
Abstract
We studied the reaction of enaminone 3 with some nitrogen nucleophiles to afford the corresponding pyrazole 4, isoxazole 5, and pyrimidine 6 derivatives, and the reactivity of enaminone 3 with heterocyclic amines to afford the corresponding fused pyrrolo[1,2-a]pyrimidine 9a, imidazo[1,2-a]pyrimidine 9b, phenylpyrrolo[1,2-a]pyrimidine 9c, and benzo[4,5]imidazo[1,2-a]pyrimidine 11 derivatives. Additionally, the electrophilic azo-coupling reaction of enaminone 3 with aromatic diazonium salts in pyridine afforded the corresponding intermediate hydrazines 13a–d, which cyclized to pyrazolo[5,1-c][1,2,4]triazine derivatives 14a–d. Moreover, addition of (E)-3-(dimethylamino)-1-(2-hydroxyphenyl)prop-2-en-1-one (3) with hydrazonoyl chloride derivatives 15a,b gave novel pyrazole derivatives 17a,b. Almost all of the synthesized heterocyclic compounds exhibited antimicrobial and in vitro anticancer activity (HepG2 and MCF-7 cell lines). Furthermore, the molecular docking of the most effective compound, i.e., 7-(4-fluorophenyl)pyrazolo[5,1-c][1,2,4]triazin-3-yl)(2-hydroxyphenyl)methanone (14c), was studied against (PDB ID: 3t88), (PDB ID: 2wje), (PDB ID: 4ynt), and (PDB ID: 1tgh) to investigate its antimicrobial activity when attached to different proteins with short bond length. Compound 14a docked with (PDB ID: 4hdq) and (PDB ID: 3pxe) with energy affinity of −9.946 and −10.55 kcal/mol, with the pyrazolo[5,1-c][1,2,4]triazine derivative involved in the pockets of the proteins. Moreover; the theoretical and investigational studies of compounds 14a,c were compatible with spectral data obtained at HF/6-31G(d) and DFT/B3LYP/6-31G(d) level.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
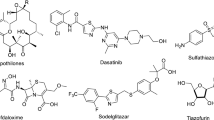
Moieties containing OH group, which include heterocyclic compounds such as pyrazole, pyrimidine, thiazole, isoxazole, and fused heterocyclic [1,2,3,4,5], have industrial and biological applications [6,7,8,9,10,11]. Some drugs include heterocycles attached to the hydroxyl group, e.g., ancistrocladidine (I), candoxatril (II), and dopamine (III) shown in Fig. 1.

Some drugs with attached OH group
Previous studies have indicated that heterocycles containing the hydroxyl group functionality exhibit many activities, including antihelmintic [12], antitubercular [13], and antitrypanosomal effects [14]. Based on the reported importance of pyridine and carboxamide derivatives, and in continuation of our interest in the exploration of novel heterocycles for biological evaluation [15,16,17], we report herein the synthesis of some heterocycles bearing the OH functional group.
We synthesize novel heterocyclic compounds from 2-hydroxyacetophenone that exhibit antimicrobial and antitumor activities, and evaluate them through molecular docking studies of the active compounds, as well as estimation of their energies, which is very important in terms of their theoretical study, chemical reactivity, and stability [18, 19].
Results and discussion
Chemistry
The reactant 1-(2-hydroxyphenyl)ethanone (1) was treated with dimethylformamide dimethylacetal (DMF-DMA) in dry tetrahydrofuran (THF) at heating reflux to afford an orange crystalline solid that was identified as (E)-3-(dimethylamino)-1-(2-hydroxyphenyl)prop-2-en-1-one (3) based on elemental analysis and Fourier-transform infrared (FT-IR), 1H and 13C nuclear magnetic resonance (NMR), and mass spectral data. For example, compound 3 was assigned the E conformation based on its 1H NMR spectrum, which showed the olefin protons as two doublets at δ 6.23 (=CH–CO) and δ 9.05 (=CH–N) with J = 7.5 Hz, as reported for such E-coupled protons. These findings are in complete agreement with recent reports [20, 21]. Also, 1H NMR revealed singlet and multiplet signals corresponding to two methyl groups at δ 3.54 and aromatic protons at δ 7.02–8.02. Its mass spectrum revealed a peak corresponding to the molecular ion at m/z 191 (M+). Reaction of enaminone (3) with nitrogen nucleophiles such as hydrazine hydrate in presence of ethanol and a few drops of triethylamine as catalyst afforded the corresponding 2-(4H-pyrazole-3-yl)phenol (4), as confirmed by the FT-IR absorption band at 3452 cm−1 due to OH. 1H NMR revealed signals at δ 8.02 due to the olefin proton of pyrazole. Its mass spectrum revealed an m/z peak at 160 (Scheme 1). Also, reaction of 3 with hydroxylamine hydrochloride in presence of K2CO3 for basic condition gave the corresponding 2-(isoxazole-5-yl)phenol (5) as confirmed by spectral data such as 1H NMR signals at 6.55 and 8.17 due to isoxazole ring. The mass spectrum showed a signal at m/z = 161.

Reaction of (E)-3-(dimethylamino)-1-(2-hydroxyphenyl)prop-2-en-1-one (3) with nitrogen nucleophiles
Finally, reaction of enaminone 3 with guanidine in ethanol reflux for 4 h afforded 2-(2,4-diaminopyrimidine-5-yl)phenol (6) as confirmed by spectral data including FT-IR absorption bands at 3403 cm−1 due to OH band and two absorption bands due to amino groups at 3345 cm−1 and 3267 cm−1, 1H NMR singlet signals at 6.23 and 8.2 ppm due to amino groups, and the singlet signal at 8.45 due to pyrimidine ring, as well as the signal at 202 in its mass spectrum (Scheme 1).
The behavior of enaminone 3 towards some heterocyclic amines was investigated as a convenient route to access synthesis of a variety of fused heterocyclic systems [22, 23]. Thus, treatment of compound 3 with 3-phenyl-1H-pyrazole-5-amine (7a), 1H-1,2,4-triazole-5-amine (7b), and 4-methyl-3-phenyl-1H-pyrazole-5-amine (7c) in refluxing pyridine furnished pyrrolo[1,2-a]pyrimidine and imidazo[1,2-a]pyrimidine derivatives 9a,b (Scheme 2). The structures of the final products were confirmed by their elemental and spectral analyses (FT-IR, 1H and 13C NMR, and mass); For example, the IR spectrum of 9a revealed the lack of a band corresponding to the carbonyl function group and showed the hydroxyl group at 2398 cm−1, while its 1H NMR spectrum showed a singlet signal due to pyrrole proton at δ 5.99. Its mass spectrum revealed a peak corresponding to its molecular ion at m/z 286 (M+). Furthermore, the reaction of enaminone 3 with aminobenzimidazole (10) in pyridine afforded 2-(benzo[4,5]imidazo[1,2-a]pyrimidin-3-yl)phenol (11), and 1H NMR revealed proton signals at 9.04 due to pyrimidine moiety. Its mass spectrum showed a signal at m/z = 261 (Scheme 2).

Reaction of enaminone (3) with heterocyclic amines
The enaminone 3 reacted with the diazonium salt of 3-phenyl-5-amino-1H-pyrazole derivatives (12a–d) [24, 25] to afford the nonisolable azo-coupling intermediates 13a–d, which cyclized via dimethylamine elimination to yield the pyrazolo[5,1-c][1,2,4]triazine derivatives 14a–d in good yields (Scheme 3). The FT-IR spectra of the latter compounds revealed, in each case, the lack of bands corresponding to endo-cyclic NH and showed carbonyl absorption bands in the region of 1675–1611 cm−1. Their mass spectra showed, in each case, a peak corresponding to the molecular ion (see “Experimental” section).

Reaction of enaminone (3) with diazonium salts
Treatment of compound 3 with hydrazonoyl halides 15a,b was also conducted in refluxing ethanol, in each case resulting in the formation of a single product as examined through thin-layer chromatography (TLC). The structures of the obtained products were established to be 4-(2-hydroxy benzoyl)-1-phenyl-1H-pyrazole-3-carbaldehyde 17a and 1-(4-(2-hydroxy benzoyl)-1-phenyl-1H-pyrazole-3-yl)ethanone 17b, respectively, through elimination of dimethylamino and cyclization to afford pyrazole derivatives. The reaction products were confirmed on the basis of their elemental analyses and spectral data. In all cases, their mass spectra showed, among other fragments, peaks corresponding to the molecular ion. The structures of compounds 17a,b were further confirmed by their independent synthesis, as outlined in Scheme 4.

Reaction of enaminone (3) with hydrazonoyl halides
Biological evaluation
Antimicrobial activity
Antimicrobial activity and structure–activity relationship (SAR)
The prepared compounds were tested for their inhibitory effects on G+ and G− bacterial strains and three antifungal strains (Table 1). In this investigation, attachment of different fused heterocyclic rings to the pyrazolo[5,1-c][1,2,4]triazine derivatives affected the antimicrobial activity. Some of the tested compounds revealed higher and moderate antimicrobial activity compared with the reference drug [26,27,28]. Compounds 6, 11, 14c exhibited high activity against all types of strain, owing to the existence of electron-withdrawing groups besides the carboxyl group. Meanwhile, compounds 9b,c showed almost the same activity. It is worth mentioning that the incorporation of withdrawing groups such as (Br, F) in compounds 14c,d induced high antimicrobial activity. Compounds 9a,b,c, 17a,b revealed moderate activity against all strains. The results listed in Table 1 indicate that compounds 6, 11, 14c showed remarkable activity. The investigation revealed that the F- and Br-atom derivatives 14c,d showed better antibacterial activities than the donating group derivatives 14a,b. Also, incorporation of pyrazolo[5,1-c][1,2,4]triazine moiety in compound 14c resulted in good antibacterial activity against Gram-negative bacteria. Moreover, compound 14c bearing substituted pyrazolo[5,1-c][1,2,4]triazine moiety emerged as the most active member against Gram-positive Bacillus subtilis (30.5 ± 0.01 mm) and fungus Syncephalastrum racemosum (26.4 ± 0.14 mm) among its fluorine group analogues and the fused pyrazolo[5,1-c][1,2,4]triazine in this study.
Molecular docking simulation of compounds 14a,c
Modeling of the molecular docking of compounds 14a (TPT) and 14c (FPT) was carried out using MOE software [29] to identify the biological activity of pyrazolo[5,1-c][1,2,4]triazin-3-yl)methanones 14a,c in accordance with the experimental data, including the docking of compound 14a,c ligands with different protein receptors such as the crystal conformation of Escherichia coli MenB in complex with substrate analog o-succinylbenzoyl-amino coenzyme A (OSB-NCoA) (PDBID: 3t88) [30], crystal conformation of tyrosine phosphatase Cps4B from Streptococcus pneumoniae TIGR4 (PDBID: 2wje) [31], crystal conformation of Aspergillus flavus flavin adenine dinucleotide (FAD) glucose dehydrogenase (PDBID: 4ynt) [32], and 1.8-Å refined structure of lipase from Geotrichum candidum (PDBID: 1tgh) [33] (Table 2; Fig. 2).

Binding models of pyrazolo[5,1-c][1,2,4]triazin-3-yl)methanones 14a,c with a Escherichia coli (PDB: 3t88), b Streptococcus pneumoniae (PDB: 2wje), c Aspergillus flavus (PDB: 4ynt), and d Geotrichum candidum (PDB: 1tgh)
According to this table, the energy affinity of compounds 14a,c with (PDB ID: 3t88) is calculated to be −9.282 and −8.6456 kcal/mol with bond distance of 2.54 Å and 2.25 Å, respectively, with different amino acids: (Thr254, Gly251, Arg230, Arg64, Thr117) and (Arg230, Pro226, Tyr65, Lys120, Gly70 Asp67, Ile69). Also the binding energy of 14a,c with (PDBID: 2wje) is −11.4219 kcal/mol and −10.255 kcal/mol with bond distance of 2.35 Å, 2.85 Å, and 2.75 Å with different amino acids: (Arg206, Tyr166, Gly205, Lys171, Ser165, Lys181, His209, Leu168, Glu212) and (Arg139, Gly205, His166, Arg206, Ser165, Tyr177). Note that all the proteins had greater energy stability with compound 14a due to the presence of methyl group and localization of electrons [34].
Antiproliferative activity
The antiproliferative activity of the various newly synthesized fused heterocyclic compounds 4, 5, 9a, 14a,b, 17a,b was studied using two human cancer cell lines, namely HepG2 hepatocellular carcinoma and MCF-7 breast cancer, based on the sulforhodamine B (SRB) colorimetric assay as described by Skehan et al. [35] and with doxorubicin as reference cytotoxic compound. The outcomes are expressed as growth inhibitory concentration (IC50) values, representing the concentration of each compound required to yield a 50% inhibition of cell growth after 72 h of incubation with respect to untreated controls (Table 3). The results indicate that most of the prepared compounds displayed excellent to modest growth inhibitory activity against the tested cancer cell lines. Investigations of the cytotoxic activity against HepG2 indicated that it was the cell line that was more sensitive to the new derivatives [36]. Compound 14a (IC50 = 11.6 ± 0.11 mg/ml and IC50 = 27.3 ± 1.3 mg/ml) was found to be the most effective derivative among the tested compounds against MCF-7 and HepG2, approaching the activity of doxorubicin (IC50 = 10.3 ± 0.8 and 28.5 ± 1.9 mg/ml) (Fig. 3). Also, compound 5 showed high activity with IC50 values of 17.2 ± 0.15 mg/ml and 27.6 ± 1.1 mg/ml, comparable to doxorubicin, against MCF-7 and Hep-G2. Furthermore, compounds 17a,b exhibited moderate activity against tumor cells due to the presence of 1-phenyl-1H-pyrazole moiety, with IC50 values of 11.5 ± 0.26 and 31.6 ± 0.88 mg/ml for compound 17a and 12.3 ± 0.32 and 33.2 ± 0.95 mg/ml for 17b.

Survival plot of HepG-2 and MCF-7 cells grown for 48 h in presence of aggregate concentrations of compounds 4,5, 9a, 14a,b, 17a,b versus doxorubicin
Molecular docking simulation of compound 14a
Kaposi’s sarcoma-related herpesvirus (KSHV) is an oncovirus that causes Kaposi’s sarcoma in acquired immunodeficiency syndrome (AIDS) patients, primary effusion lymphoma, and various categories of multicentric Castleman’s infection. KSHV encodes worthless thymidylate synthase (kTS), which shares 70% classification characteristics with hTS [37]. The three-dimensional configurations of TS have been described for numerous and diverse organisms including E. coli [38], Lactobacillus casei, B. subtilis, rat, and human [39]. To confirm the cytotoxic activity profile of the synthesized compounds, a modeling study of molecular docking was carried out. A conformational search using an implicit solvent form was carried out for the prepared compounds, monitored based on the improvement of the geometry of local minima through a quantum-mechanical (QM) method. Then, elastic docking of the compounds in the crystallographic configuration of KSHV thymidylate synthase was accomplished with a sulfonamide native ligand obtained from the Protein DataBank (PDB ID: 5H38) [40] to estimate the plausible capability of the new sulfonamide derivatives to bind in the active pocket of KSHV as a potential molecular target. The configurations obtained for our sulfonamides were found to bind in a co-crystallized ligand-like fashion with KSHV [41]. The evaluation of compound 14a for the molecular docking studies against (PDB ID: 5H38) yielded −9.946 kcal/mol with a distance of 2.32 Å and different amino acids of (Arg164, Thr178, Ala175, Ala173, Asp121, Gly118, Ser119, Thr120, Lys117, His165, Glu313), and also KRIT1-Rap1-HEG1 ternary complex encloses three NPX(Y/F) subjects, four ankyrin repeats, and a C-terminal FERM domain and this interface displayed the following FERM domain. The connection between Rap1 and its effector protein KRIT1 plays a noteworthy role in the preservation of EC junctions and regulation of cell–cell junction processes. KRIT1 associates with microtubules. Stretchy docking of compound 5c was examined with the crystallographic structure of the 4hdq complex with a uracil ligand obtained from the Protein Data Bank (PDB ID: 4hdq) [42] to approximate the plausible ability of the innovative uracil derivatives to the drug in the active pocket of 4hdq as a potential molecular target. The energy affinity with 4hdq was –10.554Kcal/mol with bond distances of 2.44 Å, 2.83 Å, and 2.91 Å and different amino acids (Arg426, Ser430, Arg 432, Tyr431, Leu526, Leu529, Ile561) (Table 4; Fig. 4).

Molecular docking of compound 14a with (PDB ID: 5H38) and (PDB ID: 4hdq)
Computational study
Molecular orbital calculations
The optimized molecular structure of fused (2-hydroxyphenyl)(7-(p-tolyl)pyrazolo[5,1-c][1,2,4]triazin-3-yl)methanone (TPT) (14a) is shown in Fig. 5, while selected bond lengths, bond angles, and dihedral angles are presented in Table 5, as calculated at B3LYP/6-31G(d) and HF/6-31G(d) level. The molecular structure of this compound is not planar. The optimized structure of 14a is compared with the crystallographic data of the most closely associated molecule, viz. (E)-3-(dimethylamino)-1-(2-hydroxyphenyl)prop-2-en-1-one(3) [43]. It is recognized that the bond lengths and angles given at HF level of theory are typically more accurate than those obtained at DFT/B3LYP/6-31G(d) level due to the exclusion of electron correlation [44]. Comparison of the theoretical data with the crystallographic data indicated slight differences in the optimized bond lengths and bond angles. Also, fused (7-(4-fluorophenyl)pyrazolo[5,1-c][1,2,4]triazin-3-yl)(2-hydroxyphenyl)methanone(FPT) (14c) showed a nonplanar structure, as shown in Fig. 5.

Optimized geometry, numbering system, and moieties of TPT (14a), X-ray of (E)-3-(dimethylamino)-1-(2-hydroxyphenyl)prop-2-en-1-one (3), and FPT (14c), at HF/6-31G(d) and DFT/B3LYP/6-31G(d) level
Table 5 reveals that the pyrazole ring of compound 14a (TPT) has an N14–N18 bond length of 1.34376 Å for HF/6-31G (d) but 1.36729 Å for DFT /B3LYP/6-31G(d), and 1.40851 Å and 1.28551 Å for 14c (FPT), respectively. Meanwhile, the N18–C17 bond length in 14a is 1.34222 Å and 1.37011 Å, while the length of the N13–C15 bond for 14c is 1.2809 Å and 1.39728 Å for HF/6-31G(d) and B3LYP/6-31G(d), respectively. The C1–O1–H1 angle of (E)-3-(dimethylamino)-1-(2-hydroxyphenyl) prop-2-en-1-one (3) [45] is 103.1(2)°, comparable to the H24–O7–C6 angle of 107.99961° and 112.031° for compound 14a and the H26–C25–C24 angle of 113.24136° and 109.03620° for compound 14c, for HF/6-31G(d) and B3LYP/6-31G(d), respectively.
Physical characterization
The physical characteristics calculated for compounds 14a,c, such as the absolute electronegativity (χ), chemical potential (Pi), absolute hardness (η), absolute softness (σ), global electrophilicity (ω), global softness (S), and additional electronic charge (ΔNmax), are presented in Table 6, obtained using the following equations and optimized via HF/6-31G(d) and DFT/B3LYP/6-31G(d) [46]. The molecular structure of these compounds is not planar. The potential activities presented by the precursor compound (2-hydroxyphenyl)(7-p-tolylpyrazolo[5,1-c][1,2,4]triazin-3-yl)methanone (TPT) (14a) are due to the presence of pyrazolo[5,1-c][1,2,4]triazine, and the difference between HF and DFT/6-31G(d) is 145.836 eV ≈ 3363.062 kcal/mol. Additionally, the difference of the dipole moment is 0.8629D, indicating easy charge separation [47] Also, the π-isoelectronic structures of (7-(4-fluorophenyl)pyrazolo[5,1-c][1,2,4]triazin-3-yl)(2-hydroxyphenyl)methanone (FPT) (14c) show a difference in energy between DFT/ B3LYP/6-31G(d) and HF/6-31G(d) of 188.73 eV ≈ 4352.41 kcal/mol, and the energy gap in DFT/B3LYP/6-31G(d) is 1.206 eV compared with 6.67 eV for HF/6-31G(d). Furthermore, the dipole moment difference (0.0152D) enables easily charge separation of FPT (14c) [48], as shown in Table 6.
Based on the results presented in Table 6, the following conclusions can be drawn:
1. The energy gap of compound TPT obtained utilizing DFT/B3LYP/6-31G(d) is 3.8 eV rather than −9.87 eV with HF/6-31G(d). This large energy difference indicates the stability of the compound due to the presence of methyl (electron-donating) group which enhances electron donation [49].
2. The energy gap of FPT obtained utilizing DFT/B3LYP/6-31G(d) is 1.20 eV rather than 6.67 eV with HF/6-31G(d). This narrow energy gap is due to the presence of electron-withdrawing fluorine group, which gives the compound stability [50].
3. The absolute electronegativity (χ) theoretically describes the affinity of an atom to abstract a mutual pair of electrons. The value of χ is 15.3744 eV ≈ 354.543 kcal/mol for TPT (14a) and −380.4262 eV ≈ −8772.8353 kcal/mol for FPT (14c). The lower value for FPT confirms its ability to attract electrons.
4. The absolute hardness (η) measures the resistance to change in the electron density around the molecule. The difference between the HF/6-31G(d) and DFT/B3LYP/6-31G(d) values of 82.178 eV ≈ 1895.1 kcal/mol for TPT (14a) and 142.016 eV ≈ 3274.97 kcal/mol for FPT (14c) indicates the higher electron density and stability of the latter compound [51].
5. The absolute softness (σ) indicates the interaction of the compound. The difference of 236.198 eV for TPT (14a) and 18.98 eV for FPT (14c) indicates their reaction activity [52].
Frontier molecular orbitals (FMOs)
FMO analysis provides a powerful guiding principle regarding the electrical and optical properties, with the highest occupied molecular orbital (HOMO) acting as an electron donor and the lowest unoccupied molecular orbital (LUMO) acting as an electron acceptor. The energy between the HOMO and LUMO is associated with the biological evaluation. Moreover, it helps to characterize the reactivity and kinetic stability of the molecule. A large energy gap between the HOMO and LUMO is reflected in high kinetic stability [53]. Figure 6 illustrates the distributions and energy levels of the HOMO, LUMO, and orbitals computed at the B3LYP/6-31G(d) level for (2-hydroxyphenyl)(7-p-tolylpyrazolo[5,1-c][1,2,4]triazin-3-yl)methanone (14a) (TPT) and (7-(4-fluorophenyl)pyrazolo[5,1-c][1,2,4]triazin-3-yl) (2-hydroxyphenyl) methanone (14c) (FPT). The positive and negative phases are indicated by red and green color, respectively. As shown in Fig. 6, the HOMO of TPT (14a) is localized over the whole molecule except the tolyl moiety, while the LUMO of the same compound is not localized on the hydroxybenzene ring and the HOMO–LUMO energy gap is −3.83 eV, Furthermore, the molecular electrostatic potential surface (ESP) lies on the more electronegative atoms such as O and N of benzene or fused pyrazolo[5,1-c][1,2,4]triazine, which easily transport electrons as shown in Fig. 6. Moreover, the HOMO and LUMO of compound FPT (14c) are distributed over the whole molecule except the p–f benzene with an energy difference of 1.20 eV. This narrow HOMO–LUMO gap indicates high excitation energies for numerous excited states and the reactivity of this compound due to the presence of the fluorine atom, which increases its activity. Also, the ESP of (7-(4-fluorophenyl)pyrazolo[5,1-c][1,2,4]triazin-3-yl)(2-hydroxyphenyl) methanone (14c) indicating a uniform distribution of surface contours on fused pyrazolo[5,1-c][1,2,4]triazine and (2-hydroxyphenyl)methanone, which is associated with the experimental and biological activity [54, 55], as shown in Fig. 6.

HOMO–LUMO energy gap and ESP contours for compounds 14a,c at B3LYP/6-31G(d) level
Experimental
General procedure
All melting points were measured on a Gallenkamp melting point apparatus. IR spectra were recorded on a Shimadzu FT-IR 8101 PC infrared spectrophotometer. 1H and 13C NMR spectra were determined in dimethylsulfoxide (DMSO)-d6 at 300 MHz on a Varian Mercury VX 300 NMR spectrometer (1H at 300 MHz, 13C at 75 MHz) using tetramethylsilane (TMS) as internal standard. Mass spectra were recorded on a Shimadzu GCMS-QP 1000 EX mass spectrometer at 70 eV. Elemental analyses were carried out at the Microanalytical Center of Cairo University, Giza, Egypt.
Materials and reagents
1-(2-Hydroxyphenyl)ethanone, dimethylformamide dimethylacetal, NH2NH2, NH2OH・HCl, triethylamine, 2-aminobenzoimidazole, and aminopyrazole were purchased from Aldrich Chemical CO. Ethanol, pyridine, toluene, THF, and piperidine were purchased from Aldrich Chemical CO. Methanol, petroleum ether, and chloroform were BDH reagents.
Preparation of (E)-3-(dimethylamino)-1-(2-hydroxyphenyl)prop-2-en-1-one (3)
Solution of 1-(2-hydroxyphenyl)ethanone (1) (10 mmol) in dry THF (50 ml) and dimethylformamide dimethylacetal (10 mmol) was refluxed for 1 h then left to cool. The orange precipitated was filtered off, washed with dry petroleum ether (40–60 °C), and dried. The isolated product was crystallized from EtOH/H2O to afford (E)-3-(dimethylamino)-1-(2-hydroxyphenyl)prop-2-en-1-one (3) [25]: orange crystals, 80% yield, m.p. = 150–152 °C, C11H13NO2 (191.23), Anal% calcd (found): C: 69.09 (69.11), H: 6.85 (6.84), N: 7.32 (7.35), IR (KBr)max/cm−1: 3210 (OH), 1683 (C=O), 158 (C=O). 1H NMR (DMSO-d6): δ 3.54 (s, 6H, H3C), 6.23 (d, 1H, HC–CO, J = 7.5 Hz), 6.9–7.5 (m, 4H, HC aromatic), 8.05 (d, 1H, HC–N, J = 7.5 Hz), 14.4 (s, 1H, HO exchangeable), 13C NMR (DMSO-d6): δ 44.3 (CH3), 117.3 (CH), 121.6 (CH), 129.6 (CH), 141 (CH), 155 (C–N), 157 (C–O), 188 (C=O), MS (m/z, r.i.%): 192 (M+, 100%), 121 (45%).
Reaction of (E)-3-(dimethylamino)-1-(2-hydroxyphenyl)prop-2-en-1-one (3) with nitrogen nucleophiles
Reaction of 3 with hydrazine hydrate (NH2NH2)
Enaminone (3) solution (0.19 g, 1 mmol) in ethanol (10 ml) was added to hydrazine hydrate (1 ml, 1 mmol), and the mixture was heated under reflux for 5 h. The reaction mixture was acidified through HCl/ice mixture, and the formed product was filtered and crystallized from ethanol. 2-(4H-Pyrazol-3-yl)phenol (4) [25]: white solid, 77% yield, m.p. = 90–94 °C, C9H8N2O (160.06), Anal% calcd (found): C: 67.49 (67.52), H: 5.03 (5.00), N: 17.49 (17.45), IR (KBr)max/cm−1: 3452 (OH), 1H NMR (DMSO-d6): δ 2.2 (d, 2H, H2C), 7.04–7.98 (m, 4H, HC aromatic), 7.99 (t, 1H, HC, J = 12.1 Hz), 11.99 (s, 1H, HO exchangeable), MS (m/z, r.i.%): 160 (M+, 100), 104 (20%), 69 (65%).
Reaction of 3 with hydroxylamine hydrochloride
An ethanolic mixture of 3 (0.19, 1 mmol) and hydroxylamine hydrochloride (0.033, 1 mmol) NH2OH in presence of potassium carbonate (0.5 g) was refluxed for 4 h and poured onto water. The solid product was filtered and crystallized from ethanol.
2-(Isoxazol-5-yl)phenol (5): pale yellow, 79% yield, m.p. = 113–115 °C, C9H7NO2 (161.16), Anal% calcd (dound): C: 67.07 (67.10), H: 4.38 (4.35), N: 8.69 (8.70), IR (KBr)max/cm−1: 3489 (OH), 1H NMR (DMSO-d6): δ 6.55 (d,1H, HC), 7.02–7.88 (m, 4H, HC aromatic), 8.17 (d, 1H,HC, J = 7.6 Hz), 11.23 (s, 1H, HO exchangeable), MS (m/z, r.i.%): 161 (M+, 100), 49 (65%), 21 (45%).
Reaction of 3 with guanidine
A mixture of 3 (0.19, 1 mmol) with guanidine (0.059, 1 mmol) in ethanol (15 ml) was refluxed for 4 h. The resulting solid was filtered off, and crystallized from EtOH/H2O to afford compound 6.
2-(2,4-Diaminopyrimidin-5-yl)phenol (6): yellow powder, 77% yield, m.p. = 296–297 °C, C10H10N4O (202.21), Anal% calcd (found): C: 59.40 (59.39), H: 4.98 (4.94), N: 27.71 (27.70), IR (KBr)max/cm−1: 3403 (OH), 3345 (NH2), 3267 (NH2), 1H NMR (DMSO-d6): δ 6.23 (s, 2H, H2N exchangeable), 7.12–7.67 (m, 4H,HC aromatic), 8.2 (s, 2H, H2N exchangeable), 8.45 (s, 1H, HC), 12.36 (s, 1H, HO exchangeable), 13C NMR (DMSO-d6): δ 106 (C–CH = N), 115 (CH), 124 (CH), 128 (CH), 129 (CH), 131 (CH), 153 (C–OH), 156 (C–N), 162 (C = N), MS (m/z, r.i.%): 202 (M+, 100), 180 (22%), 52 (35%).
General procedure for reaction of enaminone 3 with heterocyclic amines 7a,b, 10
A mixture of enaminone 3 (1.9 g, 10 mmol) and the appropriate heterocyclic amine 3-phenyl-1H-pyrazol-5-amine (7a), 1H-1,2,4-triazol-5-amine (7b), 4-methyl-3-phenyl-1H-pyrazol-5-amine (7c), and 1H-benzo[d]imidazol-2-amine (10) (10 mmol) in pyridine (25 ml) was refluxed for 12 h then left to cool. The reaction mixture was poured into cold water, and the solid product was collected by filtration, washed with water, dried, and finally recrystallized from DMF/H2O to afford the corresponding pyrazolo[1,5-a]pyrimidine and triazolo[1,5-a]pyrimidine derivatives 9a–c and 11 in 70% and 60% yield, respectively.
2-(7-Phenylpyrrolo[1,2-a]pyrimidin-4-yl)phenol (9a): dark yellow, m.p. = 200–202 °C, C19H14N2O (286.33), Anal% calcd (found): C: 79.70 (79.69), H: 4.93 (4.94), N: 9.78 (9.74), IR (KBr)max/cm−1: 3398 (OH), 1H NMR (DMSO-d6): δ 5.99 (s, 1H, HC), 6.15 (s, 1H,HC), 7.03–7.54 (m, 4H, HC aromatic), 7.74–8.03 (m, 5H, HC aromatic), 8.2 (d, 1H, HC, J = 6.1 Hz), 8.89 (d, 1H, HC, J = 7.6 Hz), 12.03 (s, 1H, HO exchangeable), 13C NMR (DMSO-d6): δ 104 (CH), 115 (CH), 121 (CH), 127.5 (CH), 129.3 (CH), 130 (CH), 134 (CH), 151 (C–OH), 157 (C=N), 166 (C–N), MS (m/z, r.i.%): 286 (M+, 100), 193 (45%), 68 (65%).
2-(Imidazo[1,2-a]pyrimidin-5-yl)phenol (9b): pale brown, m.p. = 201–204 °C, C12H9N3O (211.22), Anal% calcd (found): C: 68.24 (68.22), H: 4.29 (4.30), N: 19.89 (19.92), IR (KBr) max/cm−1: 3410 (OH), 1H NMR (DMSO-d6): δ 7.18–7.73 (m, 4H, HC aromatic), 7.8 (d, 1H, HC, J = 3.2 Hz), 8.03 (d, 1H, HC, J = 7.8 Hz), 8.15 (d, 1H,HC, J = 7.6 Hz), 8.87 (d, 1H, HC, J = 6.2 Hz), 11.99 (s, 1H, HO exchangeable), 13C NMR (DMSO-d6): δ 114 (CH), 116 (CH), 119 (CH), 129.3 (CH), 131 (CH), 148 (CH), 155 (C–OH), 158 (C=N), 166 (C-N), MS (m/z, r.i.%): 211 (M+, 100), 181 (45%), 61 (50%).
2-(Benzo[4,5]imidazo[1,2-a]pyrimidin-3-yl)phenol (11): Yellow solid, m.p. = 230–232 °C, 87% yield, C16H11N3O (261.28), Anal% calcd (found): C: 73.55 (73.53), H: 4.24 (4.30), N: 16.08 (16.11), IR (KBr)max/cm−1: 3458 (OH), 1H NMR (DMSO-d6): δ 6.86–7.23 (m,4H, HC aromatic), 7.39–8.22 (m, 4H, HC aromatic), 9.04 (s, 2H, HC), 12.4 (s, 1H, HO exchangeable), 13C NMR (DMSO-d6): δ 111 (CH), 118 (CH), 121 (CH), 123 (CH), 129.3 (CH), 131 (CH), 134 (CH), 148 (C=N), 165 (C–N), MS (m/z, r.i.%): 262 (M+1, 100), 168 (64%), 149 (12%).
General procedure for reaction of enaminone 3 with diazonium salts of heterocyclic amines 12a–d
To a stirred cold solution of enaminone 3 (0.38 g, 2 mmol) in pyridine (30 ml) was added the appropriate diazonium salt of pyrazole derivatives (2 mmol) portionwise over 30 min at 0–5 °C. After complete addition, the reaction mixture was stirred for a further 3 h at 0–5 °C. The solid that precipitated was collected by filtration, washed with water, and dried. Recrystallization from DMF/H2O afforded the corresponding fused ring system 14a–d.
(2-Hydroxyphenyl)(7-p-tolylpyrazolo[5,1-c][1,2,4]triazin-3-yl)methanone (14a), yellow solid, m.p. = 210–212 °C, C19H14N4O2 (330.34), Anal% calcd (found): C: 69.08 (69.10), H: 4.27 (4.30), N: 16.96 (16.98), IR (KBr) max/cm−1: 3389 (OH), 1675 (C=O), 1H NMR (DMSO-d6): δ 2.44 (s, 3H, CH3), 6.79 (s, 1H, HC), 6.99–7.46 (m, 4H, HC aromatic), 7.75–8.54 (m, 4H, HC aromatic), 9.80 (s, 1H, HC), 12.03 (s, 1H, HO exchangeable), 13C NMR (DMSO-d6): δ 29 (CH3), 102 (CH), 119 (CH), 125 (CH), 129 (CH), 134 (CH), 138 (CH), 139 (CH), 149 (C–N), 157 (C=N), 161 (C–OH), 189 (C=O), MS (m/z, r.i.%): 330 (M+, 100), 175 (15%), 111 (95%).
(2-Hydroxyphenyl)(7-(4-methoxyphenyl)pyrazolo[5,1-c][1,2,4]triazin-3-yl) methanone (14b): dark-yellow solid, m.p. = 214–216 °C, C19H14N4O3 (346.34), Anal% calcd (found): C: 65.89 (65.92), H: 4.07 (4.10), N: 16.18 (16.20), IR (KBr) max/cm−1:3389 (OH), 1640 (C=O), 1H NMR (DMSO-d6): δ 3.52 (s, 3H, OCH3), 6.51 (s, 1H, CH), 6.89–7.66 (m, 4H, HC aromatic), 7.81–8.05 (m, 4H, HC aromatic), 9.85 (s, 1H, HC), 12.03 (s, 1H, HO exchangeable), 13C NMR (DMSO-d6): δ 56.8 (CH3), 74.9 (CH), 115 (CH), 118 (CH), 127 (CH), 128 (CH), 133 (CH), 144 (CH), 147 (C–N), 153 (C=N), 158 (C–OCH3), 160 (C–OH), 187 (C=O), MS (m/, r.i.%): 346 (M+, 100), 225 (25%), 168 (55%).
(7-(4-Fluorophenyl)pyrazolo[5,1-c][1,2,4]triazin-3-yl)(2-hydroxyphenyl)methanone (14c), orange powder, m.p. = 220–224 °C, C18H11FN4O2 (334.30), Anal% calcd (found): C: 64.67 (64.65), H: 3.32 (3.35), N: 16.76 (16.77), F: 5.68 (5.66%), IR (KBr) max/cm−1: 3399 (OH), 1654 (C = O), 1H NMR (DMSO-d6): δ 6.87 (s, 1H,HC), 6.95–7.75 (m, 4H, HC aromatic), 7.99–8.17 (m, 4H, HC aromatic), 9.43 (s, 1H, HC), 12.02 (s, 1H, HO exchangeable), 13C NMR (DMSO-d6): δ 72 (CH), 113 (CH), 116 (CH), 118 (CH), 124 (CH), 128 (CH), 134 (CH), 144 (CH), 150 (C–N), 162 (C–F), 189 (C=O), MS (m/z, r.i.%): 334 (M+, 100).
(7-(4-Bromophenyl)pyrazolo[5,1-c][1,2,4]triazin-3-yl)(2-hydroxyphenyl)methanone (14d): orange solid, m.p. = 260–262 °C, C18H11BrN4O2 (395.21), Anal% calcd (found): C: 54.70 (54.68), H: 2.81 (2.80), N: 14.18 (14.20), Br: 20.22 (20.23%), IR (KBr) max/cm−1: 3410 (OH), 1611 (C=O), 1H NMR (DMSO-d6): δ 6.77 (s, 1H, HC), 6.88–7.88 (m, 4H, HC aromatic), 7.95–8.32 (m, 4H, HC aromatic), 9.53 (s, 1H, HC), 11.99 (s, 1H, HO exchangeable), 13C NMR (DMSO-d6): δ 74 (CH), 117 (CH), 118 (CH), 124 (CH), 132 (CH), 136 (CH), 144 (CH), 148 (C–N), 151 (C=N), 162 (C–OH), 189 (C=O), MS (m/z, r.i.%): 395 (M+, 100), 274 (15%), 121 (40%).
Reaction of compound 3 with hydrazonoyl halides
Reaction of compound 3 with hydrazonoyl halides 15a,b with reflux for 4 h yielded pyrazole derivatives 17a,b, which crystallized from EtOH/H2O in excellent yield.
4-(2-Hydroxybenzoyl)-1-phenyl-1H-pyrazole-3-carbaldehyde (17a), pale-orange solid, m.p. = 216–218 °C, C17H12N2O3 (292.29), Anal% calcd (found): C: 69.86 (69.88), H: 4.14 (4.20), N: 9.58 (9.60), IR (KBr) max/cm−1: 3446 (OH), 1668 (C = O), 1592 (C = O), 1H NMR (DMSO-d6): δ 6.76–7.34 (m, 4H, HC aromatic), 7.86–8.06 (m, 5H, HC aromatic), 8.7 (s, 1H, HC, pyrazole), 10.09 (s, 1H, HC), 12.3 (s, 1H, HO exchangeable), 13C NMR (DMSO-d6): δ 115 (CH), 127 (CH), 129 (CH), 134 (CH), 136 (CH), 145 (C=N), 161 (C–OH), 188 (C=O), 191 (C=O), MS (m/z, r.i.%): 292 (M+, 100%), 199 (20%), 121 (55%).
1-(4-(2-Hydroxybenzoyl)-1-phenyl-1H-pyrazol-3-yl)ethanone (17b), orange solid, m.p. = 230–232 °C, C18H14N2O3 (306.32), Anal% calcd (found): C: 70.58 (70.55), H: 4.61 (4.20), N: 9.15 (9.11), IR (KBr) max/cm−1: 3410 (OH), 1623 (C=O), 1611 (C=O), 1H NMR (DMSO-d6): δ 2.11 (s, 3H, H3C), 6.45–7.65 (m, 4H, HC aromatic), 7.94–8.13 (m, 5H, HC aromatic), 8.67 (s, 1H, HC, pyrazole), 12.6 (s, 1H, HO exchangeable), 13C NMR (DMSO-d6): δ 25.6 (CH3), 111 (CH), 119 (CH), 123 (CH), 129 (CH), 130 (CH), 134 (CH), 145(C=N), 162 (C–OH), 178 (C=O), 195 (C=O), MS (m/z, r.i.%): 306 (M+, 100), 213 (33%), 121 (29%).
Conclusions
A novel series of heterocycles containing a bioactive nucleus was synthesized and characterized by IR, 1H and 13C NMR, and MS analysis. Biological evaluation of the synthesized compounds elucidated their antimicrobial and antitumor activities. Furthermore, docking studies of compounds 14a,c with different proteins revealed that they are kinetically stable with short bond length. Characterization of pyrazole 14a,c utilizing DFT/ B3LYP/ 6-31G(d) and HF/6-31G(d) methods supported the stability and importance of this pyrazole. The relations between the calculated and crystallographic results indicates that the B3LYP/6-31G(d) method is better than the HF method for approximating the bond lengths, while the HOMO–LUMO gap results confirm the high kinetic stability of these compounds.
References
H.-Y. Lu, I.J. Barve, M. Selvaraju, C.-M. Sun, One-pot synthesis of unsymmetrical bis-heterocycles: benzimidazole-, benzoxazole-, and benzothiazole-linked thiazolidines. ACS Comb. Sci. 22(1), 42–48 (2020). https://doi.org/10.1021/acscombsci.9b00161
S. Mukherjee, A. Pramanik, Catalyst-free one-pot three-component synthesis of 4-hydroxy-3-pyrazolylcoumarins in ethanol at room temperature: enolisable aroylhydrazones as efficient ambident nucleophile. ACS Sustain. Chem. Eng. 8(1), 403–414 (2020). https://doi.org/10.1021/acssuschemeng.9b05682
N. Cankařová, E. Schütznerová, V. Krchňák, Traceless solid-phase organic synthesis. Chem. Rev. 119(24), 12089–12207 (2019). https://doi.org/10.1021/acs.chemrev.9b00465
E. Niknam, F. Panahi, F. Daneshgar, F. Bahrami, A. Khalafi-Nezhad, Metal–organic framework MIL-101(Cr) as an efficient heterogeneous catalyst for clean synthesis of benzoazoles. ACS Omega 3(12), 17135–17144 (2018). https://doi.org/10.1021/acsomega.8b02309
S. Kurhade, E. Diekstra, F. Sutanto, K. Kurpiewska, J. Kalinowska-Tłuścik, A. Dömling, Multicomponent reaction based synthesis of 1-tetrazolylimidazo[1,5-a]pyridines. Org. Lett. 20(13), 3871–3874 (2018). https://doi.org/10.1021/acs.orglett.8b01452
S.A. Khanum, S. Shashikanth, S. Umesha, R. Kavitha, Synthesis and antimicrobial study of novel heterocyclic compounds from hydroxybenzophenones. Eur. J. Med. Chem. 40(11), 1156–1162 (2005). https://doi.org/10.1016/j.ejmech.2005.04.005
H. Muğlu, H. Yakan, H.A. Shouaib, New 1,3,4-thiadiazoles based on thiophene-2-carboxylic acid: synthesis, characterization, and antimicrobial activities. J. Mol. Struct. 1203, 127470 (2020). https://doi.org/10.1016/j.molstruc.2019.127470
C. Tratrat, M. Haroun, A. Paparisva, A. Geronikaki, C. Kamoutsis, A. Ćirić, J. Glamočlija, M. Soković, C. Fotakis, P. Zoumpoulakis, S.S. Bhunia, A.K. Saxena, Design, synthesis and biological evaluation of new substituted 5-benzylideno-2-adamantylthiazol[3,2-b][1,2,4]triazol-6(5H)ones. Pharmacophore models for antifungal activity. Arab. J. Chem. 11, 573–590 (2018). https://doi.org/10.1016/j.arabjc.2016.06.007
J. Cramer, C.P. Sager, B. Ernst, Hydroxyl groups in synthetic and natural-product-derived therapeutics: a perspective on a common functional group. J. Med. Chem. 62(20), 8915–8930 (2019). https://doi.org/10.1021/acs.jmedchem.9b00179
R. Rani, C. Granchi, Bioactive heterocycles containing endocyclic N-hydroxy groups. Eur. J. Med. Chem. 97, 505–524 (2015). https://doi.org/10.1016/j.ejmech.2014.11.031
J. Akhtar, A.A. Khan, Z. Ali, R. Haider, M.S. Yar, Structure–activity relationship (SAR) study and design strategies of nitrogen-containing heterocyclic moieties for their anticancer activities. Eur. J. Med. Chem. 125, 143–189 (2017). https://doi.org/10.1016/j.ejmech.2016.09.023
O. Nagaraja, Y.D. Bodke, I. Pushpavathi, S. Ravi Kumar, Synthesis, characterization and biological investigations of potentially bioactive heterocyclic compounds containing 4-hydroxy coumarin. Heliyon. 6(6), e04245 (2020). https://doi.org/10.1016/j.heliyon.2020.e04245
N. Obaiah, Y.D. Bodke, S. Telkar, Synthesis of 3-[(1H-benzimidazol-2-ylsulfanyl)(aryl)methyl]-4-hydroxycoumarin derivatives as potent bioactive molecules. ChemistrySelect 5, 178–184 (2020). https://doi.org/10.1002/slct.201903472
O. Nagaraja, Y.D. Bodke, R. Kenchappa, S. Ravi Kumar, Synthesis and characterization of 3-[3-(1H-benzimidazol-2-ylsulfanyl)-3-phenyl propanoyl]-2H-chromen-2-one derivatives as potential biological agents. Chem. Data Collect. 27, 100369 (2020)
R. Kenchappa, Y.D. Bodke, Synthesis, analgesic and anti-inflammatory activity of benzofuran pyrazole heterocycles. Chem. Data Collect. 28, 100453 (2020). https://doi.org/10.1016/j.cdc.2020.100453
M. Gaba, C. Mohan, Development of drugs based on imidazole and benzimidazole bioactive heterocycles: recent advances and future directions. Med. Chem. Res. 25, 173–210 (2016). https://doi.org/10.1007/s00044-015-1495-5
A.M. Fahim, A.M. Farag, E.M.A. Yakout, G.A.M. Nawwar, E.A. Ragab, Synthesis, biological evaluation of 1,3,4-oxadiazole, triazole and uracil derivatives from poly (ethylene terephthalate) waste. Egypt J. Chem. 59, 285–303 (2016). https://doi.org/10.21608/EJCHEM.2016.1048
A.A.E.-H. Hassan, Heterocyclic synthesis via enaminones: synthesis and molecular docking studies of some novel heterocyclic compounds containing sulfonamide moiety. Int. J. Org. Chem. 4(1), 68–81 (2014). https://doi.org/10.4236/ijoc.2014.41009
F.N. Takla, A.A. Farahat, M.A.-A. El-Sayed, M.N.A. Nasr, Molecular modeling and synthesis of new heterocyclic compounds containing pyrazole as anticancer drugs. Int. J. Org. Chem. 7, 369–388 (2017). https://doi.org/10.4236/ijoc.2017.74030
S. Cunha, A.T. Gomes, Synthesis of α-aryl enaminones through reactions of β-aryl enones with benzyl azide. Tetrahedron Lett. 53(49), 6710–6713 (2012). https://doi.org/10.1016/j.tetlet.2012.09.125
A. Fahim, A.M. Farag, A. Mermer, H. Bayrak, Y. Şirin, Synthesis of novel β-lactams: antioxidant activity, acetylcholinesterase inhibition and computational studies. J. Mol. Struct. 1233, 130092 (2021). https://doi.org/10.1016/j.molstruc.2021.130092
A. Fahim, A. Mohamed, M. Ibrahim, Experimental and theoretical studies of some propiolate esters derivatives. J. Mol. Struct. 1236 (2021). https://doi.org/10.1016/j.molstruc.2021.130281
A. Mohamed, A. Fahim, M. Ibrahim, Theoretical investigation on hydrogen bond interaction between adrenaline and hydrogen sulfide. J. Mol. Model. 26, 354 (2020). https://doi.org/10.1007/s00894-020-04602-2
A.S. Shawali, A new convenient synthesis of 3-hetaryl-pyrazolo[5,1-c][1,2,4]triazines. J. Adv. Res. 3, 185–188 (2012). https://doi.org/10.1016/j.jare.2011.07.004
K.M. Dawood, S.M. Moghazy, A.M. Farag, Convenient synthesis of azolopyrimidine, azolotriazine, azinobenzimidazole and 1,3,4-thiadiazole derivatives. Arab. J. Chem. 10, S2782–S2789 (2017). https://doi.org/10.1016/j.arabjc.2013.10.029
A.M. Fahim, E.H.I. Ismael, Synthesis, antimicrobial activity and quantum calculations of Novel sulphonamide derivatives, Egypt. J. Chem. 62(8), 1427–1440 (2019). https://doi.org/10.21608/EJCHEM.2019.6870.1575
A.M. Fahim, A.M. Farag, E.M.A. Yakout, G.A.M. Nawwar, E.A. Ragab, Sun degradation and synthesis of new antimicrobial and antioxidant utilisingpoly(ethylene terephthalate) waste. Int. J. Environ. Waste Manag. 22, 239–259 (2018). https://doi.org/10.1504/IJEWM.2018.094111
E.M. Akl, S. Dacrory, M.S. Abdel-Aziz, S. Kamel, A.M. Fahim, Preparation and characterization of novel antibacterial blended films based on modified carboxymethyl cellulose/phenolic compounds. Polym. Bull. 78, 1061–1085 (2021). https://doi.org/10.1007/s00289-020-03148-w
G. Hagelueken, H. Huang, I.L. Mainprize, C. Whitfield, J.H. Naismith, Crystal structures of wzb of Escherichia coli and cpsb of Streptococcus pneumoniae, representatives of two families of tyrosine phosphatases that regulate capsule assembly. J. Mol. Biol. 392, 678–688 (2009). https://doi.org/10.1016/j.jmb.2009.07.026
A. Aboelnaga, A.M. Fahim, T.H. El-Sayed, Computer aid screening for potential antimalarial choroquinone compounds as Covid 19 utilizing computational calculations and molecular docking study. OnLine J. Biol. Sci. 20(4), 207–220 (2020). https://doi.org/10.3844/ojbsci.2020.207.220
K.K. Masibi, O.E. Fayemi, A.S. Adekunle, A.M. Al-Mohaimeed, A.M. Fahim, B.B. Mamba, E.E. Ebenso, Electrochemical detection of endosulfan using an AONP-PANI-SWCNT modified glassy carbon electrode. Materials 14, 723 (2021). https://doi.org/10.3390/ma14040723
C.A. Schiffer, I.J. Clifton, V.J. Davisson, D.V. Santi, R.M. Stroud, Crystal structure of human thymidylate synthase: a structural mechanism for guiding substrates into the active site. Biochemistry 34, 16279–16287 (1995). https://doi.org/10.1021/bi00050a007
A. Barakat, S.M. Soliman, H.A. Ghabbour, M.A. Al-Majid, M.S. Islam, A.A. Ghfar, Molecular structure, spectroscopic and DFT computational studies of arylidene-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione. Curr. Comput.-Aided Drug Des. 6, 110 (2016). https://doi.org/10.3390/cryst6090110
A.M. Farag, A.M. Fahim, Synthesis, biological evaluation and DFT calculation of novel pyrazole and pyrimidine derivatives. J. Mol. Struct. 1179, 304–314 (2019). https://doi.org/10.1016/j.molstruc.2018.11.008
P. Skehan, R. Strong, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J.T. Warren, H. Bokesch, S. Kenney, M. Boyd, New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 82, 1107–1112 (1990). https://doi.org/10.1093/jnci/82.13.1107
A.M. Fahim, A.M. Farag, M.R. Shabban, E.A. Ragab, Regioselective synthesis and DFT study of novel fused heterocyclic utilizing thermal heating and Microwave Irradiation. Afinidad 75, 148–159 (2018)
A.M. Fahim, A.M. Farag, Synthesis, antimicrobial evaluation, molecular docking and theoretical calculations of novel pyrazolo[1,5-a]pyrimidine derivatives. J. Mol. Struct. 1199, 127025 (2020). https://doi.org/10.1016/j.molstruc.2019.127025
A.M. Fahim, Microwave-assisted synthesis of pyrazolo[1,5-a]pyrimidine, triazolo[1,5-a]pyrimidine, pyrimido[1,2-a]benzimdazole, triazolo[5,1-c] [1,2,4]triazine and imidazo[2,1-c][1,2,4]triazine. Curr. Microw. Chem. 5(2), 111–119 (2018). https://doi.org/10.2174/2213335605666180425144009
A.M. Fahim, E.M.A. Yakout, G.A. Nawwar, Facile synthesis of in-vivo insecticidal and antimicrobial evaluation of bis heterocyclic moiety from pet waste. Online J. Biol. Sci. 14, 196–208 (2014). https://doi.org/10.3844/ojbssp.2014.196.208
A.M. Fahim, M.S. Elshikh, N.M. Darwish, Synthesis, antitumor activity, molecular docking and DFT study of Novel pyrimidiopyrazole derivatives. Curr. Comput. Aided Drug Des. 16(4), 486–499 (2020). https://doi.org/10.2174/1573409915666190710094425
A.M. Fahim, A.M. Farag, G.A.M. Nawwar, E.M.A. Yakout, E.A. Ragab, Synthesis and DFT calculations of aza-Michael adducts obtained from degradation poly(methyl methacrylate) plastic wastes. Int. J. Environ. Waste Manag. 24(4), 337–353 (2019). https://doi.org/10.1504/IJEWM.2019.103641
A.R. Gingras, W. Puzon-McLaughlin, M.H. Ginsberg, The structure of the ternary complex of Krev interaction trapped 1 (KRIT1) bound to both the Rap1 GTPase and the heart of glass (HEG1) cytoplasmic tail. J. Biol. Chem. 288, 23639–23649 (2013). https://doi.org/10.1074/jbc.M113.462911
A.M. Fahim, Microwave-assisted regioselective synthesis and biological evaluation of pyrano[2,3-c]pyridine derivatives utilizing DMAP as a catalyst. Online J. Biol. Sci. 17, 394–403 (2017). https://doi.org/10.3844/ojbsci.2017.394.403
S. Dacrory, A.M. Fahim, Synthesis, anti-proliferative activity, computational studies of tetrazole cellulose utilizing different homogenous catalyst. Carbohydr. Polym. 229, 115537 (2020). https://doi.org/10.1016/j.carbpol.2019.115537
A.M. Fahim, B. Wasiniak, J.P. Łukaszewicz, Molecularly imprinted polymer and computational study of (E)-4-(2-cyano-3-(dimethylamino)acryloyl)benzoic acid from poly(ethylene terephthalate) plastic waste. Curr. Anal. Chem. 16(2), 119–137 (2020). https://doi.org/10.2174/1573411015666190131123843
M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Hratchian, H.P. Li, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery, J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J.M. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, O. Farkas, J.B. Foresman, J.V. Ortiz, J. Cioslowski, D.J. Fox, Gaussian 09, Revision a.1 (Gaussian Inc., Wallingford, 2009).
E.A. Zayed, M.A. Zayed, A.M. Fahim, F.A. El-Samahy, Synthesis of novel macrocyclic Schiff’s-base and its complexes having N2O2 group of donor atoms. Characterization and anticancer screening are studied. Appl. Organometal. Chem. 31, e3694 (2017). https://doi.org/10.1002/aoc.3694
A.M. Fahim, A.M. Farag, G.A.M. Nawwar, E.M.A. Yakout, E.A. Ragab, Chemistry of terephthalate derivatives: a review. Int. J. Environ. Waste Manag. 24(3), 273–301 (2019). https://doi.org/10.1504/IJEWM.2019.103104
R. Dennington, T. Keith, J. Millam, GaussView, Version 5 (SemichemInc, Shawnee Mission, 2009).
S. Mondal, S.M. Mandal, T.K. Mondal, C. Sinha, Spectroscopic characterization, antimicrobial activity, DFT computation and docking studies of sulfonamide Schiff bases. J. Mol. Struct. 1127, 557–567 (2017). https://doi.org/10.1016/j.molstruc.2016.08.011
O. Trott, A.J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010). https://doi.org/10.1002/jcc.21334
G.M. Morris, R. Huey, W. Lindstrom, M.F. Sanner, R.K. Belew, D.S. Goodsell, A.J. Olson, AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30(16), 2785–2791 (2009). https://doi.org/10.1002/jcc.21256
R. Almog, C.A. Waddling, F. Maley, G.F. Maley, P. Van Roey, The Crystal structure of a deletion mutant of human thymidylate synthase D (7e29) andits ternary complex with Tomudex and dUMP. Prot. Sci. 10, 988–996 (2001). https://doi.org/10.1110/ps.47601
K. Fukui, Role of frontier orbitals in chemical reactions. Science 218, 747–754 (1982). https://doi.org/10.1126/science.218.4574.747
E. Runge, E.K.U. Gross, Density-functional theory for time-dependent systems. Phys. Rev. Lett. 52, 997–1000 (1984). https://doi.org/10.1103/PhysRevLett.52.997
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Fahim, A.M., Tolan, H.E.M., Awad, H. et al. Synthesis, antimicrobial and antiproliferative activities, molecular docking, and computational studies of novel heterocycles. J IRAN CHEM SOC 18, 2965–2981 (2021). https://doi.org/10.1007/s13738-021-02251-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-021-02251-7