
Abstract
Betamethasone dipropionate topical formulations are using to treat anti-inflammatory skin diseases such as dermatitis, eczema, and psoriasis. The current research study confers the eco-friendly toward green chemistry and stability-indicating RP-UHPLC method for the simultaneous determination of chlorocresol (CCL) and betamethasone dipropionate (BTD) in topical formulations (cream and ointment). The CCL and BTD were accurately quantitated by employing beclomethasone dipropionate (BCD) as an internal standard. The developed method was optimized utilizing QbD-based Box–Behnken Design (BBD) prior to method validation for the intended purpose. The critical quality attributes (CQAs) and critical method parameters (CMPs) identified and executed 15 design of experiments (DoEs). The foremost influencing factors were fine-tuned and optimized using graphical and numerical evaluation. The chromatographic separation was accomplished on Acquity UPLC BEH C18, 100 mm × 2.1 mm, 1.7 µm column with potassium phosphate buffer (0.02 M) and acetonitrile (ACN) using gradient elution with a runtime of 8 min. The set flow rate and injection volumes were 0.4 mL/min and 5 µL, respectively. The detection was made at 240 nm and maintained column oven temperature at 40 °C. The analytical method was validated as per the current ICH guideline Validation of Analytical Procedures: Text and Methodology Q2(R1). The linearity ranges for CCL, BTD, and BCD were 20.4–61.1, 10.3–30.8, and 10.4–31.1 µg/mL with correlation coefficients of > 0.999. As revealed, the method was superior accuracy with % recovery for CCL 98.6–101.5 and BTD 99.6–101.6 at three different levels. The results dictate the fitness of the method for the routine quality control determination of CCL and BTD from its commercial topical formulations. The proposed method with a low flow rate and less runtime benefits to analyze high throughput quality control samples with less solvent consumption, and it helps the environment by supporting the green chemistry concept.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
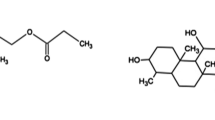
Betamethasone dipropionate (BTD) is a dipropionate ester of betamethasone, in which 17, 21 hydroxy hydrogens are replaced by propanoyl groups. BTD is a synthetic adrenocorticoid used to treat anti-inflammatory skin conditions such as dermatitis, eczema, and psoriasis [1]. It has a glucocorticoid activity in high and mineralocorticoid activity in low. The chemical name is Pregna-1,4-diene-3,20-dione, 9-fluoro-11-hydroxy-16-methyl-17,21-bis(1-oxopropoxy)-, (11β,16β); or 9-Fluoro-11β,17,21-trihydroxy-16β-methylpregna-1,4-diene-3,20-dione 17,21 dipropionate with the empirical formula of C28H37FO7, and molecular weight of 504.59 [2]. The chemical structure is shown in Fig. 1a. BTD is a white crystalline, odorless powder. It is insoluble in water, freely soluble in acetone, ACN, and chloroform, and sparingly soluble in alcohol.

Chemical structure of betamethasone dipropionate a and chlorocresol b
Chlorocresol (CCL) is a monochlorinated m-cresol. It is used as an antimicrobial preservative in cosmetics and pharmaceutical formulations [3]. The chemical name is 4-Chloro-3-methylphenol or p-Chlorocresol with the empirical formula of C7H7ClO and molecular weight of 142.58 [2]. The chemical structure is shown in Fig. 1b. CCL appears as a pinkish to white crystalline solid with a phenolic odor. It is very soluble in alcohol and ACN, freely soluble in fatty oils, and slightly soluble in water.
In the reported preceding literature, several analytical methods presented for the quantification of BTD those are [2, 4,5,6,7,8,9,10,11], for CCL [12,13,14,15,16,17], and the simultaneous determination of CCL and BTD [18,19,20]. The reported methods for quantification of CCL and BTD are not economical (long runtime, THF, and IPA usage in mobile phase). A detailed comparison of reported analytical methods and the proposed method is tabulated in Table 1. The reported methods have complex sample extraction procedures and quantification of components without accounting for the dead volume generated from the sample matrix. Degradation studies play a vital role in proving the stability-indicating capability of the chromatographic method. The absence of forced degradation studies and QbD-based optimization was limiting their application. The objective of the current study is to develop a precise, specific, accurate, stability-indicating, rugged, and robust UHPLC analytical method by utilizing the QbD approach. In recent days, the QbD approach has been used effectively for the risk assessment or optimization of chromatographic conditions.
Generally, chromatographic methods are developed using a trial-and-error or one-factor-at-a-time (OFAT) approach, in which one of the parameters is varied in consecutive experiments until a sufficient retention time, symmetry, good peak shape of the analyte, and a resolution between impurities and components present in the sample matrix are achieved [21,22,23,24,25,26,27,28,29,30,31,32,33]. In the current study, BBD was utilized for the optimization of RP-UPLC chromatographic parameters. The BBD design was developed by George E. P. Box and Donald Behnken in 1960 [34]. It is a fixed three-level equally spaced design with numeric and categoric factors as a variable. A minimum of three variables is required to conduct any BBD study. This design is used to produce higher-order response surfaces with fewer experimental runs than the factorial design. Experimental runs are conducted at midpoints of the edges and at the center point. QbD-based method optimization is helpful to the user to understand the individual and interaction effect of the variables on each CQA. Identification of CQAs and CMPs is crucial for the QbD-based method optimization. The CQAs include retention time (Rt), resolution (Rs) of components, and other system suitability parameters (tailing factor and plate count). Out of all CQAs, Rt and Rs significantly impact the performance of the analytical method. Hence, the current study was optimized by monitoring the Rt and Rs responses.
The QbD-based test method optimization was executed for determining the probability of risk(s) or failure(s)’ [21]. To meet the objective, a cause–effect relation between the CMPs and CQAs was evaluated. The CMPs making include %v/v of solvent (ACN in mobile phase-A) (mL) (A), flow rate (mL/min) (B), and column oven temperature (°C) (C). These CMPs was evaluated for 2 CQAs viz., Rt of CCL (R1), Rt of BTD (R2), Rt of BCD (R3); Rs between CCL and BTD (R4), and Rs between BTD and BCD (R5) at a standard concentration for all experimental trails.
Green chemistry is defined as “The design, development, and implementation of chemical products and processes that reduce or eliminate the use and generation of hazardous substances” [35]. The green chemistry concept is well-promoted by Paul T. Anastas, also known as the “Father of Green Chemistry” [36]. The green chemistry concept includes 12 Principles [37]. Out of all, the current study focused on minimizing the usage of an organic solvent such as methanol or ACN in the mobile phase. Additionally, low flow rate and short analysis time help to minimize the usage of an organic modifier. Several green chemistry and green chromatographic analytical techniques were reported in various publications [38,39,40].
To the best of our understanding, the current research study is the first-ever QbD-based optimized and validated UHPLC method with a green chemistry concept for the quantification of CCL and BTD in topical formulations. The proposed method has clear improvements over formerly published test methods, including the execution of a QbD approach prior to method validation. The predefined objectives, its cost-effectiveness with shorter runtime (8 min) using UHPLC and no interference from its formulation excipients for the quantification of CCL and BTD, accurately estimated anticipated components from its sample matrix using BCD as an internal standard with superior accuracy and precision. Further, various stress conditions were conducted to verify the peak purity of CCL, BTD, and BCD in degradation studies.
Materials and methods
Instrumentation and software
Waters Acquity H-Class UHPLC equipped with Quaternary Solvent Manager (QSM), sample manager—Flow Through Needle (FTN), and column manager with eCord and TUV/PDA detector (Waters Corporation, Milford, Massachusetts, USA) were used for the analysis. The output signal was monitored through empower 3 data acquisition software (Waters Corporation, Milford, Massachusetts, USA). The QbD-based method optimization was carried out using the Design-Expert® software version 12 (Stat-Ease Inc, Minneapolis, USA). For the optimization of chromatographic conditions, different Acquity UPLC analytical columns were used, those were HSS C18, 100 mm × 2.1 mm, 1.8 µm; BEH C18, 100 mm × 2.1 mm, 1.7 µm; Cortecs UPLC T3, 100 mm × 2.1 mm, 1.6 µm; BEH C8, 100 mm × 2.1 mm, 1.7 µm; HSS C18, 50 mm × 2.1 mm, 1.8 µm; BEH C18, 50 mm × 2.1 mm, 1.7 µm; and BEH Phenyl, 100 mm × 2.1 mm, 1.7 µm from Waters Corporation (Milford, Massachusetts, USA). The Mettler Toledo analytical balance (Model: XPE205, Columbus, Ohio, USA) was used to weigh standards and samples. The sonicator from Bransonic (Model: CPX8800H, Danbury, Connecticut, USA) was used for the standard preparations. The water bath from PolyScience (Model: WB28, W. Touhy Avenue Niles, Illinois, USA), vortex from Scientific Industries Inc (Model: SI-P236, Bohemia, New York, USA), and centrifuge from Beckman Coulter (Model: Allegra X-14R, Beckman Coulter, Brea, California, USA) were used for the sample preparation. The oven from Sheldon Manufacturing Inc (Model: 1330FMS, Cornelius, Oregon, USA) and photostability chamber from Luzchem (Model: LZC-ICH2, Ottawa, Ontario, Canada) were used to expose degradation samples as per ICH recommendations.
Materials and reagents
The BTD (Purity: 99.7%) reference standard was obtained from USP (Rockville, Maryland, USA). The CCL (Purity: 99.9%) was procured from Sigma-Aldrich (St. Louis, Missouri, USA). The BCD (Purity: 100.0%) was purchased from Millipore-Sigma (Laramie, Wyoming, USA). The analytical grade potassium phosphate monobasic (Purity > 99.0%), sodium hydroxide (Purity ≥ 99.0%), and ACN (Purity ≥ 99.9%) from EMD Millipore (Burlington, Massachusetts, USA), methanol (Purity > 99.8%) from VWR (Radnor, Pennsylvania, USA) were purchased. The in-house purified water from the ELGA lab water system (pH between 5.0 and 6.4, Conductivity < 1.5 µS/cm and TOC < 500 ppb) (High Wycombe, Bucks, UK) was used throughout the UHPLC analysis. The hydrochloric acid (Purity: 36.5–38.0%) from Avantor (Radnor, Pennsylvania, USA) and hydrogen peroxide (Purity: 30–32%) from EMD Millipore (Billerica, Massachusetts, USA) were purchased and used for the degradation studies. The 0.2 µm Nylon, polytetrafluoroethylene (PTFE), and polyvinylidene difluoride (PVDF) syringe filters were obtained from Whatman (GE Healthcare Life Sciences) (Little Chalfont, Buckinghamshire, UK).
Chromatographic conditions
The chromatographic separation was accomplished by employing UPLC BEH C18, 100 mm × 2.1 mm, 1.7 µm (Waters Acquity) column with linear gradient elution. The mobile phase-A(MP-A) contains 0.02 M potassium phosphate monobasic and ACN as 70:30%v/v. Mobile phase-B(MP-B) contains 0.02 M potassium phosphate monobasic and ACN as 30:70%v/v, filtered using 0.2 µm Nylon membrane filter and degassed in a sonicator. The gradient program was T (min) /%B: 0.0/40, 6.0/70, 6.1/40, 8.0/40. The injection volume and flow rate were set as 5.0 µL and 0.4 mL/min, respectively. The set column oven temperature was 40 °C, and UV detection was made at 240 nm.
Preparation of diluent
ACN was utilized for the preparation of standard stock solutions and intermediate standard solutions. The final standard solution was prepared using water and ACN in the ratio of 50:50%v/v. Similarly, ACN was utilized for the extraction of components from the sample matrix. The final sample solution was prepared using water and ACN in the ratio of 50:50%v/v.
Preparation of standard solution
Preparation of standard stock solutions for CCL, BTD, and BCD
Prepared individual standard stock solutions of CCL, BTD, and BCD in ACN w.r.t 800 µg/mL, 400 µg/mL, and 400 µg/mL concentration. The concentration of BTD and BCD was corrected as betamethasone and beclomethasone, respectively.
Standard solution
Transferred 5.0 mL of CCL, BTD, and BCD standard stock solutions into 25 mL of VF, dilute to volume with ACN. Diluted 5.0 mL into 20 mL with diluent (water/ACN 50:50%v/v) and mixed well.
Preparation of sample solutions
Accurately weighed 4.0 g of BTD topical formulation USP, 0.05% into 50-mL glass centrifuge tube, added 5.0 mL of BCD standard stock (Internal standard), 20.0 mL of ACN, and closed with the cap firmly. The sample solution was kept in a water bath at 60 °C with an intermittent vortex every 5 min till 30 min. After that the sample solution was transferred into an ice bath (~ 0 °C), vortexed intermittently every 5 min till 15 min, and centrifuged for 20 min at room temperature with 3000 RPM. Transferred 5.0 mL of centrifuged sample solution into 20 mL VF and made up to the volume with diluent and filtered with 0.2 µm PVDF filter.
Statistical analysis
Based on initial method development experiments, the CMPs are %v/v of Organic solvent in MP-A (ACN)(mL), flow rate (mL/min), and column temp (°C) were selected. A total of 15 experiments were carried out by 33 BBD employing three numeric factors at three different levels, including three center points. The standard and sample solutions contain 40 µg/mL of CCL and 20 µg/mL of BTD and BCD were used for the design of experiments. The data from the experiments were evaluated against two CQAs viz., Rt of CCL (R1), Rt of BTD (R2), Rt of BCD (R3), Rs between CCL and BTD (R4), and Rs between BTD and BCD (R5) using Design-Expert® software. The best fitted quadratic polynomial second-order equation was produced for each response based on CMPs significance from an analysis of variance (ANOVA). The statistical analysis was presented for individual and interaction variables against the coefficient estimate of the responses [41, 42].
where R is the response to be examined, b0 is the intercept, b1–b33 are the factor regression coefficients.
The generated model was examined and compared for distinct boundaries of correlation coefficient (R2), adjusted R2, predicted R2, and adequate precision. The response surface analysis (RSA) was accomplished by producing 2D-contour plots and 3D-response surface plots. The optimum chromatographic conditions were anticipated from numerical and graphical optimization. The optimized chromatographic conditions were marked in a graphical overlay plot of the design space region.
Results and discussion
Method development
Optimization of chromatographic conditions
The purpose of the current study is to develop a simple, rapid, and specific method for the simultaneous determination of CCL and BTD in its topical formulations. The main challenge in the method development study is to accomplish acceptable resolution between the analyte peaks within a short runtime. Numerous experiments were conducted to optimize both the mobile phase and stationary phases. Photo-diode array (PDA) detector offers the benefit of quantifying the analyte at its maximum wavelength, thus improving the sensitivity of the analytical method. Further, the PDA detector helps to identify the peak homogeneity of each component. Each component was analyzed in a PDA detector and observed maximum absorbance for CCL at 227 nm, BTD, and BCD at 240 nm. The detection wavelength was selected as 240 nm for the quantification of cited components. Based on the USP monograph, water/ACN 2:1%v/v was chosen to evaluate the separation in isocratic mode with different UPLC columns such as HSS C18, 100 mm × 2.1 mm, 1.8 µm; BEH C18, 100 mm × 2.1 mm, 1.7 µm; Cortecs UPLC T3, 100 mm × 2.1 mm, 1.6 µm; BEH Phenyl, 100 mm × 2.1 mm, 1.7 µm; and BEH C8, 100 mm × 2.1 mm, 1.7 µm. The obtained chromatograms showed the BTD, BCD peaks were eluted lately with less resolution and asymmetric peak shape.
Alternatively, methanol was used in place of ACN in the mobile phase, and analysis was carried out using HSS C18 Columns (100 mm × 2.1 mm, 1.8 µm, and 50 mm × 2.1 mm, 1.8 µm); and BEH C18 Columns (100 mm × 2.1 mm, 1.7 µm, and 50 mm × 2.1 mm, 1.7 µm). No improvement was observed in methanol as an organic modifier. Moreover, comparatively better peak shapes and retention times were observed in ACN. Therefore, continued with ACN for further development studies. Potassium phosphate buffer (0.02 M) and ACN mixture in the ratio of 50:50%v/v was used as mobile phase to separate the components. Two different columns, such as HSS C18 and BEH C18 columns in 100 mm length with a flow rate of 0.5 mL/min and 50 mm length with a flow rate of 0.3 mL/min, were screened for the separation. All the peaks were eluted within the 6 min on HSS C18 and BEH C18 100 mm length columns, but the BCD was eluted as a broad peak. On the other hand, all the peaks were eluted within the 6 min on both HSS C18 and BEH C18 50 mm length columns, but the CCL peak was eluted closely to the unretained peak, further BCD eluted as a broad peak. Overall, 100 mm length columns were comparatively better than 50 mm length columns in terms of resolution. Therefore, the BEH C18 column in 100 mm length was continued for the next set of experiments.
To reduce the overall runtime, ACN composition was increased from 50 to 70%v/v, with a flow rate of 0.4 mL/min. The chromatograms showed that CCL was eluted closely to the unretained peak, and the BCD was eluted as a broad peak. To reduce the peak broadening of the lately eluting components, a gradient program was proposed to incorporate using a mixture of potassium phosphate buffer (0.02 M) and ACN in the ratio of 70:30%v/v as mobile phase-A, and a mixture of potassium phosphate buffer (0.02 M) and ACN in the ratio of 30:70%v/v as mobile phase-B with a gradient program T (min) /%B: 0.0/40, 6.0/70, 6.1/40, 8.0/40. The flow rate was set as 0.4 mL/min, and the column temperature was set as 40 °C. The obtained chromatograms showed a better resolution between the BTD and BCD peaks (Rs: 7.8), and CCL was eluted at 1.6 min and is well-separated from the unretained peak. The BCD (Rt: 5.4 min) peak was eluted with good symmetry (USP Tailing factor: 1.1) without broadening.
Optimization of diluent
In general, topical formulations contain a low amount of active components, w.r.t inactive ingredients. Selection of diluent is a crucial step while achieving better sample extraction during the sample preparation. Most of the topical formulations are semi-solid, and their ingredients are insoluble in aqueous solutions. Both components, BTD and CCL were insoluble in aqueous solutions and soluble in organic solvents like methanol and ACN. Initially, 0.1% acetic acid in methanol was adopted from the USP monograph and used as extraction solvent and diluent. The chromatograms showed that the peaks were distorted. Pure organic solvents such as ACN and methanol were evaluated as an extraction solvent, and a mixture of organic solvent (ACN or Methanol) and water 50:50 v/v was evaluated as diluent. The obtained chromatograms showed better peak shape in both solvents. However, the sample solutions extracted in ACN and diluted with water and ACN in the ratio of 50:50 v/v was showed a better peak shape. Moreover, obtained sample solution from the ACN was clear compared with methanol extracted sample solutions. The chromatograms and the extracted sample solutions indicated that ACN is the suitable solvent for the extraction, and water/ACN 50:50v/v is the best diluent for the preparation of sample solutions.
The sample preparation procedure was evaluated for the effective extraction of components from the sample matrix for superior accuracy. The sample solution was prepared by taking 4 g of sample in a 50-mL glass centrifuge tube, adding 5.0 mL of BCD standard stock solution and 20 mL of ACN. The sample solution was kept in a water bath at 60 °C, vortexed every 5 min till 30 min. The sample solution was transferred to an ice bath and vortexed for every 5 min till 15 min, and then centrifuged the solution at room temperature (RT) for 20 min with 3000 RPM. Transferred 5.0 mL of the supernatant solution into 20 mL VF and diluted with diluent and mixed well. The resulted solution was filtered through a 0.2-µm syringe filter and injected into the chromatograph. The obtained recoveries for BTD was 99.6% and for CCL was 99.8%, and found to be satisfactory for finalizing extraction solvent and diluent. The obtained sample solution contains 40 µg/mL of CCL, 20 µg/mL of BTD (betamethasone), and BCD (beclomethasone). Different injection volumes were evaluated from 2.0 µL to 10.0 µL and selected 5.0 µL as the optimum injection volume based on analyte peak responses. The method conditions have been optimized using QbD-based BBD to achieve a robust analytical method. The summary of method development experiments for the optimization of mobile phase, stationary phase, and diluents is shown in Table 2.
Method optimization using Box–Behnken design—Response Surface Analysis (RSA)
The software recommended best-fitting quadratic model was used for each CQA response viz., Rt of CCL (R1), Rt of BTD (R2), Rt of BCD (R3), Rs between CCL and BTD (R4), and Rs between BTD and BCD (R5). The experimental design for the optimized factors and responses are shown in Table 3. ANOVA F test proved the significance for a selected quadratic model with p < 0.05 for all the CQAs. The chosen responses showed superior R2 values from 0.9989 to 0.9999. Illustrate the best fit of polynomials to the response data (p < 0.0001 in most cases). All the five responses showed “lack of fit” values were insignificant (p > 0.05 in all cases), implying that the selected model is apt for all variables. The difference between predicted R2 and the adjusted R2 values was obtained < 0.2; it revealed an excellent fit of the data due to the closeness of these values. The adequate precision for all the responses was found between 72.9259 and 356.9978; it indicates a high signal-to-noise ratio. The model F and P values, lack of fit, R2, adjusted R2 and predicted R2, and adequate precision values are summarized in ANOVA results Table 4.
The statistical analysis was accomplished for a coefficient estimate of the responses against factors and is given in Table 5. The quadratic polynomial second-order equation was generated by the software for all the responses with intercept b0, and CMPs interaction coefficients b1–b33 are shown in Table 6. The quadratic polynomial second-order equation was evaluated for the application within design space with change in different factor variables and found that observed values and predicted values were within 0.5% of relative error. The results are summarized in Table 7.
The Box–Cox diagnostics current lambda was less than 1 and indicates that no transformation of data is required for all the measured responses. The 2D contour plots and 3D surface model graphs were drawn to acknowledge the interaction between the variables in the experimental design. Figure 2a–e describes the normal plot and 3D response surface plots for all the responses. The interaction graphs, contour plots, and 3D surface plots indicate that responses R1, R2, R3, R4, and R5 were decreased with increasing %v/v organic solvent (ACN) in MP-A (CMP-A). Responses R1, R2, and R3 decreased, R4 increased, and R5 did not affect with the increase in flow rate (CMP-B). Responses R1, R2, R3, and R5 decreased; R4 increased with the increase in column oven temperature (CMP-C). Responses R1, R2, R3, and R4 have a higher AB interaction effect than AC and BC. Response R5 has a higher BC interaction effect than AB and AC.

Normal plot, 3D response surface plot for the response R1 a, R2 b, R3 c, R4 d, and R5 e
The optimum chromatographic conditions were identified using the numerical and graphical optimization method with desired conditions of lower response values R1, R2, and R3 and higher resolution values of R4 and R5. The graphical overlay plots dictate that CMP-A (%v/v organic solvent) (ACN) (mL), CMP-B (flow rate) (mL/min), and CMP-C (column temperature) (°C) were 301.21 mL, 0.396761 mL/min, and 39.676 °C, respectively. The obtained values were similar to the optimized chromatographic conditions from the method development. The desirability and predicted responses are shown in Fig. 3. The overlay plot of post-analysis prediction point values were two-sided with 95% confidence and 99% population. The statistical analysis data for the point of prediction and its responses are tabulated in Table 8. The overlay plots are shown in Fig. 4. The optimized method has been validated in accordance with ICH guidelines.

Desirability and predicted response graphs

Overlay plots predicted responses from the design space region
Method validation
System suitability
The system suitability of the analytical method was assessed in every analysis to ensure adequate separation and reproducibility of the current optimized method. The examined system suitability parameters were USP tailing, USP plate count, USP resolution between BTD and BCD, and %RSD for the peak area ratio of analytes. The system suitability results are tabulated in Table 9. The results were indicative that the method was suitable for the determination of CCL and BTD in topical formulations.
Linearity
The analytical method linearity was determined by injecting seven solutions from 50 to 150% of the nominal concentration of components prepared in a final diluent. The linearity solutions were injected twice into the UPLC and recorded the peak responses, plotted graph between concentration and mean area of the analyte, determined the intercept, slope, correlation coefficient, and residual sum of squares. The linear regression equations for CCL (y = 6274.567x + 1245.940, r2 = 0.9999), BTD (y = 30,599.279x−13,178.800, r2 = 0.9999) and BCD (y = 29,290.277x−11,308.464, r2 = 0.9999) were established. The linearity data indicates components have a linear response from 50 to 150% of concentration. The linearity results of are shown in Table 9.
Precision
The method precision was demonstrated by preparing six (6) individual sample solutions from the homogenous cream and ointment formulations sample. The precision was evaluated for % RSD from the six sample solutions. Intermediate precision was accomplished by another analyst with a different system, a different column in a different day using the same homogenous sample from the precision experiment. The % difference between method precision and intermediate precision results was found less than 2.0%. The precision results are summarized in Table 9.
Accuracy
The accuracy of the test method was determined by spiking the known amount of analyte into the placebo, prepared the samples as per the proposed method. The accuracy experiment was executed for three different levels (50%, 100% & 150% of target concentration) and each level with triplicate preparations. Calculate the amount added, the amount found, % recovered, and standard deviation (SD) for each recovery level. The results are tabulated in Table 9.
Specificity
Specificity of the test method was demonstrated by injecting the blank, standard, as such sample, placebo, individual impurities, spiked sample, and forced degradation samples. The degradation studies were carried out for acid, base, peroxide, thermal, and photolytic conditions. No interference was observed at the retention time of analyte peaks from the placebo, impurities, and degradation peaks. Peak purity was evaluated for standard, as such sample, spiked sample, and degradation samples found that purity threshold was greater than purity angle with no purity flag. The degradation conditions, % degradation, and peak purity results are tabulated in Table 10. The overlayed UHPLC chromatograms of the blank, placebo, standard, sample, and spiked sample with impurities which indicate the specificity of the method for the cream formulation are shown in Fig. 5, and for ointment formulation, it is shown in Fig. 6.

UHPLC chromatogram for the betamethasone dipropionate cream formulation, contains 40 µg/mL CCL; 20 µg/mL BTD; and 20 µg/mL BCD. Injection volume 5.0 µL; Column oven temperature 40 °C and UV detection at 240 nm

UHPLC chromatogram for the betamethasone dipropionate ointment formulation, contains 20 µg/mL BTD; and 20 µg/mL BCD; Injection volume 5.0 µL; Column oven temperature 40 °C; and UV detection at 240 nm
Robustness
Robustness was conducted with varying ± 10% of flow rate (i.e., 0.36 & 0.44 mL/min), ± 2% gradient program for organic variation, ± 5 °C for column oven temperature (i.e., 35 °C & 45 °C) and ± 2 nm for wavelength detection from the actual method conditions. The optimized method was found robust within the range was selected, and the results were found within limits.
Solution stability and filter evaluation study
The solution stability of sample and standard solutions were established at refrigerator (2–8 °C) and room temperature (~ 25 °C) conditions by reinjecting the solutions from the precision experiment after 24 and 48 h against the freshly prepared standard solution. Calculated the % difference from initial assay values and found less than 2% in both the storage conditions up to 48 h. The data concluded that standard and sample solutions were stable up to 48 h at refrigerator and room temperature. The filter evaluation studies were conducted utilizing the 0.2 µm Nylon, PVDF, and PTFE syringe filter with a discarding volume of 0 mL, 3 mL, and 5 mL. The identified suitable filter for the simultaneous determination of CCL and BTD from its topical formulation was 0.2 µm PVDF and PTFE syringe filter with 5-mL discarded volume.
Conclusion
A simple, rapid, and stability-indicating UHPLC method was developed with a runtime of 8 min for the real-time quantification of CCL and BTD in topical formulations. BCD was used as an internal standard to quantify CCL and BTD for better accuracy. The developed method was optimized using QbD-based 33 level BBD. CMPs organic ratio, flow rate, and column oven temperature were varied to study the CQAs such as retention time (R1: Rt of CCL; R2: Rt of BTD; and R3: Rt of BCD) and resolution (R4: Rs between CCL and BTD; and R5: Rs between BTD and BCD). The main and interaction effects of CMPs were evaluated statistically using quadratic polynomial equation and drawn 2D-contour plots and 3D-response surface plots. The best possible analytical conditions were identified by a numerical optimization method. The CMPs and CQAs were located in the overlay plot of the analytical design space region. The proposed method is capable to determine two components (both CCL and BTD) simultaneously with less solvent consumption, with the usage of low flow rate and less run time, and supporting the green chemistry concept for the healthy environment. The developed method was validated as per ICH guidelines and found specific, precise, accurate, rugged, robust, and stability-indicating to determine CCL and BTD in the topical formulation, and the method can be used in quality control laboratories.
Data availability
All the experimental data was included in the manuscript.
References
D.A.W. Downie, G.D. Cains, R.D. Peek, Aust. J. Derm. 19, 114–117 (1978)
Betamethasone Dipropionate, USP43-NF38 2S, 546 (2020)
C.R. Raymond, J.S. Paul, E.Q. Marian, Handbook of pharmaceutical excipients, 6th edn. (Pharmaceutical press, London, 2009), P. 168
E.R.M. Kedor-Hackmann, E.A.S. Gianotto, M.I.R.M. Santoro, Drug Dev. Ind. Pharm. 24, 553–555 (1998)
L. Mei, W. Nian, J. Pharm. Biomed. Anal. 19, 945–954 (1999)
M. Shou, W.A. Galinada, Y.C. Wei, Q. Tang, R.J. Markovich, A.M. Rustum, J. Pharm. Biomed. Anal. 50, 356–361 (2009)
D.B. Suryakant, J.R. Sadhana, J. AOAC Int. 94, 106–109 (2011)
R. Chinmoy, C. Jitamanyu, R.R. Rammohan, Arch. Appl. Sci. Res. 5, 15–24 (2013)
L.T. Shankar, K.D. Rajesh, T.B. Madhusudan, Asian J. Chem. 26, 6465–6468 (2014)
B. Mayank, P.B. Jadav, Pharm. Chem. J. 2, 17–21 (2015)
C.E. Milena, A.S. Ana Laura, B. Rudy, B.A. Magali, Cri. Rev. Anal. Chem. 50, 111–124 (2020)
L. Brealey, K.A. Proctor, J. Pharm. Pharmacol. 7, 830–835 (1955)
R. Gatti, P. Roveri, D. Bonazzi, V. Cavrini, J. Pharm. Biomed. Anal. 16, 405–412 (1997)
S. Saleem, M.S. Muneera, O.A. Thusleem, T. Muhammad, V.K.A. Anand, J. Chromatogr, Science 47, 178–183 (2009)
Z. Lun-Yi, A.M. Barbero, H.F. Frasch, Open Anal. Chem. J. 4, 10–17 (2010)
J.G. Lee, S. Shin, H. Shin, Y. Huh, S. Lee, D. Kim, S. Lee, Y. Kim, S.B. Han, J. Lee, J.H. Park, S.W. Kwon, J. Pharm. Investig. 42, 47–50 (2012)
Z.M. Turabi, O.A. Khatatbeh, Int. J. Pharm. Sci. Drug Res. 6, 140–144 (2014)
S.E. Johnston, N.L. Gill, Y.C. Wei, R. Markovich, A.M. Rustum, J. Chromatogr. Sci. 48, 733–741 (2010)
S.D. Bendre, P.J. Ghule, Int. Res. J. Pharm. 7, 74–83 (2016)
S.T. Hassib, M.A. Mahrouse, E.F. Elkady, R.M. Sayed, J. Chromatogr. Sci. 56, 716–723 (2018)
S.P. Tulshidas, S.D. Ashwini, Chromatographia 82, 579–590 (2019)
T. Dongala, L.N.R. Katakam, A.K. Palakurthi, N.K. Katari, Anal. Chem. Lett. 9, 697–710 (2019)
S.R. Chaudhari, A.A. Shirkhedkar, J. Anal. Sci. Technol. 10, 10 (2019). https://doi.org/10.1186/s40543-019-0170-8
V.B. Subramanian, N.K. Katari, T. Dongala, S.B. Jonnalagadda, Biomed. Chromatogr. 34, e4719 (2020)
A.K. Palakurthi, T. Dongala, R.K. Yalavarthi, J. Anireddy, Biomed. Chromatogr. 34, e4755 (2020)
M. Patel, C. Kothari, J. Anal. Sci. Technol. 11, 29 (2020). https://doi.org/10.1186/s40543-020-00228-4
V.M. Marisetti, N.K. Katari, Chromatographia 84, 359–369 (2021)
S. Pirsa, F.M. Nejad, Sens. Rev. 37, 155–164 (2021). https://doi.org/10.1108/SR-10-2016-0217
S.K. Muchakayala, K. Pavithra, N.K. Katari, V.M. Marisetti, T. Dongala, R.V.K. Vegesna, Anal. Methods 13, 3705–3723 (2021). https://doi.org/10.1039/D1AY01096D
S. Kalantari, L. Roufegarinejad, S. Pirsa, M. Gharekhani, Main Group Chem. 19, 61–80 (2020). https://doi.org/10.3233/MGC-190821
S. Pirsa, Ş Tağı, M. Rezaei, J. Electron. Mater. 50, 3406–3414 (2021). https://doi.org/10.1007/s11664-021-08855-2
M. Alizadeh, S. Pirsa, N. Faraji, Food Anal. Methods 10, 2092–2101 (2017). https://doi.org/10.1007/s12161-016-0747-4
S. Pirsa, M. Alizadeh, N. Ghahremannejad, Curr. Anal. Chem. 12, 457–464 (2016). https://doi.org/10.2174/1573411012666151009195422
Box-Behnken Design. https://en.wikipedia.org/wiki/Box–Behnken_design Accessed August 13, 2021
Green chemistry pocket guide. https://www.acs.org/content/dam/acsorg/greenchemistry/redesign/principles/the-12-principles-of-green-chemistry-pocket-guide.pdf. Accessed May 27, 2021
Paul Anastas. https://en.wikipedia.org/wiki/Paul_Anastas Accessed May 27, 2021
V.K. Ahluwalia, M. Kidwai, Basic Principles of Green Chemistry. In: New Trends in Green Chemistry. (Springer, Dordrecht, 2004), P. 5–14. https://doi.org/10.1007/978-1-4020-3175-5_3
N. Alizadeh, A.A. Ataei, S. Pirsa, J. Iran. Chem. Soc. 12, 1585–1594 (2015). https://doi.org/10.1007/s13738-015-0631-y
S. Pirsa, Nanosci. Nanotechnol. - Asia 6, 119–127 (2016)
S. Pirsa, J. Sep. Sci. 40, 1724–1730 (2017). https://doi.org/10.1002/jssc.201601393
S. Pirsa, E. Banafshechin, S. Amiri, A. Rahimirad, J. Ghafarzadeh, J. Iran. Chem. Soc. 18, 1167–1177 (2021). https://doi.org/10.1007/s13738-020-02100-z
N. Alizadeh, S. Pirsa, A. Mani-Varnosfaderani, M.S. Alizadeh, IEEE Sens. J. 15, 4130–4136 (2015). https://doi.org/10.1109/JSEN.2015.2411515
Acknowledgements
Authors would like to thank Douglas Pharma US Inc management for approving the publication of this article.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
SKM performed a literature search, development of the RP-UHPLC method, data interpretation, QbD experimentation and data analysis, method validation, and original draft preparation. TD involved in QbD experiments planning, data analysis, and original draft preparation. VMM participated in manuscript review and editing. GV supported us to improve the quality of draft during revision and to address the comments in systemic way. RVKV helped with resources and proofreading. NKK provided guidance on the selection of journal and supervision of the entire process. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest/ competing interests.
Ethical approval
This article does not contain any studies with animals performed by any of the authors.
Consent for publication
We authorize to publish the article without any conflict.
Rights and permissions
About this article

Cite this article
Muchakayala, S.K., Katari, N.K., Dongala, T. et al. Eco-friendly and green chromatographic method for the simultaneous determination of chlorocresol and betamethasone dipropionate in topical formulations using Box–Behnken design. J IRAN CHEM SOC 19, 1397–1412 (2022). https://doi.org/10.1007/s13738-021-02388-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-021-02388-5