos. 2007;35:246–55.
Maeda K, Ikeda Y, Fujita T, et al. Identification of the rate-determining process in the hepatic clearance of atorvastatin in a clinical cassette microdosing study. Clin Pharmacol Ther. 2011;90:575–81.
Treiber A, Schneiter R, Delahaye S, et al. Inhibition of organic anion transporting polypeptide-mediated hepatic uptake is the major determinant in the pharmacokinetic interaction between bosentan and cyclosporin A in the rat. J Pharmacol Exp Ther. 2004;308:1121–9.
Kirchheiner J, Brockmöller J, Meineke I, et al. Impact of CYP2C9 amino acid polymorphisms on glyburide kinetics and on the insulin and glucose response in healthy volunteers. Clin Pharmacol Ther. 2002;71:286–96.
Obach RS, Walsky RL, Venkatakrishnan K, et al. The utility of in vitro cytochrome P450 inhibition data in the prediction of drug–drug interactions. J Pharmacol Exp Ther. 2006;316:336–48.
Kajosaari LI, Niemi M, Neuvonen M, et al. Cyclosporine markedly raises the plasma concentrations of repaglinide. Clin Pharmacol Ther. 2005;78:388–99.
Ogilvie BW, Zhang D, Li W, et al. Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of CYP2C8: implications for drug–drug interactions. Drug Metab Dispos. 2006;34:191–7.
Niemi M, Pasanen MK, Neuvonen PJ. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev. 2011;63:157–81.
Gibbs MA, Thummel KE, Shen DD, et al. Inhibition of cytochrome P-450 3A (CYP3A) in human intestinal and liver microsomes: comparison of Ki values and impact of CYP3A5 expression. Drug Metab Dispos. 1999;27:180–7.
Sekar VJ, Lefebvre E, De Pauw M, et al. Pharmacokinetics of darunavir/ritonavir and ketoconazole following co-administration in HIV-healthy volunteers. Br J Clin Pharmacol. 2008;66:215–22.
Kim JW, Yi S, Kim TE, et al. Increased systemic exposure of fimasartan, an angiotensin II receptor antagonist, by ketoconazole and rifampicin. J Clin Pharmacol. 2013;53:75–81.
Acknowledgments
The author wishes to thank Dr. Yuan-Sheng Zhao from Mount Sinai School of Medicine for his fruitful discussions during the preparation of this paper.
Conflict of interest
No funding has been received for the conduct of this study. The author has no conflicts of interest that are directly relevant to the content of this study.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Hu, ZY. Disposition Pathway-Dependent Approach for Predicting Organic Anion-Transporting Polypeptide-Mediated Drug–Drug Interactions. Clin Pharmacokinet 52, 433–441 (2013). https://doi.org/10.1007/s40262-013-0045-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-013-0045-x