
Abstract
Background
Cancer stem cells (CSCs), also known as tumor-initiating cells (TICs), are characterized by high self-renewal and multi-lineage differentiation capacities. CSCs are thought to play indispensable roles in the initiation, progression and metastasis of many types of cancer. Leukemias are thought to be initiated and maintained by a specific sub-type of CSC, the leukemia stem cell (LSC). An important feature of LSCs is their resistance to standard therapy, which may lead to relapse. Increasing efforts are aimed at developing novel therapeutic strategies that selectively target LSCs, while sparing their normal counterparts and, thus, minimizing adverse treatment-associated side-effects. These LSC targeting therapies aim to eradicate LSCs through affecting mechanisms that control their survival, self-renewal, differentiation, proliferation and cell cycle progression. Some LSC targeting therapies have already been proven successful in pre-clinical studies and they are now being tested in clinical studies, mainly in combination with conventional treatment regimens.
Conclusions
A growing body of evidence indicates that the selective targeting of LSCs represents a promising approach to improve disease outcome. Beyond doubt, the CSC hypothesis has added a new dimension to the area of anticancer research, thereby paving the way for shaping a new trend in cancer therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Normal hematopoietic stem cells
The hematopoietic system is organized in a hierarchical order of cell populations with different capacities to proliferate, differentiate and self-renew. Hematopoietic stem cells (HSCs) are found at the apex of this system and are defined by their ability to self-renew and to give rise to all hematopoietic lineages. HSCs can be identified through the expression of CD34 on their surface and a lack of expression of CD38 and other markers associated with terminal differentiation, such as CD2, CD3, CD4, CD8, CD14, CD15, CD16, CD19, CD20, CD56, CD66b and glycophorin A (CD235a). In healthy individuals, HSCs represent less than 0.1 % of the hematopoietic compartment in the bone marrow (BM). HSCs give rise to multi-potent hematopoietic progenitor cells (HPCs) that can differentiate into lineage-committed precursors and, ultimately, mature blood cells. En route to differentiation, the cells acquire lineage-specific markers and the capacity to perform lineage-specific functions [1, 2]. Hematopoiesis is a tightly regulated process that is controlled by complex cellular signaling networks, transcription factors, cytokines and interactions with the BM microenvironment. Alterations that perturb this process may result in hematologic malignancies such as leukemia.
2 Leukemia stem cells
In analogy to the normal hematopoietic system, it has been found that different types of leukemia, which are characterized by a predominance of immature abnormally proliferating blood cells, can originate from and be maintained by leukemia stem cells (LSCs). Specifically, it has been shown for myeloid leukemias (both chronic and acute) that the process of leukemogenesis is driven by a hierarchical route and is sustained by a small, self-renewing population of LSCs. LSCs give rise to leukemic progenitors with a reduced capacity to self-renew which, in turn, yield leukemic blasts without a capacity to self-renew. Similar to their normal counterparts, acute myeloid leukemia (AML) and chronic myeloid leukemia (CML)-derived LSCs express CD34, but not CD38, in most patients. It has also been reported that some patients may exhibit CD34+CD38+ [3] or CD34− [4] surface marker phenotypes. Although these markers alone may not be sufficient to specifically identify LSCs, they have been used to define a clinically relevant population of cells that correlate with a poor disease outcome [5]. In addition to the phenotypic markers mentioned, LSCs may also be defined by their function. They are, for example, capable of both self-renewing and giving rise to leukemic progeny. The functional characterization of LSCs is typically performed in vitro using colony forming assays, or in vivo using xenotransplantation models.
Another important feature of LSCs is their ability to remain in a quiescent state. It has been shown that quiescent LSCs from both AML and CML patients are able to retain the self-renewal capacity in vivo and to recapitulate leukemias in xenografted animal models [6]. Furthermore, it has been found that quiescent LSCs may be protected in osteoblast-rich areas (niches) of the BM [7–9]. Importantly, quiescent LSCs are able to evade the action of chemotherapeutic agents [10], which is due to the fact that most of these agents are aimed at targeting rapidly cycling cells. Thus, LSCs represent a chemotherapy-resistant reservoir of cells that may give rise to relapse.
Recently, a growing number of antigens associated with the LSC phenotype has been described for AML. These antigens provide the possibility to distinguish LSCs from HSCs and include CD123, CD25, CD32, CD44, CD47, CD90, CD96, CD117, TIM-3, CLL1, ALDH1, CD99 and IL-1RAP [11–17]. IL-1RAP and CD26 are LSC-associated markers that can be used to distinguish LSCs from HSCs in CML [18, 19]. Although not all of these markers are expressed by LSCs in all patients, it has been reported that patients with a higher number of cells that express these markers exhibit a poorer outcome [20–22].
In view of the critical role played by LSCs in the pathogenesis of hematologic malignancies, increasing efforts have been devoted at developing approaches to eradicate them. The fundamental tenet of these approaches is to establish methods that effectively target neoplastic LSCs and spare normal HSCs. This becomes particularly important since several commonalities exist between HSCs and LSCs.
3 Survival of pre-leukemic HSCs after chemotherapy and their persistence during remission
LSCs are heterogeneous in terms of origin. Although progenitor-derived LSCs are similar to committed progenitors from which they have emerged, LSCs execute a self-renewal-associated program that is also present in normal HSCs [23]. It has been shown that pre-leukemic HSCs are resistant to chemotherapy, thus representing a potential reservoir for disease relapse in at least some AML patients. Pre-leukemic HSCs are genetically distinct from normal germ-line HSCs, but they may be considered as antecedent to leukemic cells [24, 25]. Recently, a pattern of acquired mutations in AML cells and a concomitant persistence of pre-leukemic HSCs in patients with a complete remission have been reported [25]. The authors concluded that mutations in “landscaping” genes involved in chromosomal modifications may occur early in HSCs, whereas mutations in “proliferative” genes may occur late. The so-called pre-leukemic HSCs (stem/progenitor cells) may persist during remission and contribute to relapse through the acquisition of additional genetic and/or epigenetic alterations.
4 Targeting leukemia stem cells
In the past, several strategies have been developed to eliminate LSCs. In Fig. 1 different approaches are depicted that can be used for the treatment of leukemia via LSC targeting. These approaches aim to specifically kill LSCs by interfering with different molecular mechanisms that underlie their survival, growth and multiplication. Accordingly, adverse effects associated with treatment are minimized and therapeutic efficacies are maximized. Overall, it has been shown that CSC targeting therapies exhibit a higher efficacy in cancer elimination than conventional anticancer therapies [26]. In the following sections, the targeting of LSCs through different routes will be discussed.

Different strategies for the treatment of hematologic malignancies through the targeting of LCSs. These approaches cover a diverse range of therapeutic modalities that affect a variety of mechanisms underlying the survival, growth, differentiation, self-renewal and amplification of LSCs
4.1 Cell surface antigens
Although LSCs share a CD34+CD38− phenotype with HSCs, there are also cell surface antigens that are selectively expressed by LSCs. These antigens demarcate a specific LSC phenotype and have, therefore, the potential to be used as targets for therapies directed against hematologic malignancies.
4.1.1 CD33
CD33 was the first antigen to be approved by the US FDA as target for the treatment of AML. CD33 is an attractive target since in normal hematopoiesis its expression is restricted to advanced stages of myeloid differentiation and, therefore, it is not expressed on normal HSCs. CD33 is highly expressed by LSCs in AML. The strategy applied to target CD33 implies the use of CD33-specific monoclonal antibodies (mAbs) conjugated with cytotoxic agents, including radioisotopes, ricin, gelonin and calicheamicin. Gemtuzumab ozogamicin (GO) is composed of calicheamicin linked to the humanized anti-CD33 antibody hP67.6. This antibody-drug conjugate was found to inhibit the growth of AML colony forming cells (CFCs) and to significantly reduce the in vivo growth of AML-derived HL-60 xenografts. These antitumor effects were found to be triggered by a selective cytotoxicity against CD33+ AML stem cells [27]. Toxic side-effects caused by this drug have, however, led to its withdrawal from the market. The high potential of CD33 as a therapeutic target has, nevertheless, inspired researchers to design new trials in which GO is combined with other chemotherapeutic agents [28]. Additionally, tumor-specific chimeric antigen receptor-modified T-cell (CART) approaches are currently being developed to target CD33 expressing cells [29, 30].
4.1.2 CD123
Another antigen highly expressed on bulk and CD34+CD38− AML cells is the alpha subunit of the interleukin-3 receptor (IL-3Rα). IL3 binding is known to lead to receptor activation and to promote cell proliferation and survival. Based on this knowledge, a human interleukin-3 diphtheria toxin fusion protein (DT388IL3) has been engineered with the purpose to target and kill AML cells. Treatment of AML xenografts with DT388IL3 has indeed led to the elimination of leukemias from three of six poor prognosis AML patient samples [31]. Combination treatments of cytarabine or tyrosine kinase inhibitors (TKIs) with DT388IL3 have been shown to act synergistically in eliminating AML and CML stem/progenitor cells that express IL-3R [32]. Anti-CD123 monoclonal antibody-based strategies, such as those using the neutralizing antibody 7G3, have also been tested. Ex vivo and in vivo treatments with 7G3 revealed a selective targeting of AML cells, while avoiding their normal counterparts. Importantly, tests in primates have shown that a chimeric variant of 7G3 (CSL360) exhibits no significant toxic side-effects [33]. The mechanisms of action of this anti-CD123 antibody may involve (i) inhibition of IL3-mediated signaling, (ii) complement-dependent cytotoxicity (CDC) or (iii) recruitment of effector cells resulting in antibody-dependent cellular cytotoxicity (ADCC). To improve the ADCC effect, a second generation anti-CD123 mAb, called CSL362, has been developed. This antibody showed an increased in vivo cell killing capacity in mouse xenograft models [34]. Application of the CART strategy (see above) against CD123 has also yielded promising results in in vivo xenograft AML models [35, 36].
4.1.3 CD44
The adhesion molecule CD44 mediates interactions between hematopoietic cells and their BM microenvironment in endosteal regions. CD44 is a transmembrane glycoprotein with a ubiquitous expression pattern and can be alternatively spliced into many variant isoforms (CD44v). The expression of CD44v is associated with a poor prognosis in AML. CD44-specific mAbs are able in vitro to reverse differentiation blocks, to inhibit proliferation and to induce apoptosis of AML cells [37, 38]. One CD44-specific mAb (H90) has been shown to effectively and selectively eliminate LSCs in vivo by reducing the trafficking and homing of LSCs into its supportive microenvironments [39].
4.1.4 CD25
CD25, the alpha chain of the IL-2 receptor (IL-2Rα), has been reported to serve as a LSC marker. In support of this, gene expression profiles of CD25+ AML cells have revealed correlations with those of LSCs. Currently, daclizumab (a humanized anti-CD25 mAb) that blocks the formation of the high affinity IL-2 receptor on T-cells, has been tested with some success in phase II trials in patients with newly diagnosed T-cell lymphoma and refractory T-cell and B-cell lymphomas [40]. Given the significance of CD25 as a marker for LSCs, anti-CD25 therapies are of potential use for the treatment of AML. CD25 expression has been related to an adverse prognosis in AML patients, and its association with internal tandem duplication (ITD) mutations in the Fms-like tyrosine kinase-3 (FLT3) gene has been found to be indicative for a poor overall and relapse-free survival [21, 41, 42]. CD25 is regulated by STAT5, and its high expression likely results from STAT5 hyper-activation. Therefore, approaches that inhibit the JAK-STAT pathway are of major interest for the treatment of CD25+ AML patients [43].
4.1.5 Other phenotypic markers with therapeutic potential
Other phenotypic markers that have been considered as therapeutic targets for LSCs including CD47, CCL-1 and IL-1RAP. CD47 contributes to phagocytosis and is expressed by macrophages and dendritic cells. A high expression has been reported in AML-derived LSCs. Treatment with B6H12.2 or BRIC126 antibodies has been found to result in an enhancement of phagocytic activity and an inhibition of LSC engraftment in immune-deficient mice [13]. The C-type lectin-like molecule-1 (CLL-1) plays a role in immune regulation, is highly expressed in leukemic blasts and LSCs, and its inhibition by 21.16 and 1075.7 antibodies can decrease the size of HL-60 xenografts [44]. More recently, a novel bispecific antibody, αCLL1-αCD3, has been generated that can recruit cytotoxic T-cells (CTLs) to CLL-1+ AML cells, leading to a potent and selective cytotoxicity in vitro [45]. The interleukin 1 receptor accessory protein (IL-1RAP) has been found to be expressed in AML and CML stem cells. A chimeric anti-IL-1RAP mAb (81.2) of the human IgG1 subtype has been found to elicit ADCC activity in both leukemic blasts and stem/progenitor cells [46].
Taken together, targeting cell surface antigens, in particular through receptor-specific antibodies, holds promise as a therapeutic approach to eliminate LSCs (Supplementary Fig. 1). All above-mentioned antigens also represent attractive targets for CART strategies that have the capacity to kill aggressive acute lymphoblastic leukemia (ALL) cells in vivo. The major concern raised for this type of therapy may be the emergence of sub-clones of cells that do not express the target antigen(s). The putative presence of these cells emphasizes the need to develop alternative approaches for the treatment of ALL.
4.2 Differentiation therapy
Differentiation therapy has been introduced since the early 1980s. This approach is based on forcing immature cancer cells to terminally differentiate and to, ultimately, die (Supplementary Fig. 2).
4.2.1 Retinoids
Retinoids are chemically related to vitamin A and its metabolite all-trans retinoic acid (ATRA). The biological effects of retinoids are mediated via activating the retinoic acid receptors (RARα, RARβ and RARγ) [47]. ATRA has been shown to efficiently induce the differentiation of acute promyelocytic leukemia (APL) cells, which are associated with a balanced translocation between chromosomes 15 and 17. As a consequence of this translocation a PML- RARα fusion protein is formed that triggers a differentiation arrest at the promyelocyte stage [48]. Recently, it has been shown that AML cells can also be induced to differentiate via a RAR-independent, RXR-dependent mechanism. The induction of myeloid differentiation by RXR-specific ligands requires the expression of PU.1, a member of the ETS family of transcription factors. Liver X receptor (LXR) ligands have been found to consistently enhance the RXR ligand effects, leading to differentiation or cytotoxicity in primary AML cells, but not in normal progenitor cells [49].
A combination of retinoic acid (RA) and arsenic trioxide (ATO) is considered effective in the treatment of APL. This combination has been found to increase APL cell death and to reduce the number of bone marrow blasts through the induction of oxidative stress and interference with the translation of mutant oncoproteins [50, 51]. In this context, El Hajj et al. [47] found that RA and ATO can synergistically trigger the proteasomal degradation of mutant nucleophosmin-1 (NPM1), the most frequently mutated protein in AML. They showed that combined RA/ATO treatment can cause the differentiation, growth arrest and apoptosis of AML cells harboring NPM1 mutations, as well as a reduction of bone marrow blasts in NPM1 mutant AML patients. Additionally, they found that ATO can abrogate the stem cell capacity of LSCs that express the PML-RARα fusion protein. It appears that the high rate of complete molecular remission (CMR) and long-term relapse-free survival (RFS) induced by ATO in APL patients may be attributed to its ability to efficiently target LSCs [52].
4.2.2 Iron metabolism disruptors
Disruption of the iron metabolism may lead to differentiation in AML cells. Iron chelators are able to trigger the differentiation of leukemia blasts via the induction of reactive oxygen species (ROS). Iron chelators with well-established differentiation-inducing capacities in AML cells include deferoxamine (DFO) and deferasirox (DFX) [53]. Interestingly, it has been found that eltrombopag (EP), which is a thrombopoietin receptor (TPO-R) agonist, can reduce free intracellular iron levels in leukemic cells in a dose-dependent manner and induce the differentiation of these cells through a mechanism independent of TPO-R [54].
4.3 Mitochondria and other metabolic pathways
HSCs and LSCs are considered to be metabolically dormant since they persist in low energy states. These cells favor incomplete oxidation of glucose through uncoupling glycolysis and oxidative phosphorylation, which leads to low levels of endogenous ROS. The necessity to maintain low amounts of endogenous ROS in LSCs, however, inevitably renders these cells sensitive and vulnerable to changes in their intracellular redox state. This distinctive characteristic of LSCs provides an ideal opportunity for therapeutic intervention. Mitochondria are critical for the generation of energy and the regulation of redox homeostasis. These organelles control the production of free radical species such as ROS and reactive nitrogen species (RNS). These free radical species are known to play important roles in cellular homeostasis, and their excessive production may result in cell death [55, 56]. Based on these characteristics, priming of mitochondria for apoptosis has been used as a parameter to determine the sensitivity of AML cells to chemotherapeutic agents [57]. Mitochondrial priming may be assessed through the expression of Bcl-2 (B-cell lymphoma 2) family member proteins and their accompanying Bcl-2 homology 3 (BH3)-only family members. Cellular stress associated with chemotherapy may activate pro-apoptotic signals that induce death in cells expressing high levels of BH3-only family members (primed for death through mitochondrial priming). In contrast, cells with high amounts of anti-apoptotic proteins do not die when they are exposed to therapeutic agents. Mitochondrial priming has been shown to dictate the efficacy of topoisomerase II inhibitors in vitro. Importantly, mitochondrial priming in AML samples derived from patients prior to the onset of therapy has been correlated with the efficacy of induction chemotherapy (5-year complete remission, CR). These studies have also indicated that normal HSCs rely primarily on Mcl-1, while LSCs rely on Bcl-2. This difference has provided a rationale for using the Bcl-2 inhibitor ABT-737 [57]. Additional studies in blast crisis CML (bcCML) have emphasized the significance of the Bcl-2 family members in LSCs [58]. Stem cells derived from bcCML over-express several anti-apoptotic Bcl-2 family member splice isoforms, such as Bcl-2L, Mcl-1L, Bcl-xL, and Bfl-1 L, compared to chronic phase CML progenitors and normal HSCs. Increased Bcl-xL expression levels have been found to be associated with BCR-ABL fusion protein expression in CML, suggesting another mechanism for over-expression of Bcl-2L, Mcl-1L and Bfl-1L. A pan-active Bcl-2 protein family antagonist, sabutoclax, has been found to affect the survival of CML-derived LSCs. Sabutoclax treated LSCs exhibit a decreased survival, a decreased colony forming capacity and a capacity for serial xenotransplantation. Importantly, dual treatment with sabutoclax and the TKI dasatinib may result in tumor shrinkage in established CML xenograft models compared to dasatinib alone. Thus, the dependence of CML-derived LSCs on pro-survival Bcl-2 family member signaling is a key factor to be considered for patients exhibiting resistance to TKIs [59]. Studies on AML-derived cell lines using a specific inhibitor of Bcl-2 and Bcl-xL (ABT-737) have revealed an up-regulation of Mcl-1 [60], which can be phosphorylated and stabilized through the MAPK/ERK pathway. Combinations of MEK inhibitors and ABT-737 prevents the up-regulation of Mcl-1, leading to an increased apoptotic rate in LSCs from primary AML patient samples [60]. The relevance of preventing Mcl-1 accumulation as a result of acquired resistance to Bcl-2 inhibition has been underscored by studies in which the efficacy of a BH3 mimetic, obutoclax, was found to be improved by sorafenib as a multi-kinase inhibitor [61]. It was found that this combination therapy results in an increased killing of AML cells in vitro and a prolonged survival of AML xenotransplants in vivo.
In addition to targeting Bcl-2 family members that reside in mitochondria and play critical roles in deciding whether a cell will live or die, it was recently found that mitochondrial translation also represents a target for ablating AML cells and LSCs [62]. Tigecycline, a US FDA-approved antimicrobial compound identified in a chemical screen, was found to effectively reduce the engraftment capacity of LSCs in NOD/SCID mice with minimal effects on normal HSCs. Tigecycline potently blocks mitochondrial ribosomes, resulting in inhibition of mitochondrial translation. Compared to HSCs, LSCs have been found to possess a larger mitochondrial mass, a higher mitochondrial DNA copy number, and an increased rate of oxygen consumption. The sensitivity to tigecycline of AML cells is directly associated with a higher mitochondrial mass. This notion suggests that assessment of the mitochondrial mass may be used to identify patients that benefit from therapies targeting mitochondrial function. However, prior to using mitochondrial mass assessment and mitochondrial translation inhibition in the treatment of individual patients, it is important to exactly determine to what extent disease outcomes correlate with the mitochondrial mass [62, 63].
Aerobic glycolysis or the “Warburg effect” entails that most tumors predominantly favor glycolysis over oxidative phosphorylation in their mitochondria for energy production [64]. This process is followed by lactic acid fermentation in the cytosol. These processes provide a growth advantage of tumor cells over normal cells. The M2 isoform of pyruvate kinase (PK) plays an essential role in maintaining aerobic glycolysis. PKM2 catalyzes the final and rate limiting step of glycolysis (Fig. 2). PKM2 deletion has been found to increase oxidative phosphorylation over glycolysis. Interestingly, HPCs are only affected under stress conditions that drive HPC proliferation. PKM2 deletion does not affect leukemia initiation, but rather appears to be important for the progression of leukemias driven by BCR-ABL and MLL-AF9 fusion proteins [65]. PKM2 exists either as an active tetramer or as an inactive monomer/dimer induced by allosteric regulators such as metabolites or ROS, as well as by phosphotyrosine modifications [66]. Such modifications limit the flux of glucose carbons in the form of pyruvate to mitochondria for energy production and, instead, shunt them towards anabolic processes. Some pharmacological PKM2 activators have been generated that are able to decrease aerobic glycolysis in cancer cells and to suppress tumor growth [66, 67]. CD34+ AML cells exhibit no significant differences in total PKM2 levels compared to the bulk of AML cells. However, CD34+ progenitor cells have been found to exhibit significantly increased levels of phosphorylated PKM2 at Tyr105, which is known to promote the less active non-tetramer [68]. Consistently, PKM2 activity has been found to be reduced in CD34+ progenitor cells compared to blasts [69]. These data suggest that AML stem and progenitor cells may contain more inactive PKM2 tetramers than mature proliferating AML cells. Therefore, perturbation of the metabolic state of AML cells with PKM2 activators may be considered as a means to disrupt LSC homeostasis, thereby impairing the growth of more differentiated AML cells.

The Warburg effect or aerobic glycolysis in tumor cells. Normal differentiated cells generate energy through glycolysis (at low oxygen levels) and mitochondrial oxidative phosphorylation (at high oxygen levels). However, rapidly proliferating tumor cells generate energy mainly through glycolysis, followed by lactic acid fermentation in the cytosol. This metabolic change is observed at both low and high oxygen levels. Pyruvate kinase plays a leading role in the establishment and maintenance of aerobic glycolysis adaptation
4.4 Epigenetic regulators
Growing evidence indicates that epigenetic alterations may contribute to malignancy. These alterations result in gene silencing and activation as a consequence of a series of chromatin remodeling events [70]. Chromatin remodeling is driven by a variety of modifications, including histone methylation, histone acetylation and DNA methylation. It has been found that specific epigenetic profiles allow the stratification of AML patients since they are often associated with specific clinical features of the disease. Recent studies have focused on investigating the epigenome of stem (CD34+CD38−), progenitor (CD34+CD38+) and mature (CD34−) AML cells. No consistent differences in DNA methylation patterns were found [71]. In contrast, differences in chromatin (histone) states associated with H3K4me3(K4) and H3K27me3(K27) were recognized in purified stem, progenitor and mature AML cell subpopulations. Interestingly, enrichment for ERK/MAPK, hypoxia and NRF2-mediated oxidative stress response pathways were noted in the lysine 4 (K4) regions of undifferentiated populations. These pathways are consistent with those found to drive LSCs towards a low ROS condition and may be encountered in hypoxic areas of the BM [58, 72]. Obviously, larger cohorts are required to define epigenetic profiles indicative of candidate druggable pathways with a potential for ablating chemo-resistant LSCs.
Global epigenetic modifiers such as histone deacetylase (HDAC) and DNA methyltransferase (DNMT) inhibitors are currently being tested for their efficacy in AML, alone or in combination with other therapeutic modalities. Patients undergoing treatment with sodium valproate and 5′-azacitidine (VAL-AZA) who achieved a complete remission (CR) or a CR with incomplete blood count recovery (CRi), have been found to show significant decreases in LSCs, albeit that a complete eradication of LSCs did not seem to occur [73]. Interestingly, an expansion of LSCs was observed just prior to the occurrence of an overt relapse. The inability to ablate LSCs with DNMT inhibitors and HDAC inhibitors, as well as the intra/inter-patient heterogeneity found in epigenetic marks, suggests a need to design inhibitors targeting specific patterns of epigenetic alterations in individual AML patients. Examples of newly emerging therapeutic targets are DOT1L, LSD1, EZH2, IDH1/2, Menin and Brd4 [74–78]. In leukemia, DOT1L regulates genes that are critical for LSC self-renewal and survival. It has also been found to be required for the initiation and maintenance of MLL-AF9 fusion protein-associated leukemias. Therefore, specific DOT1L inhibitors, such as EPZ004777, are currently under investigation [78]. The tumor suppressor Menin is another critical factor in MLL-driven leukemias, and is a highly specific binding partner of MLL and various MLL fusion proteins. Menin is known to be required for the regulation of MLL target genes, including HOXA9 and MEIS1 [79]. Disruption of interactions between Menin and the MLL fusion proteins is essential for MLL-induced leukemia initiation. Since the Menin-MLL interaction represents a potential therapeutic target, small molecule inhibitors with the capacity to affect this interaction have been developed and are currently under evaluation. LSD1 (KDM1/AOF2), a lysine-specific demethylase that is able to demethylate H3K4 and H3K9, is required for sustaining LSCs in MLL-AF9 fusion protein-driven leukemias. LSD1 is highly expressed in AML. Thus, LSD1 may serve as a therapeutic target that can be used for evaluating the effects of both reversible and irreversible LSD1 inhibitors [80]. It has been found that LSD1 targeting may lead to restoration of RARα2 expression in a subset of AML cells in which loss of RARα2 expression is associated with a reduction in H3K4me2 on the RARA2 gene promoter, and increased sensitivity to ATRA leading to the differentiation and, ultimately, death of AML cells [77]. DZNep (3-deazaneplanocin A), a histone methyltransferase inhibitor, can disrupt the polycomb repressive complex 2 (PRC2), resulting in apoptosis of AML cells [81, 82]. PRC2 is known to mediate gene silencing through H3K27 trimethylation. EZH2 plays a key role in the methyltransferase activity of the PRC2 complex. DZNep treatment may also induce apoptosis in LSC subpopulations via reactivation of the thioredoxin binding protein 2 (TXNIP), which leads to enhanced ROS levels [82]. TXNIP reactivation by DZNep is triggered by PRC2 depletion and a subsequent decrease in H3K27me3. Small molecule inhibitors of EZH2 are currently under investigation and represent promising anti-leukemia stem cell agents. Isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) are frequently mutated in AML cells, thereby inducing global changes in DNA methylation [83]. IDH1-R132, IDH2-R140 and IDH2-R172 mutations are commonly observed in AML, resulting in an aberrant production of 2-hydroxyglutarate (2HG). 2HG affects a DNA demethylase called Tet methylcytosinedeoxygenase 2 (TET2) and generates DNA hypermethylation as well as other changes that affect the quiescence and self-renewal capacities of HSCs associated with leukemic transformation [84, 85] Importantly, new small molecule inhibitors are now available targeting specific IDH mutations. These inhibitors provide a therapeutic opportunity to target AML cells harboring these mutations.
4.5 Cellular signaling pathways
Intricate signaling pathways are known to regulate the differentiation, survival, proliferation and cell cycle progression of LSCs. A detailed understanding of these pathways has led to the identification of LSC-specific signaling molecules, some of which have been proposed as useful therapeutic targets.
4.5.1 Nuclear factor kappa B
Nuclear factor kappa B (NF-kB) comprises a conserved family of transcription factors with five members (Rel A or p65, Rel B, Rel C, p50 and p52) that form hetero-complexes among each other. In untransformed or unstimulated cells, NF-kB is sequestered in the cytoplasm through binding to inhibitory proteins such as IkB. Upon a variety of different stimuli, IkB may be phosphorylated by the IKK complex, resulting in NF-kB translocation to the nucleus where it can regulate genes involved in cellular growth and proliferation, the inhibition of apoptosis, and multidrug resistance [86, 87].
NF-kB is constitutively activated in LSCs, but not in normal HSCs [88], and represents one of the first unique therapeutic targets identified in LSCs. Recently, it was found that NF-kB activity can be maintained through autocrine secretion of TNF-α in LSCs [89]. The small molecule inhibitor parthenolide (PTL), a known inhibitor of NF-kB, has been found to preferentially induce apoptosis in LSCs and progenitor cells in AML via increasing ROS and activating p53 [90]. PTL has also shown efficacy against LSCs from pediatric ALL patients [91]. Recently, a water-soluble analog of PTL, DMAPT, has been generated [92] and this analog is currently being evaluated in clinical trials. PTL’s ability to target LSCs while sparing HSCs has been employed to identify diverse chemical agents with similar biological activities using chemogenomic approaches. These agents include celastrol, 4-hydroxy-nonenal (4-HNE) [93] and AR-42 [94]. An alternative approach to inhibit NF-kB using the IKK inhibitor AS602869 has resulted in the induction of apoptosis in primary AML blasts and progenitor cells. Inhibition of NF-kB has been found to be associated with mitochondrial transmembrane potential failure without affecting normal cells [86]. AS602869 has also shown activity against imatinib-resistant primary CML cells [95]. Taken together, these data indicate a therapeutic potential of targeting NF-kB in leukemia.
4.5.2 PI3K/AKT/mTOR pathway
The PI3K/AKT/mTOR network represents an intracellular signaling pathway that plays an important role in normal and malignant hematopoiesis. Phosphatidylinositol 3 kinase (PI3K) can be activated in response to growth factors and several hematopoietic cytokines leading to the activation of downstream kinases such as the serine/threonine kinase Akt (also known as protein kinase B or PKB) that, in turn, can activate the mTOR (mammalian target of rapamycin) complex. Activation of this network contributes to cellular survival, growth, apoptosis and proliferation. Malignant hematopoietic cells may exhibit alterations in this pathway, which may result in its constitutive activation and, thus, increased survival, growth and/or chemotherapy resistance [96, 97].
In AML, constitutive PI3K activation has been observed in 50 % of the cases, whereas constitutive Akt/PKB phosphorylation has been observed in nearly 80 % of the cases. These activated states are associated with a poor overall survival (OS). The PI3K/AKT pathway plays a key role in the proliferation and survival of AML progenitor cells and blasts. Thus, PI3K inhibition in these cells, using compounds such as LY29402, may result in apoptosis. The exploitation of this agent in combination with a topoisomerase II inhibitor called VP16 has resulted in a reduction in cell proliferation, an induction of apoptosis, a down-regulation of p-AKT and a constitutive NF-kB activity in primary AML cells [98]. Wortmannin is a cell permeable irreversible inhibitor of PI3K that also inhibits DNA-dependent protein kinase (DNA-PK), mTOR and ataxia telangiectasia mutated (ATM) kinase [99]. It has been found that AML cell treatment with wortmannin up-regulates p53 and down-regulates the multidrug resistance-associated protein 1 (MRP1) [100]. Wortmannin also sensitizes cells to MRP1 substrate drugs such as etoposide [101], suggesting that PI3K/Akt activation leads to an increased chemoresistance in AML. Perifosine, another PI3K/Akt inhibitor, is known to target cellular membranes and to modulate membrane permeability as well as lipid composition. These functions ultimately trigger the differentiation and growth inhibition of AML-derived cell lines and primary AML cells. Perifosine also reduces the clonogenic activity of AML progenitors without having any impact on normal CD34+ cells [102]. Interestingly, perifosine has also been found to induce protective autophagy in CML cells [103]. Rapamycin (sirolimus) is a macrolide derived from the bacterium Streptomyces hygroscopicus that acts as an allosteric inhibitor of mTOR. This inhibitor has been shown to block the growth of AML-derived cell lines and to reduce the colony forming capacity of CD34+ AML cells while sparing normal progenitors [104, 105]. Rapamycin has also been found to elicit an efficient clinical response in 4 out of 9 refractory/relapsed AML patients tested [86]. In addition, a combinatory strategy based on common and novel anti-leukemia agents in conjunction with PI3K/mTOR/Akt inhibitors has shown efficacy against LSCs in AML and CML. Some examples of these anti-leukemic agents include etoposide [105], clofarabine [106], the water soluble analog of parthenolide [107], PIM kinase inhibitors [108] and dasatinib [109]. Based on the anti-leukemia activities observed for inhibitors of the PI3K/Akt/mTOR pathway, dual inhibitors for PI3K and mTOR have been developed such as NVP-BEZ235, GDC-0980 and XL-765 [110–112], of which NVP-BEZ235 has been found to exhibit activity against AML blasts and progenitor cells with a minimal toxicity to normal progenitor cells.
4.5.3 Reactive oxygen species
Reactive oxygen species (ROS) contain hydrogen peroxide (H2O2), hydroxyl radicals (OH-) and superoxide anion radicals (O2-). These agents are essential for various biological processes in normal cells where they act as second messengers. Physiologically, ROS are reduced by anti-oxidizing agents including superoxide dismutase (SOD), glutathione (GSH) and catalase. High ROS levels may, however, confer cellular damage to DNA, proteins and lipids. In hematopoietic cells it has been found that deficiencies in forkhead transcription factors (FOXOs) may give rise to increased ROS levels and, consequently, defective long-term repopulating activities, terminal differentiation and cell cycle progression [113]. Germline loss of FOXO3 may also result in p38/MAPK activation in HSC compartments. This observation suggests that low oxygen levels in the BM may provide a long-term protection of HSCs from ROS, whereas high oxygen levels may limit the lifespan of HSCs [114]. In AML, constitutively activated Akt inhibits FOXO and increases ROS levels. Enhanced ROS signaling causes damage to nuclear and mitochondrial DNA, compromises the DNA repair system and, thereby, increases mutation rates [115]. CML stem and progenitor cells contain high levels of ROS and concomitant oxidative DNA damage compared to their normal counterparts, and it has been found that increased ROS levels may contribute to the generation of imatinib-resistant LSC clones [116]. In recent years, ROS perturbation has gained interest as a strategy to eliminate cancer cells [55, 56]. As such, anti-LSC activity via ROS up-regulation has already been observed using agents such as (i) PTL [90] and/or piperlongumine through transient GSH depletion [117], (ii) DZNep through inhibition of thioredoxin [82], (iii) auranofin (AF), which is clinically used for the treatment of rheumatoid arthritis, through inhibition of thioredoxin reductase [118], (iv) retinoid fenretinide through the oxidative/endoplasmic reticulum stress-mediated pathway [119] and (v) the iron chelator deferasirox through induction of cellular differentiation [53].
4.5.4 Heat shock proteins
Heat shock proteins (HSPs) constitute a family of highly conserved proteins involved in catalyzing the proper folding and, thereby, aggregation of proteins. Hsp90 and Hsp70 are highly sought-after therapeutic targets in cancer and, consequently, small molecule inhibitors targeting these HSPs have recently been developed. Hsp90 has housekeeping functions and ensures the proper folding of proteins involved in cellular growth, differentiation, DNA damage and apoptosis. Specific co-chaperones and post-translational modifications can shunt Hsp90 into an alternative form (known as oncogenic or tumor enriched Hsp90) with specialized oncogenic functions, including blocking the degradation of oncogenic proteins that are often associated with Hsp90. Oncogenic Hsp90 forms have been found to sustain BCR-ABL activity and active STAT5 signaling in CML [120]. In AML, Hsp90 is highly expressed which correlates with high levels of CD34 and Bcl2, autonomous growth, increased colony formation and a poor survival [121]. In addition, bioinformatic analyses have revealed that Hsp90 is enriched in patients harboring FLT3 internal tandem duplications (FLT3-ITD) [122]. Hsp90 has also been correlated with the disease state in CML and, thus, has been proposed to serve as a risk factor [123]. In vitro exposure of primary CD34+ AML cells and AML-derived cell lines to the Hsp90 inhibitor 17-N-allylamino-17-demethoxygeldanamycin (17-AAG) has been found to inhibit growth, apoptosis and cell cycle arrest without affecting normal CD34+ cells [124]. Interestingly, a phase I clinical trial for 17-DMAG (a synthetic analog of 17-AAG) has reported 3 complete remissions out of 17 patients that were evaluated [125]. Several small molecule Hsp90 inhibitors are currently under investigation for their efficacy in different tumor types. One of these, PU-H71, which is a novel agent that preferentially binds to Hsp90, has been shown to possess potent anticancer activities in animal models for several types of tumors [126–128]. This compound is currently being tested in a phase I clinical trial for metastatic solid tumors and lymphomas (www.clinicaltrials.gov).
4.5.5 Bcl-6
Bcl-6 is a transcriptional repressor that has been found to play an important role in the acquisition of chemo-resistance by CML and ALL stem cells. Bcl-6 is a member of the BTB-POZ protein family whose members mediate transcriptional silencing. In BCR-ABL+ B-ALL and CML, Bcl-6 has been found to be essential for the acquisition of imatinib resistance through p53 and ARF-mediated pathways. Bcl-6 inhibition by RI-BPI (peptidomimetic inhibitor) has been found to lead to the ablation of LSCs in B-ALL and CML cases in which the LSCs could not be eliminated by imatinib [129–131]. These observations suggest that simultaneous targeting of BCR-ABL and Bcl-6 may represent a therapeutic strategy to ablate LSCs and to prevent TKI resistance and relapses in BCR-ABL+ leukemias.
4.6 Self-renewal pathways
Self-renewal pathways have also been suggested as candidate targets to ablate LSCs. The canonical Wnt (Wnt/β-catenin), Notch and Hedgehog pathways are self-renewal pathways with a high potential to serve as therapeutic targets in leukemia.
4.6.1 Wnt/β-catenin pathway
β-catenin, when activated by Wnt, is protected from degradation and accumulates within cells. This process leads to translocation of β-catenin to the nucleus where genes associated with self-renewal are regulated [132]. The Wnt/β-catenin pathway is active in HSCs, but not in granulocyte macrophage progenitors (GMPs), as well as in a variety of leukemias. Activation of β-catenin signaling by, for example GSK3β mis-splicing, results in the transformation of progenitor cells [133, 134]. Microarray-based expression analyses have revealed that Wnt/β-catenin pathway deregulation may contribute to CML progression driven by BCR-ABL kinase activity [135, 136]. In AML, the expression of β-catenin has been correlated with a poor prognosis. Since β-catenin is not required for adult HSC self-renewal, targeting the Wnt/β-catenin pathway may be a therapeutic option for eradicating LSCs in AML and CML patients [137, 138]. In pre-clinical studies on AML cases with MLL translocations, β-catenin targeting has revealed a successful killing of LSCs [139]. Interestingly, it has been found that iron chelators such as acyl hydrazones can inhibit Wnt signaling via destabilization of β-catenin and decrease the expression of Wnt in samples derived from AML patients [140]. Homoharringtonine has shown the capacity to disrupt WNT/β-catenin signaling, thereby reducing the growth and inducing the apoptosis of LSCs in AML and CML, alone or in combination with other agents [137]. In CML, the application of Wnt/β-catenin inhibitors such as indomethacin or AV65 has been found to enhance the degradation of β-catenin and to reduce the number of LSCs [138]. It has, however, also been found that despite the presence of an aberrant Wnt/β-catenin pathway, LSCs may not be dependent on this pathway for their survival in all leukemias [141]. Therefore, the identification of predictors of response to Wnt/β-catenin inhibition is of particular importance for the execution of successful clinical trials.
4.6.2 Notch pathway
The notch signaling pathway is evolutionarily conserved and plays an important role in processes such as self-renewal, cell fate, proliferation and survival. The Delta and Jagged ligand family members bind to Notch receptors, which leads to their proteolytic cleavage mediated by metalloproteases and the γ-secretase complex. This cleavage results in release of the intracellular domain into the nucleus where it controls the transcription of its target genes. Notch1 plays a central role in T-cell acute lymphocytic leukemia (T-ALL) and, based on this, γ-secretase inhibitors (GSIs) are currently being tested for their efficacy in both pre-clinical models and clinical trials [142]. However, GSIs also inhibit the cleavage of other proteins such as CD44. In AML, gene expression profiling studies have revealed over-expression of Jagged-2 in LSCs. Consistently, treatment of AML cells with GSIs has been found to lead to decreased colony formation [19]. In addition, the Notch signaling network has been demonstrated to be silenced in human AML samples and in LSCs derived from animal models. Interestingly, Notch inactivation has been shown to cooperate in vivo with loss of TET2 (frequently mutated in AML) [143]) for the induction of AML-like diseases [144]. These data suggest that Notch receptor agonists provide therapeutic potential for AMLs in which the Notch pathway is silenced.
4.6.3 Hedgehog pathway
The Hedgehog (Hh) pathway is an important mediator of normal organ development and hematopoiesis, cell fate determination, differentiation, proliferation and survival. In mammals, Hh signaling has been found to be ligand-mediated and to be initiated by Hh ligand binding to its surface receptor Patched (PTCH). In a resting state, PTCH is expressed on the plasma membrane and acts to repress Smo activity. Upon binding, PTCH is internalized and Smo is no longer repressed, which results in the activation of Gli transcription factors. Loss of Smo has been suggested to cause decreased transformation of HSCs expressing the BCR-ABL fusion protein [145]. Additionally, inhibition of Hh signaling by cyclopamine (a Smo inhibitor) supports the notion that Hh is required for the maintenance of CML stem cells [9]. Unlike CML, Hh signaling is not essential for MLL-AF9 fusion protein-driven AML. Importantly, it has been shown that Hh inhibition has no major effect on normal hematopoiesis.
4.7 Microenvironment disruption
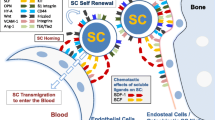
The BM microenvironment comprises a variety of cellular elements that, in combination with non-cellular matrix elements, provide numerous signals for regulating the activity of HSCs. Similarly, LSCs send to and receive signals from their microenvironment. Molecules such as CD44 and N-cadherin, as well as those from the CXCR4-CXCL12 axis, have been shown to regulate LSC survival and BM retention.
During normal hematopoiesis, HSCs interact trough CXCR4 with the stromal cell-derived factor 1 (SDF1/CXCL12) to remain within their BM niche. Several lines of evidence imply that LSCs use the same axis to home to their niche [146]. The BCR-ABL fusion protein is able to inhibit CXCR4 expression in CML cells, and it has been found that treatment of these cells with TKIs can restore CXCR4 expression, thereby promoting their migration to the BM, triggering cell cycle arrest and enhancing the quiescence and survival of CML stem/progenitor cells [147]. In AML, high levels of CXCR4 expression have been associated with a poor prognosis. Recent studies have also revealed that CXCR4 serine339 phosphorylation is correlated with chemo-resistance and retention of leukemic cells in the BM [148]. Inhibition of CXCR4-CXCL12 interactions with CXCR4 antagonizing agents such as perixafor has been found to mobilize leukemic cells and to reduce drug resistance in animal models [149]. Additional strategies to inhibit the CXCR4/SDF1 axis in AML cells include the use of neutralizing CXCR4 antibodies, neutralizing CXCL12 antibodies, or the application of AMD3465 (a CXCR4 antagonist) [150].
Other molecules that regulate interactions of LSCs with their microenvironment are surface antigens such as CD44 (discussed above) for which monoclonal antibodies have been shown to reduce the homing, retention and/or maintenance of LSCs in the BM. Recent evidence has revealed that a hypoxic microenvironment is of particular importance for maintaining normal BM functions, in which the hypoxia-inducible factor 1α (HIF-1α) makes an essential contribution to the survival and quiescence of HSCs. Even though HIF-1α plays an important role in normal HSCs, it has been found that treatment of BM with the HIF-1α inhibitor echinomycin may selectively hamper the colony forming capacity of lymphoma cells without affecting normal murine BM cells. In addition, it has been revealed that echinomycin shows activity against LSCs in AML [151]. E-selectin is another important adhesion molecule that is now being pursued as a therapeutic target. Primary AML blasts and LSCs express an E-selectin ligand [5] thought to enhance the survival of leukemia cells and to increase their chemo-resistance in the BM niche. Blocking E-selectin interactions results in enhanced sensitivity to chemotherapeutic agents and the mobilization of LSCs. Currently, E-selectin inhibitors and dual E-selectin-CXCR4 inhibitors are under investigation to mobilize and sensitize LSCs to chemotherapeutic agents.
In Table 1 the therapeutic approached described above, along with the agents used in each approach for targeting LSCs, are listed. This table provides a snapshot of the treatment modalities that may target LSCs and that are available for the (pre)clinical management of hematologic malignancies.
4.8 CML and tyrosine kinase inhibitors
As outlined above, tyrosine kinase inhibitors (TKIs) represent a group of small molecules that can specifically block the kinase activities of its target proteins. Imatinib, a first generation TKI, is a 2-phenylamiropyrimidine (PAP) derivative that competitively blocks the adenosine triphosphate (ATP) binding site of the BCR-ABL fusion protein, thereby preventing ATP from interacting with its catalytic domain [152]. This inhibitory activity impairs BCR-ABL auto-phosphorylation and its downstream signaling, leading to reduced proliferation and apoptosis-mediated cell death. Imatinib treatment results in durable cytogenetic remissions in CML with minimal damage to normal cells. However, one-third of the patients may develop resistance or intolerance to imatinib. These patients can be treated with second generation TKIs, i.e., nilotinib, dasatinib or bosutinib [138, 153]. Nilotinib is an aminopyrimidine that has been engineered to increase its binding to the ATP pocket of BCR-ABL. It is 20 to 50 times more potent than imatnib and is active in imatinib-resistant cells harboring various kinase domain mutations, except T315I. Dasatinib can bind to both the active and inactive conformations of BCR-ABL and is able to inhibit downstream Src family kinases. Dasatinib is 300 times more active than imatinib but, similar to nilotinib, is inactive in cells harboring the T315I mutation. Its adverse effects are similar to the other TKIs mentioned [154].
Despite the positive clinical effects of TKIs, there are important clinical aspects that need to be considered, i.e., long-term tolerability, resistance in case of the T315I mutation and interruption during pregnancy. Although imatinib, nilotinib and dasatinib are able to reduce the number of tumor cells, induce apoptosis and decrease self-renewal, it has been found that they fail to kill quiescent CD34+CD38−lin− cell populations in animal models [155–157]. A recent assessment of CML mouse models and patient-derived stem cells has revealed that CML-derived stem cells may not be dependent on the presence of BCR-ABL for their survival. These data suggest that therapeutic approaches should also be aimed at targeting BCR-ABL independent mechanisms governing LSC resistance and survival [158, 159].
The development of resistance against TKIs may be either BCR-ABL dependent or independent. Dependent mechanisms involve mutations in the ATP binding domain of BCR-ABL and/or amplification of its encoding fusion gene. Independent mechanisms include reduced expression of the cation transporter hOCT1, which is involved in the influx of imatinib into the cell, increased expression of efflux pumps such as P-glycoprotein (P-gp), sequestration of imatinib by the serum alpha-1-acid glycoprotein (AGP) in plasma, activation of BCR-ABL independent signaling pathways (Src, STAT, Wnt/beta catenin, among others) and LSC-quiescence [153].
5 Conclusion and future perspectives
Although substantial progress has been made in recent years in the development of anti-cancer therapeutic approaches, treatment failure is still a common problem. The main reason of this failure is therapeutic resistance leading to relapse. It has now become widely accepted that current therapies are often capable of eradicating the general population of cancer cells and shrinking the bulk of the tumor, but are not able to uproot a particularly stubborn subpopulation of cells called CSCs. These cells are characterized by high degrees of tumorigenic activity, self-renewal capacity, propagation and multi-lineage differentiation potential through symmetrical and asymmetrical cell divisions that result in a hierarchical heterogeneity among the cells within a tumor. CSCs arise from normal cells with acquired mutations that lead to the deregulation of self-renewal or the dedifferentiation of differentiated cells. While the exact mechanisms underlying the birth and burst of CSCs are not yet fully understood, they are believed to play critically important roles in the pathogenesis of cancer. A large body of information has revealed that these auto-regenerating tumor-initiating cells (TICs) may be the main suspects of tumor formation in a variety of tumor types and, in addition, may be responsible for tumor progression, metastasis, drug resistance and, importantly recurrence. The dormancy of CSCs is an attribute that turns them refractory to conventional antitumor drugs, the action of which largely depends on cell cycle functions. CSCs have a property of robustness, which is defined by several biological features including a slow cell cycle progression, a raised capacity for the efflux of cytotoxic anticancer drugs through ATP-binding cassette transporters, a rapid response to the repair of damaged DNA, a resistance to oxidative stress and an adaptation to hyper-inflammatory or hypo-nutritious microenvironments. All these characteristics may account for the development of therapeutic resistance and subsequent disease relapse.
Targeting CSCs, rather than the bulk of the tumor cells, is a highly promising scenario for eradicating hematopoietic malignancies and for preventing disease relapse (Fig. 3). This type of treatment eradicates the causes rather than the symptoms and, therefore, may trigger a long-lasting clinical response. The identification of key cell surface markers and the appreciation of molecular attributes involved in controlling the CSC phenotype have made major contributions to the design of CSC targeted treatment options. Although these novel options have shown promise in the treatment of cancer, some of them turned out to be non-specific and, therefore, to elicit off-target effects. Thus far, multiple strategies have been developed to selectively target LSCs and to alter their supporting microenvironment. These strategies include targeting LSC-specific cell surface markers, modulating various cellular signaling cascades, adjusting micro-environmental signals, blocking drug efflux pumps, activating anti-apoptotic pathways and inducing cellular differentiation. Some of these CSC targeting strategies have turned out to be successful in preclinical and clinical studies, mostly in combination with traditional anticancer strategies, while others are still under preclinical and clinical evaluation. Although LSC targeting seems to be a promising approach for the treatment of leukemia, there are still challenges that need to be met. LSC cell surface marker profiles may not be universal, i.e., not all LSCs may express certain markers whereas other markers may also be expressed by non-LSCs. Therefore, the identification of novel markers has resulted in repeatedly redefining LSC populations. The currently identified markers can be applied to recognize LSC-rich subpopulations, but may not be suitable to precisely isolate and define all LSCs within a tumor mass. Some cellular signaling pathways responsible for the maintenance of self-renewal and stemness are shared by LCSs and normal HSCs. So, by manipulating these pathways it is difficult to selectively destroy LSCs. Furthermore, the inhibition of a specific signal transduction pathway in LSCs may become ineffective owing to the activation of (an) alternative pathway(s). This phenomenon, which is called adaptive response, underscores the importance of simultaneously interrupting multiple signaling pathways (Supplementary Fig. 3).

Cancer stem cell targeted cancer therapy versus conventional cancer therapy. Conventional cancer therapy targets tumor cells in general, but does not selectively kill CSCs. Therefore, there is a high chance for relapse. When the therapeutic approach is targeted towards CSCs, these tumor-initiating cells are killed. As a result, the tumor loses the capacity to regenerate and, thus, is more unlikely to recur
The development of therapeutic approaches which selectively target LSCs, while minimizing toxicity to normal HSCs, is urgently needed. Most treatment modalities have been tested in vitro or in vivo in animal models. Clearly, precisely controlled clinical trials need to be carried out before the results can be translated to the clinic. Characterization of LSCs at the single cell level, unraveling the genetic, epigenetic and biochemical mechanisms that regulate distinct features of LSCs, as well as advancing our understanding of the role played by stem cell niches in controlling the biological function of LCSs may offer novel clues for the design of better LSC targeted therapeutic approaches. These modalities may include combinations with other existing or novel anticancer therapeutic modalities. In the end, combination therapy may turn out to be the most powerful option for the management and eradication of different leukemias. There is no doubt, however, that the CSC concept has opened up new frontiers in the area of anticancer research and shaped a new doctrine of cancer therapy.
Abbreviations
- HSCs:
-
Hematopoietic stem cells
- HPCs:
-
Hematopoietic progenitor cells
- LSCs:
-
Leukemic stem cells
- AML:
-
Acute myeloid leukemia
- CML:
-
Chronic myeloid leukemia
- ALL:
-
Acute lymphoblastic leukemia
- CML:
-
Chronic myeloid leukemia
- CFCs:
-
Colony-forming cells
- CDC:
-
Complement-dependent cytotoxicity
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- ATP:
-
Adenosine triphosphate
- FLT3:
-
Fms-like tyrosine kinase 3
- ATRA:
-
All-trans retinoic acid
- LXR:
-
Liver X receptor
- DFO:
-
Deferoxamine
- DFX:
-
Deferasirox
- EP:
-
Eltrombopag
- TPO-R:
-
Thrombopoietin receptor
- RNS:
-
Reactive nitrogen species
- BCL-2:
-
B-cell lymphoma 2
- HDAC:
-
Histone deacetylase
- DNMT:
-
DNA methyltransferase
- PRC2:
-
Polycomb repressive complex 2
- NF-kB:
-
Nuclear factor kappa B
- PTL:
-
Parthenolide
- PI3K:
-
Phosphatidylinositol 3 kinase
- DNA-PK:
-
DNA-dependent protein kinase
- MRP1:
-
Multidrug resistance-associated protein 1
- FOXOs:
-
Forkhead transcription factors
- HSPs:
-
Heat shock proteins
- GMPs:
-
Granulocyte macrophage progenitors
- Hh:
-
Hedgehog
- PTCH:
-
Patched
- TICs:
-
Tumor-initiating cells
- TKIs:
-
Tyrosine kinase inhibitors
References
S.J. Szilvassy, The biology of hematopoietic stem cells. Arch. Med. Res. 34, 446–460 (2003). doi:10.1016/j.arcmed.2003.06.004
A.W. Wognum, A.C. Eaves, T.E. Thomas, Identification and isolation of hematopoietic stem cells. Arch. Med. Res. 34, 461–475 (2003). doi:10.1016/j.arcmed.2003.09.008
D.C. Taussig, F. Miraki-Moud, F. Anjos-Afonso, D.J. Pearce, K. Allen, C. Ridler, D. Lillington, H. Oakervee, J. Cavenagh, S.G. Agrawal, T.A. Lister, J.G. Gribben, D. Bonnet, Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood 112, 568–575 (2008). doi:10.1182/blood-2007-10-118331
D.C. Taussig, J. Vargaftig, F. Miraki-Moud, E. Griessinger, K. Sharrock, T. Luke, D. Lillington, H. Oakervee, J. Cavenagh, S.G. Agrawal, T.A. Lister, J.G. Gribben, D. Bonnet, Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(-) fraction. Blood 115, 1976–1984 (2010). doi:10.1182/blood-2009-02-206565
S. Aref, O. Salama, Y. Al-Tonbary, M. Fouda, A. Menessy, M. El-Sherbiny, L and E selectins in acute myeloid leukemia: expression, clinical relevance and relation to patient outcome. Hematology 7, 83–87 (2002). doi:10.1080/10245330290028579
A.J. Gentles, S.K. Plevritis, R. Majeti, A.A. Alizadeh, Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA 304, 2706–2715 (2010). doi:10.1001/jama.2010.1862
F. Ishikawa, S. Yoshida, Y. Saito, A. Hijikata, H. Kitamura, S. Tanaka, R. Nakamura, T. Tanaka, H. Tomiyama, N. Saito, M. Fukata, T. Miyamoto, B. Lyons, K. Ohshima, N. Uchida, S. Taniguchi, O. Ohara, K. Akashi, M. Harada, L.D. Shultz, Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat. Biotechnol. 25, 1315–1321 (2007). doi:10.1038/nbt1350
T. Holyoake, X. Jiang, C. Eaves, A. Eaves, Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood 94, 2056–2064 (1999)
C. Zhao, A. Chen, C.H. Jamieson, M. Fereshteh, A. Abrahamsson, J. Blum, H.Y. Kwon, J. Kim, J.P. Chute, D. Rizzieri, M. Munchhof, T. VanArsdale, P.A. Beachy, T. Reya, Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 458, 776–779 (2009). doi:10.1038/nature07737
M.L. Guzman, C.F. Swiderski, D.S. Howard, B.A. Grimes, R.M. Rossi, S.J. Szilvassy, C.T. Jordan, Preferential induction of apoptosis for primary human leukemic stem cells. Proc. Natl. Acad. Sci. U. S. A. 99, 16220–16225 (2002). doi:10.1073/pnas.252462599
C.T. Jordan, D. Upchurch, S.J. Szilvassy, M.L. Guzman, D.S. Howard, A.L. Pettigrew, T. Meyerrose, R. Rossi, B. Grimes, D.A. Rizzieri, S.M. Luger, G.L. Phillips, The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 14, 1777–1784 (2000)
N. Hosen, C.Y. Park, N. Tatsumi, Y. Oji, H. Sugiyama, M. Gramatzki, A.M. Krensky, I.L. Weissman, CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc. Natl. Acad. Sci. U. S. A. 104, 11008–11013 (2007). doi:10.1073/pnas.0704271104
R. Majeti, M.P. Chao, A.A. Alizadeh, W.W. Pang, S. Jaiswal, K.D. Gibbs Jr., N. van Rooijen, I.L. Weissman, CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 138, 286–299 (2009). doi:10.1016/j.cell.2009.05.045
Y. Kikushige, T. Shima, S. Takayanagi, S. Urata, T. Miyamoto, H. Iwasaki, K. Takenaka, T. Teshima, T. Tanaka, Y. Inagaki, K. Akashi, TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell 7, 708–717 (2010). doi:10.1016/j.stem.2010.11.014
M.P. Scolnik, R. Morilla, M.M. de Bracco, D. Catovsky, E. Matutes, CD34 and CD117 are overexpressed in AML and may be valuable to detect minimal residual disease. Leuk. Res. 26, 615–619 (2002)
A. Chavez-Gonzalez, E. Dorantes-Acosta, D. Moreno-Lorenzana, A. Alvarado-Moreno, L. Arriaga-Pizano, H. Mayani, Expression of CD90, CD96, CD117, and CD123 on different hematopoietic cell populations from pediatric patients with acute myeloid leukemia. Arch. Med. Res. 45, 343–350 (2014). doi:10.1016/j.arcmed.2014.04.001
J.M. Gerber, B.D. Smith, B. Ngwang, H. Zhang, M.S. Vala, L. Morsberger, S. Galkin, M.I. Collector, B. Perkins, M.J. Levis, C.A. Griffin, S.J. Sharkis, M.J. Borowitz, J.E. Karp, R.J. Jones, A clinically relevant population of leukemic CD34(+)CD38(-) cells in acute myeloid leukemia. Blood 119, 3571–3577 (2012). doi:10.1182/blood-2011-06-364182
M. Jaras, P. Johnels, N. Hansen, H. Agerstam, P. Tsapogas, M. Rissler, C. Lassen, T. Olofsson, O.W. Bjerrum, J. Richter, T. Fioretos, Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL-1 receptor accessory protein. Proc. Natl. Acad. Sci. U. S. A. 107, 16280–16285 (2010). doi:10.1073/pnas.1004408107
H. Herrmann, I. Sadovnik, S. Cerny-Reiterer, T. Rulicke, G. Stefanzl, M. Willmann, G. Hoermann, M. Bilban, K. Blatt, S. Herndlhofer, M. Mayerhofer, B. Streubel, W.R. Sperr, T.L. Holyoake, C. Mannhalter, P. Valent, Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood 123, 3951–3962 (2014). doi:10.1182/blood-2013-10-536078
M.E. Chamuleau, G.J. Ossenkoppele, A. van Rhenen, L. van Dreunen, S.M. Jirka, A. Zevenbergen, G.J. Schuurhuis, A.A. van de Loosdrecht, High TRAIL-R3 expression on leukemic blasts is associated with poor outcome and induces apoptosis-resistance which can be overcome by targeting TRAIL-R2. Leuk. Res. 35, 741–749 (2011). doi:10.1016/j.leukres.2010.12.032
M. Terwijn, N. Feller, A. van Rhenen, A. Kelder, G. Westra, S. Zweegman, G. Ossenkoppele, G.J. Schuurhuis, Interleukin-2 receptor alpha-chain (CD25) expression on leukaemic blasts is predictive for outcome and level of residual disease in AML. Eur. J. Cancer 45, 1692–1699 (2009). doi:10.1016/j.ejca.2009.02.021
M. Terwijn, W. Zeijlemaker, A. Kelder, A.P. Rutten, A.N. Snel, W.J. Scholten, T. Pabst, G. Verhoef, B. Lowenberg, S. Zweegman, G.J. Ossenkoppele, G.J. Schuurhuis, Leukemic stem cell frequency: a strong biomarker for clinical outcome in acute myeloid leukemia. PLoS One 9, e107587 (2014). doi:10.1371/journal.pone.0107587
A.V. Krivtsov, D. Twomey, Z. Feng, M.C. Stubbs, Y. Wang, J. Faber, J.E. Levine, J. Wang, W.C. Hahn, D.G. Gilliland, T.R. Golub, S.A. Armstrong, Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 442, 818–822 (2006). doi:10.1038/nature04980
L.I. Shlush, S. Zandi, A. Mitchell, W.C. Chen, J.M. Brandwein, V. Gupta, J.A. Kennedy, A.D. Schimmer, A.C. Schuh, K.W. Yee, J.L. McLeod, M. Doedens, J.J. Medeiros, R. Marke, H.J. Kim, K. Lee, J.D. McPherson, T.J. Hudson, A.M. Brown, F. Yousif, Q.M. Trinh, L.D. Stein, M.D. Minden, J.C. Wang, J.E. Dick, Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 506, 328–333 (2014). doi:10.1038/nature13038
M.R. Corces-Zimmerman, W.J. Hong, I.L. Weissman, B.C. Medeiros, R. Majeti, Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. U. S. A. 111, 2548–2553 (2014). doi:10.1073/pnas.1324297111
S. Babashah, Cancer stem cells: emerging concepts and future perspectives in translational oncology (Springer International Publishing, Switzerland, 2015)
P.R. Hamann, L.M. Hinman, I. Hollander, C.F. Beyer, D. Lindh, R. Holcomb, W. Hallett, H.R. Tsou, J. Upeslacis, D. Shochat, A. Mountain, D.A. Flowers, I. Bernstein, Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug. Chem. 13, 47–58 (2002)
A. Takeshita, Efficacy and resistance of gemtuzumab ozogamicin for acute myeloid leukemia. Int. J. Hematol. 97, 703–716 (2013). doi:10.1007/s12185-013-1365-1
A. Dutour, V. Marin, I. Pizzitola, S. Valsesia-Wittmann, D. Lee, E. Yvon, H. Finney, A. Lawson, M. Brenner, A. Biondi, E. Biagi, R. Rousseau, In vitro and in vivo antitumor effect of anti-CD33 chimeric receptor-expressing EBV-CTL against CD33 acute myeloid leukemia. Adv. Hematol. 2012, 683065 (2012). doi:10.1155/2012/683065
I. Pizzitola, V. Agostoni, E. Cribioli, M. Pule, R. Rousseau, H. Finney, A. Lawson, A. Biondi, E. Biagi, V. Marin, In vitro comparison of three different chimeric receptor-modified effector T-cell populations for leukemia cell therapy. J. Immunother. 34, 469–479 (2011). doi:10.1097/CJI.0b013e31821e763b
M. Feuring-Buske, A.E. Frankel, R.L. Alexander, B. Gerhard, D.E. Hogge, A diphtheria toxin-interleukin 3 fusion protein is cytotoxic to primitive acute myeloid leukemia progenitors but spares normal progenitors. Cancer Res. 62, 1730–1736 (2002)
H.P. Kim, A.E. Frankel, D.E. Hogge, A diphtheria toxin interleukin-3 fusion protein synergizes with tyrosine kinase inhibitors in killing leukemic progenitors from BCR/ABL positive acute leukemia. Leuk. Res. 34, 1035–1042 (2010). doi:10.1016/j.leukres.2009.12.008
L. Jin, E.M. Lee, H.S. Ramshaw, S.J. Busfield, A.G. Peoppl, L. Wilkinson, M.A. Guthridge, D. Thomas, E.F. Barry, A. Boyd, D.P. Gearing, G. Vairo, A.F. Lopez, J.E. Dick, R.B. Lock, Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell 5, 31–42 (2009). doi:10.1016/j.stem.2009.04.018
S.J. Busfield, M. Biondo, M. Wong, H.S. Ramshaw, E.M. Lee, S. Ghosh, H. Braley, C. Panousis, A.W. Roberts, S.Z. He, D. Thomas, L. Fabri, G. Vairo, R.B. Lock, A.F. Lopez, A.D. Nash, Targeting of acute myeloid leukemia in vitro and in vivo with an anti-CD123 mAb engineered for optimal ADCC. Leukemia 28, 2213–2221 (2014). doi:10.1038/leu.2014.128
I. Pizzitola, F. Anjos-Afonso, K. Rouault-Pierre, F. Lassailly, S. Tettamanti, O. Spinelli, A. Biondi, E. Biagi, D. Bonnet, Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia 28, 1596–1605 (2014). doi:10.1038/leu.2014.62
S. Gill, S.K. Tasian, M. Ruella, O. Shestova, Y. Li, D.L. Porter, M. Carroll, G. Danet-Desnoyers, J. Scholler, S.A. Grupp, C.H. June, M. Kalos, Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 123, 2343–2354 (2014). doi:10.1182/blood-2013-09-529537
Z. Gadhoum, J. Delaunay, E. Maquarre, L. Durand, V. Lancereaux, J. Qi, J. Robert-Lezenes, C. Chomienne, F. Smadja-Joffe, The effect of anti-CD44 monoclonal antibodies on differentiation and proliferation of human acute myeloid leukemia cells. Leuk. Lymphoma 45, 1501–1510 (2004). doi:10.1080/1042819042000206687
G. Song, X. Liao, L. Zhou, L. Wu, Y. Feng, Z.C. Han, HI44a, an anti-CD44 monoclonal antibody, induces differentiation and apoptosis of human acute myeloid leukemia cells. Leuk. Res. 28, 1089–1096 (2004). doi:10.1016/j.leukres.2004.02.005
L. Jin, K.J. Hope, Q. Zhai, F. Smadja-Joffe, J.E. Dick, Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med. 12, 1167–1174 (2006). doi:10.1038/nm1483
J.L. Berkowitz, J.E. Janik, D.M. Stewart, E.S. Jaffe, M. Stetler-Stevenson, J.H. Shih, T.A. Fleisher, M. Turner, N.E. Urquhart, G.H. Wharfe, W.D. Figg, C.J. Peer, C.K. Goldman, T.A. Waldmann, J.C. Morris, Safety, efficacy, and pharmacokinetics/pharmacodynamics of daclizumab (anti-CD25) in patients with adult T-cell leukemia/lymphoma. Clin. Immunol. 155, 176–187 (2014). doi:10.1016/j.clim.2014.09.012
J. Cerny, H. Yu, M. Ramanathan, G.D. Raffel, W.V. Walsh, N. Fortier, L. Shanahan, E. O’Rourke, J. Bednarik, B. Barton, A. Kroll-Desrosiers, S. Hao, B. Woda, L. Hutchinson, A.M. Evens, A.G. Rosmarin, R. Nath, Expression of CD25 independently predicts early treatment failure of acute myeloid leukaemia (AML). Br. J. Haematol. 160, 262–266 (2013). doi:10.1111/bjh.12109
G.J.R. J. N. Allan, E. J. Feldman, J. M. Scandura, E. K. Ritchie, L. Lam, W. Xie, H-T Hsu, D. C. Hassane, M. L. Guzman, In 56th ASH Annual Meeting and Exposition, (San Francisco, CA) (2014)
Z. Guo, A. Wang, W. Zhang, M. Levit, Q. Gao, C. Barberis, M. Tabart, J. Zhang, D. Hoffmann, D. Wiederschain, J. Rocnik, F. Sun, J. Murtie, C. Lengauer, S. Gross, B. Zhang, H. Cheng, V. Patel, L. Schio, F. Adrian, M. Dorsch, C. Garcia-Echeverria, S.M. Huang, PIM inhibitors target CD25-positive AML cells through concomitant suppression of STAT5 activation and degradation of MYC oncogene. Blood 124, 1777–1789 (2014). doi:10.1182/blood-2014-01-551234
X. Zhao, S. Singh, C. Pardoux, J. Zhao, E.D. Hsi, A. Abo, W. Korver, Targeting C-type lectin-like molecule-1 for antibody-mediated immunotherapy in acute myeloid leukemia. Haematologica 95, 71–78 (2010). doi:10.3324/haematol.2009.009811
H. Lu, Q. Zhou, V. Deshmukh, H. Phull, J. Ma, V. Tardif, R.R. Naik, C. Bouvard, Y. Zhang, S. Choi, B.R. Lawson, S. Zhu, C.H. Kim, P.G. Schultz, Targeting human C-type lectin-like molecule-1 (CLL1) with a bispecific antibody for immunotherapy of acute myeloid leukemia. Angew. Chem. Int. Ed. Engl. 53, 9841–9845 (2014). doi:10.1002/anie.201405353
M. Askmyr, H. Agerstam, N. Hansen, S. Gordon, A. Arvanitakis, M. Rissler, G. Juliusson, J. Richter, M. Jaras, T. Fioretos, Selective killing of candidate AML stem cells by antibody targeting of IL1RAP. Blood 121, 3709–3713 (2013). doi:10.1182/blood-2012-09-458935
D.E. Berardi, C. Flumian, P.B. Campodonico, A.J. Urtreger, M.I. Diaz Bessone, A.N. Motter, E.D. Bal de Kier Joffe, E.F. Farias, L.B. Todaro, Myoepithelial and luminal breast cancer cells exhibit different responses to all-trans retinoic acid. Cell. Oncol. 38, 289–305 (2015). doi:10.1007/s13402-015-0230-z
A. Kakizuka, W.H. Miller Jr., K. Umesono, R.P. Warrell Jr., S.R. Frankel, V.V. Murty, E. Dmitrovsky, R.M. Evans, Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell 66, 663–674 (1991)
P.V. Sanchez, S.T. Glantz, S. Scotland, M.T. Kasner, M. Carroll, Induced differentiation of acute myeloid leukemia cells by activation of retinoid X and liver X receptors. Leukemia 28, 749–760 (2014). doi:10.1038/leu.2013.202
S. Grant, ATRA and ATO team up against NPM1. Blood 125, 3369–3371 (2015). doi:10.1182/blood-2015-04-636217
H. El Hajj, Z. Dassouki, C. Berthier, E. Raffoux, L. Ades, O. Legrand, R. Hleihel, U. Sahin, N. Tawil, A. Salameh, K. Zibara, N. Darwiche, M. Mohty, H. Dombret, P. Fenaux, H. de The, A. Bazarbachi, Retinoic acid and arsenic trioxide trigger degradation of mutated NPM1, resulting in apoptosis of AML cells. Blood 125, 3447–3454 (2015). doi:10.1182/blood-2014-11-612416
X. Zheng, A. Seshire, B. Ruster, G. Bug, T. Beissert, E. Puccetti, D. Hoelzer, R. Henschler, M. Ruthardt, Arsenic but not all-trans retinoic acid overcomes the aberrant stem cell capacity of PML/RARalpha-positive leukemic stem cells. Haematologica 92, 323–331 (2007)
Y. Jiang, Z.H. Xue, W.Z. Shen, K.M. Du, H. Yan, Y. Yu, Z.G. Peng, M.G. Song, J.H. Tong, Z. Chen, Y. Huang, M. Lubbert, G.Q. Chen, Desferrioxamine induces leukemic cell differentiation potentially by hypoxia-inducible factor-1 alpha that augments transcriptional activity of CCAAT/enhancer-binding protein-alpha. Leukemia 19, 1239–1247 (2005). doi:10.1038/sj.leu.2403734
M. Roth, B. Will, G. Simkin, S. Narayanagari, L. Barreyro, B. Bartholdy, R. Tamari, C.S. Mitsiades, A. Verma, U. Steidl, Eltrombopag inhibits the proliferation of leukemia cells via reduction of intracellular iron and induction of differentiation. Blood 120, 386–394 (2012). doi:10.1182/blood-2011-12-399667
M. Szwed, A. Laroche-Clary, J. Robert, Z. Jozwiak, Efficacy of doxorubicin-transferrin conjugate in apoptosis induction in human leukemia cells through reactive oxygen species generation. Cell. Oncol. 39, 107–118 (2016). doi:10.1007/s13402-015-0256-2
J. Liu, X. Wei, Y. Wu, Y. Wang, Y. Qiu, J. Shi, H. Zhou, Z. Lu, M. Shao, L. Yu, L. Tong, Giganteaside D induces ROS-mediated apoptosis in human hepatocellular carcinoma cells through the MAPK pathway. Cell. Oncol. 39, 333–342 (2016). doi:10.1007/s13402-016-0273-9
T.T. Vo, J. Ryan, R. Carrasco, D. Neuberg, D.J. Rossi, R.M. Stone, D.J. Deangelo, M.G. Frattini, A. Letai, Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 151, 344–355 (2012). doi:10.1016/j.cell.2012.08.038
D.J. Goff, A. Court Recart, A. Sadarangani, H.J. Chun, C.L. Barrett, M. Krajewska, H. Leu, J. Low-Marchelli, W. Ma, A.Y. Shih, J. Wei, D. Zhai, I. Geron, M. Pu, L. Bao, R. Chuang, L. Balaian, J. Gotlib, M. Minden, G. Martinelli, J. Rusert, K.H. Dao, K. Shazand, P. Wentworth, K.M. Smith, C.A. Jamieson, S.R. Morris, K. Messer, L.S. Goldstein, T.J. Hudson, M. Marra, K.A. Frazer, M. Pellecchia, J.C. Reed, C.H. Jamieson, A pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell 12, 316–328 (2013). doi:10.1016/j.stem.2012.12.011
A.S. Corbin, A. Agarwal, M. Loriaux, J. Cortes, M.W. Deininger, B.J. Druker, Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Invest. 121, 396–409 (2011). doi:10.1172/JCI35721
M. Konopleva, M. Milella, P. Ruvolo, J.C. Watts, M.R. Ricciardi, B. Korchin, T. McQueen, W. Bornmann, T. Tsao, P. Bergamo, D.H. Mak, W. Chen, J. McCubrey, A. Tafuri, M. Andreeff, MEK inhibition enhances ABT-737-induced leukemia cell apoptosis via prevention of ERK-activated MCL-1 induction and modulation of MCL-1/BIM complex. Leukemia 26, 778–787 (2012). doi:10.1038/leu.2011.287
M. Rahmani, M.M. Aust, E. Attkisson, D.C. Williams Jr., A. Ferreira-Gonzalez, S. Grant, Inhibition of Bcl-2 antiapoptotic members by obatoclax potently enhances sorafenib-induced apoptosis in human myeloid leukemia cells through a Bim-dependent process. Blood 119, 6089–6098 (2012). doi:10.1182/blood-2011-09-378141
M. Skrtic, S. Sriskanthadevan, B. Jhas, M. Gebbia, X. Wang, Z. Wang, R. Hurren, Y. Jitkova, M. Gronda, N. Maclean, C.K. Lai, Y. Eberhard, J. Bartoszko, P. Spagnuolo, A.C. Rutledge, A. Datti, T. Ketela, J. Moffat, B.H. Robinson, J.H. Cameron, J. Wrana, C.J. Eaves, M.D. Minden, J.C. Wang, J.E. Dick, K. Humphries, C. Nislow, G. Giaever, A.D. Schimmer, Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 20, 674–688 (2011). doi:10.1016/j.ccr.2011.10.015
Y. Jitkova, M. Gronda, R. Hurren, X. Wang, C.A. Goard, B. Jhas, A.D. Schimmer, A novel formulation of tigecycline has enhanced stability and sustained antibacterial and antileukemic activity. PLoS One 9, e95281 (2014). doi:10.1371/journal.pone.0095281
K. Goetze, C.G. Fabian, A. Siebers, L. Binz, D. Faber, S. Indraccolo, G. Nardo, U.G. Sattler, W. Mueller-Klieser, Manipulation of tumor metabolism for therapeutic approaches: ovarian cancer-derived cell lines as a model system. Cell. Oncol. (Dordr) 38, 377–385 (2015). doi:10.1007/s13402-015-0237-5
Y.H. Wang, W.J. Israelsen, D. Lee, V.W. Yu, N.T. Jeanson, C.B. Clish, L.C. Cantley, M.G. Vander Heiden, D.T. Scadden, Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 158, 1309–1323 (2014). doi:10.1016/j.cell.2014.07.048
D. Anastasiou, Y. Yu, W.J. Israelsen, J.K. Jiang, M.B. Boxer, B.S. Hong, W. Tempel, S. Dimov, M. Shen, A. Jha, H. Yang, K.R. Mattaini, C.M. Metallo, B.P. Fiske, K.D. Courtney, S. Malstrom, T.M. Khan, C. Kung, A.P. Skoumbourdis, H. Veith, N. Southall, M.J. Walsh, K.R. Brimacombe, W. Leister, S.Y. Lunt, Z.R. Johnson, K.E. Yen, K. Kunii, S.M. Davidson, H.R. Christofk, C.P. Austin, J. Inglese, M.H. Harris, J.M. Asara, G. Stephanopoulos, F.G. Salituro, S. Jin, L. Dang, D.S. Auld, H.W. Park, L.C. Cantley, C.J. Thomas, M.G. Vander Heiden, Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol. 8, 839–847 (2012). doi:10.1038/nchembio.1060
K.M. Parnell, J.M. Foulks, R.N. Nix, A. Clifford, J. Bullough, B. Luo, A. Senina, D. Vollmer, J. Liu, V. McCarthy, Y. Xu, M. Saunders, X.H. Liu, S. Pearce, K. Wright, M. O’Reilly, M.V. McCullar, K.K. Ho, S.B. Kanner, Pharmacologic activation of PKM2 slows lung tumor xenograft growth. Mol. Cancer Ther. 12, 1453–1460 (2013). doi:10.1158/1535-7163.MCT-13-0026
T. Hitosugi, S. Kang, M.G. Vander Heiden, T.W. Chung, S. Elf, K. Lythgoe, S. Dong, S. Lonial, X. Wang, G.Z. Chen, J. Xie, T.L. Gu, R.D. Polakiewicz, J.L. Roesel, T.J. Boggon, F.R. Khuri, D.G. Gilliland, L.C. Cantley, J. Kaufman, J. Chen, Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci. Signal. 2, ra73 (2009). doi:10.1126/scisignal.2000431
E.M. Sturgill, M.L. Guzman, In Proceedings of the 105th Annual Meeting of the American Association for Cancer Research, (San Diego, CA) (2014)
A. Ferraro, Altered primary chromatin structures and their implications in cancer development. Cell. Oncol. 39, 195–210 (2016). doi:10.1007/s13402-016-0276-6
J. Yamazaki, M.R. Estecio, Y. Lu, H. Long, G.G. Malouf, D. Graber, Y. Huo, L. Ramagli, S. Liang, S.M. Kornblau, J. Jelinek, J.P. Issa, The epigenome of AML stem and progenitor cells. Epigenetics 8, 92–104 (2013). doi:10.4161/epi.23243
E.D. Lagadinou, A. Sach, K. Callahan, R.M. Rossi, S.J. Neering, M. Minhajuddin, J.M. Ashton, S. Pei, V. Grose, K.M. O’Dwyer, J.L. Liesveld, P.S. Brookes, M.W. Becker, C.T. Jordan, BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 12, 329–341 (2013). doi:10.1016/j.stem.2012.12.013
C. Craddock, L. Quek, N. Goardon, S. Freeman, S. Siddique, M. Raghavan, A. Aztberger, A. Schuh, D. Grimwade, A. Ivey, P. Virgo, R. Hills, T. McSkeane, J. Arrazi, S. Knapper, C. Brookes, B. Davies, A. Price, K. Wall, M. Griffiths, J. Cavenagh, R. Majeti, I. Weissman, A. Burnett, P. Vyas, Azacitidine fails to eradicate leukemic stem/progenitor cell populations in patients with acute myeloid leukemia and myelodysplasia. Leukemia 27, 1028–1036 (2013). doi:10.1038/leu.2012.312
M.J. Chang, H. Wu, N.J. Achille, M.R. Reisenauer, C.W. Chou, N.J. Zeleznik-Le, C.S. Hemenway, W. Zhang, Histone H3 lysine 79 methyltransferase Dot1 is required for immortalization by MLL oncogenes. Cancer Res. 70, 10234–10242 (2010). doi:10.1158/0008-5472.CAN-10-3294
P. Jo, K. Jung, M. Grade, L.C. Conradi, H.A. Wolff, J. Kitz, H. Becker, J. Ruschoff, A. Hartmann, T. Beissbarth, A. Muller-Dornieden, M. Ghadimi, R. Schneider-Stock, J. Gaedcke, CpG island methylator phenotype infers a poor disease-free survival in locally advanced rectal cancer. Surgery 151, 564–570 (2012). doi:10.1016/j.surg.2011.08.013
K.M. Bernt, N. Zhu, A.U. Sinha, S. Vempati, J. Faber, A.V. Krivtsov, Z. Feng, N. Punt, A. Daigle, L. Bullinger, R.M. Pollock, V.M. Richon, A.L. Kung, S.A. Armstrong, MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 20, 66–78 (2011). doi:10.1016/j.ccr.2011.06.010
T. Schenk, W.C. Chen, S. Gollner, L. Howell, L. Jin, K. Hebestreit, H.U. Klein, A.C. Popescu, A. Burnett, K. Mills, R.A. Casero Jr., L. Marton, P. Woster, M.D. Minden, M. Dugas, J.C. Wang, J.E. Dick, C. Muller-Tidow, K. Petrie, A. Zelent, Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 18, 605–611 (2012). doi:10.1038/nm.2661
S.R. Daigle, E.J. Olhava, C.A. Therkelsen, C.R. Majer, C.J. Sneeringer, J. Song, L.D. Johnston, M.P. Scott, J.J. Smith, Y. Xiao, L. Jin, K.W. Kuntz, R. Chesworth, M.P. Moyer, K.M. Bernt, J.C. Tseng, A.L. Kung, S.A. Armstrong, R.A. Copeland, V.M. Richon, R.M. Pollock, Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20, 53–65 (2011). doi:10.1016/j.ccr.2011.06.009
I. Maillard, J.L. Hess, The role of menin in hematopoiesis. Adv. Exp. Med. Biol. 668, 51–57 (2009)
D.P. Mould, A.E. McGonagle, D.H. Wiseman, E.L. Williams, A.M. Jordan, Reversible inhibitors of LSD1 as therapeutic agents in acute myeloid leukemia: clinical significance and progress to date. Med. Res. Rev. 35, 586–618 (2014). doi:10.1002/med.21334
W. Fiskus, Y. Wang, A. Sreekumar, K.M. Buckley, H. Shi, A. Jillella, C. Ustun, R. Rao, P. Fernandez, J. Chen, R. Balusu, S. Koul, P. Atadja, V.E. Marquez, K.N. Bhalla, Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood 114, 2733–2743 (2009). doi:10.1182/blood-2009-03-213496
J. Zhou, C. Bi, L.L. Cheong, S. Mahara, S.C. Liu, K.G. Tay, T.L. Koh, Q. Yu, W.J. Chng, The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood 118, 2830–2839 (2011). doi:10.1182/blood-2010-07-294827
A. Chaturvedi, M.M. Araujo Cruz, N. Jyotsana, A. Sharma, H. Yun, K. Gorlich, M. Wichmann, A. Schwarzer, M. Preller, F. Thol, J. Meyer, R. Haemmerle, E.A. Struys, E.E. Jansen, U. Modlich, Z. Li, L.M. Sly, R. Geffers, R. Lindner, D.J. Manstein, U. Lehmann, J. Krauter, A. Ganser, M. Heuser, Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 122, 2877–2887 (2013). doi:10.1182/blood-2013-03-491571
M.E. Figueroa, S. Lugthart, Y. Li, C. Erpelinck-Verschueren, X. Deng, P.J. Christos, E. Schifano, J. Booth, W. van Putten, L. Skrabanek, F. Campagne, M. Mazumdar, J.M. Greally, P.J. Valk, B. Lowenberg, R. Delwel, A. Melnick, DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 17, 13–27 (2010). doi:10.1016/j.ccr.2009.11.020
O. Abdel-Wahab, R.L. Levine, Metabolism and the leukemic stem cell. J. Exp. Med. 207, 677–680 (2010). doi:10.1084/jem.20100523
C. Frelin, V. Imbert, E. Griessinger, A.C. Peyron, N. Rochet, P. Philip, C. Dageville, A. Sirvent, M. Hummelsberger, E. Berard, M. Dreano, N. Sirvent, J.F. Peyron, Targeting NF-kappaB activation via pharmacologic inhibition of IKK2-induced apoptosis of human acute myeloid leukemia cells. Blood 105, 804–811 (2005). doi:10.1182/blood-2004-04-1463
K. Vazquez-Santillan, J. Melendez-Zajgla, L. Jimenez-Hernandez, G. Martinez-Ruiz, V. Maldonado, NF-kappaB signaling in cancer stem cells: a promising therapeutic target? Cell. Oncol. 38, 327–339 (2015). doi:10.1007/s13402-015-0236-6
M.L. Guzman, S.J. Neering, D. Upchurch, B. Grimes, D.S. Howard, D.A. Rizzieri, S.M. Luger, C.T. Jordan, Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 98, 2301–2307 (2001)
Y. Kagoya, A. Yoshimi, K. Kataoka, M. Nakagawa, K. Kumano, S. Arai, H. Kobayashi, T. Saito, Y. Iwakura, M. Kurokawa, Positive feedback between NF-kappaB and TNF-alpha promotes leukemia-initiating cell capacity. J. Clin. Invest. 124, 528–542 (2014). doi:10.1172/JCI68101
M.L. Guzman, R.M. Rossi, L. Karnischky, X. Li, D.R. Peterson, D.S. Howard, C.T. Jordan, The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood 105, 4163–4169 (2005). doi:10.1182/blood-2004-10-4135
P. Diamanti, C.V. Cox, J.P. Moppett, A. Blair, Parthenolide eliminates leukemia-initiating cell populations and improves survival in xenografts of childhood acute lymphoblastic leukemia. Blood 121, 1384–1393 (2013). doi:10.1182/blood-2012-08-448852
M.L. Guzman, R.M. Rossi, S. Neelakantan, X. Li, C.A. Corbett, D.C. Hassane, M.W. Becker, J.M. Bennett, E. Sullivan, J.L. Lachowicz, A. Vaughan, C.J. Sweeney, W. Matthews, M. Carroll, J.L. Liesveld, P.A. Crooks, C.T. Jordan, An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood 110, 4427–4435 (2007). doi:10.1182/blood-2007-05-090621
D.C. Hassane, M.L. Guzman, C. Corbett, X. Li, R. Abboud, F. Young, J.L. Liesveld, M. Carroll, C.T. Jordan, Discovery of agents that eradicate leukemia stem cells using an in silico screen of public gene expression data. Blood 111, 5654–5662 (2008). doi:10.1182/blood-2007-11-126003
M.L. Guzman, N. Yang, K.K. Sharma, M. Balys, C.A. Corbett, C.T. Jordan, M.W. Becker, U. Steidl, O. Abdel-Wahab, R.L. Levine, G. Marcucci, G.J. Roboz, D.C. Hassane, Selective activity of the histone deacetylase inhibitor AR-42 against leukemia stem cells: a novel potential strategy in acute myelogenous leukemia. Mol. Cancer Ther. 13, 1979–1990 (2014). doi:10.1158/1535-7163.MCT-13-0963
N. Lounnas, C. Frelin, N. Gonthier, P. Colosetti, A. Sirvent, J.P. Cassuto, F. Berthier, N. Sirvent, P. Rousselot, M. Dreano, J.F. Peyron, V. Imbert, NF-kappaB inhibition triggers death of imatinib-sensitive and imatinib-resistant chronic myeloid leukemia cells including T315I Bcr-Abl mutants. Int. J. Cancer 125, 308–317 (2009). doi:10.1002/ijc.24294
S.K. Tasian, D.T. Teachey, S.R. Rheingold, Targeting the PI3K/mTOR Pathway in Pediatric Hematologic Malignancies. Front. Oncol. 4, 108 (2014). doi:10.3389/fonc.2014.00108
T. Skorski, P. Kanakaraj, M. Nieborowska-Skorska, M.Z. Ratajczak, S.C. Wen, G. Zon, A.M. Gewirtz, B. Perussia, B. Calabretta, Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromosome-positive cells. Blood 86, 726–736 (1995)
C. Billottet, V.L. Grandage, R.E. Gale, A. Quattropani, C. Rommel, B. Vanhaesebroeck, A. Khwaja, A selective inhibitor of the p110delta isoform of PI 3-kinase inhibits AML cell proliferation and survival and increases the cytotoxic effects of VP16. Oncogene 25, 6648–6659 (2006). doi:10.1038/sj.onc.1209670
A.M. Martelli, C. Evangelisti, F. Chiarini, J.A. McCubrey, The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget 1, 89–103 (2010)
P.L. Tazzari, A. Cappellini, F. Ricci, C. Evangelisti, V. Papa, T. Grafone, G. Martinelli, R. Conte, L. Cocco, J.A. McCubrey, A.M. Martelli, Multidrug resistance-associated protein 1 expression is under the control of the phosphoinositide 3 kinase/Akt signal transduction network in human acute myelogenous leukemia blasts. Leukemia 21, 427–438 (2007). doi:10.1038/sj.leu.2404523
E. Pastwa, T. Poplawski, U. Lewandowska, S.B. Somiari, J. Blasiak, R.I. Somiari, Wortmannin potentiates the combined effect of etoposide and cisplatin in human glioma cells. Int. J. Biochem. Cell Biol. 53, 423–431 (2014). doi:10.1016/j.biocel.2014.06.007
V. Papa, P.L. Tazzari, F. Chiarini, A. Cappellini, F. Ricci, A.M. Billi, C. Evangelisti, E. Ottaviani, G. Martinelli, N. Testoni, J.A. McCubrey, A.M. Martelli, Proapoptotic activity and chemosensitizing effect of the novel Akt inhibitor perifosine in acute myelogenous leukemia cells. Leukemia 22, 147–160 (2008). doi:10.1038/sj.leu.2404980
Y. Tong, Y.Y. Liu, L.S. You, W.B. Qian, Perifosine induces protective autophagy and upregulation of ATG5 in human chronic myelogenous leukemia cells in vitro. Acta Pharmacol. Sin. 33, 542–550 (2012). doi:10.1038/aps.2011.192
C. Recher, O. Beyne-Rauzy, C. Demur, G. Chicanne, C. Dos Santos, V.M. Mas, D. Benzaquen, G. Laurent, F. Huguet, B. Payrastre, Antileukemic activity of rapamycin in acute myeloid leukemia. Blood 105, 2527–2534 (2005). doi:10.1182/blood-2004-06-2494
Q. Xu, J.E. Thompson, M. Carroll, mTOR regulates cell survival after etoposide treatment in primary AML cells. Blood 106, 4261–4268 (2005). doi:10.1182/blood-2004-11-4468
F. Chiarini, A. Lonetti, G. Teti, E. Orsini, D. Bressanin, A. Cappellini, F. Ricci, P.L. Tazzari, A. Ognibene, M. Falconi, P. Pagliaro, I. Iacobucci, G. Martinelli, S. Amadori, J.A. McCubrey, A.M. Martelli, A combination of temsirolimus, an allosteric mTOR inhibitor, with clofarabine as a new therapeutic option for patients with acute myeloid leukemia. Oncotarget 3, 1615–1628 (2012)
D.C. Hassane, S. Sen, M. Minhajuddin, R.M. Rossi, C.A. Corbett, M. Balys, L. Wei, P.A. Crooks, M.L. Guzman, C.T. Jordan, Chemical genomic screening reveals synergism between parthenolide and inhibitors of the PI-3 kinase and mTOR pathways. Blood 116, 5983–5990 (2010). doi:10.1182/blood-2010-04-278044
K. Meja, C. Stengel, R. Sellar, D. Huszar, B.R. Davies, R.E. Gale, D.C. Linch, A. Khwaja, PIM and AKT kinase inhibitors show synergistic cytotoxicity in acute myeloid leukaemia that is associated with convergence on mTOR and MCL1 pathways. Br. J. Haematol. 167, 69–79 (2014). doi:10.1111/bjh.13013
F. Pellicano, M.T. Scott, G.V. Helgason, L.E. Hopcroft, E.K. Allan, M. Aspinall-O’Dea, M. Copland, A. Pierce, B.J. Huntly, A.D. Whetton, T.L. Holyoake, The antiproliferative activity of kinase inhibitors in chronic myeloid leukemia cells is mediated by FOXO transcription factors. Stem Cells 32, 2324–2337 (2014). doi:10.1002/stem.1748
N. Chapuis, J. Tamburini, A.S. Green, C. Vignon, V. Bardet, A. Neyret, M. Pannetier, L. Willems, S. Park, A. Macone, S.M. Maira, N. Ifrah, F. Dreyfus, O. Herault, C. Lacombe, P. Mayeux, D. Bouscary, Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as a new therapeutic strategy for acute myeloid leukemia. Clin. Cancer Res. 16, 5424–5435 (2010). doi:10.1158/1078-0432.CCR-10-1102
X. Ding, F. Li, J. McKnight, C. Schmidt, K. Strooisma, H. Shimizu, K. Faber, J.A. Ware, B. Dean, A supported liquid extraction-LC-MS/MS method for determination of GDC-0980 (Apitolisib), a dual small-molecule inhibitor of class 1A phosphoinositide 3-kinase and mammalian target of rapamycin, in human plasma. J. Pharm. Biomed. Anal. 100, 150–156 (2014). doi:10.1016/j.jpba.2014.08.001
P. Yu, A.D. Laird, X. Du, J. Wu, K.A. Won, K. Yamaguchi, P.P. Hsu, F. Qian, C.T. Jaeger, W. Zhang, C.A. Buhr, P. Shen, W. Abulafia, J. Chen, J. Young, A. Plonowski, F.M. Yakes, F. Chu, M. Lee, F. Bentzien, S.T. Lam, S. Dale, D.J. Matthews, P. Lamb, P. Foster, Characterization of the activity of the PI3K/mTOR inhibitor XL765 (SAR245409) in tumor models with diverse genetic alterations affecting the PI3K pathway. Mol. Cancer Ther. 13, 1078–1091 (2014). doi:10.1158/1535-7163.MCT-13-0709
Z. Tothova, D.G. Gilliland, FoxO transcription factors and stem cell homeostasis: insights from the hematopoietic system. Cell Stem Cell 1, 140–152 (2007). doi:10.1016/j.stem.2007.07.017
K. Ito, A. Hirao, F. Arai, K. Takubo, S. Matsuoka, K. Miyamoto, M. Ohmura, K. Naka, K. Hosokawa, Y. Ikeda, T. Suda, Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 12, 446–451 (2006). doi:10.1038/nm1388
F. Zhou, Q. Shen, F.X. Claret, Novel roles of reactive oxygen species in the pathogenesis of acute myeloid leukemia. J. Leukoc. Biol. 94, 423–429 (2013). doi:10.1189/jlb.0113006
M. Nieborowska-Skorska, P.K. Kopinski, R. Ray, G. Hoser, D. Ngaba, S. Flis, K. Cramer, M.M. Reddy, M. Koptyra, T. Penserga, E. Glodkowska-Mrowka, E. Bolton, T.L. Holyoake, C.J. Eaves, S. Cerny-Reiterer, P. Valent, A. Hochhaus, T.P. Hughes, H. van der Kuip, M. Sattler, W. Wiktor-Jedrzejczak, C. Richardson, A. Dorrance, T. Stoklosa, D.A. Williams, T. Skorski, Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood 119, 4253–4263 (2012). doi:10.1182/blood-2011-10-385658
S. Pei, M. Minhajuddin, K.P. Callahan, M. Balys, J.M. Ashton, S.J. Neering, E.D. Lagadinou, C. Corbett, H. Ye, J.L. Liesveld, K.M. O’Dwyer, Z. Li, L. Shi, P. Greninger, J. Settleman, C. Benes, F.K. Hagen, J. Munger, P.A. Crooks, M.W. Becker, C.T. Jordan, Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J. Biol. Chem. 288, 33542–33558 (2013). doi:10.1074/jbc.M113.511170
X. Chen, X. Shi, C. Zhao, X. Li, X. Lan, S. Liu, H. Huang, N. Liu, S. Liao, D. Zang, W. Song, Q. Liu, B.Z. Carter, Q.P. Dou, X. Wang, J. Liu, Anti-rheumatic agent auranofin induced apoptosis in chronic myeloid leukemia cells resistant to imatinib through both Bcr/Abl-dependent and -independent mechanisms. Oncotarget 5, 9118–9132 (2014)
Y. Du, Y. Xia, X. Pan, Z. Chen, A. Wang, K. Wang, J. Li, J. Zhang, Fenretinide targets chronic myeloid leukemia stem/progenitor cells by regulation of redox signaling. Antioxid. Redox Signal. 20, 1866–1880 (2014). doi:10.1089/ars.2012.4935
K. Moulick, J.H. Ahn, H. Zong, A. Rodina, L. Cerchietti, E.M. Gomes DaGama, E. Caldas-Lopes, K. Beebe, F. Perna, K. Hatzi, L.P. Vu, X. Zhao, D. Zatorska, T. Taldone, P. Smith-Jones, M. Alpaugh, S.S. Gross, N. Pillarsetty, T. Ku, J.S. Lewis, S.M. Larson, R. Levine, H. Erdjument-Bromage, M.L. Guzman, S.D. Nimer, A. Melnick, L. Neckers, G. Chiosis, Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat. Chem. Biol. 7, 818–826 (2011). doi:10.1038/nchembio.670
P. Flandrin, D. Guyotat, A. Duval, J. Cornillon, E. Tavernier, N. Nadal, L. Campos, Significance of heat-shock protein (HSP) 90 expression in acute myeloid leukemia cells. Cell Stress Chaperones 13, 357–364 (2008). doi:10.1007/s12192-008-0035-3
H. Reikvam, K.J. Hatfield, E. Ersvaer, R. Hovland, J. Skavland, B.T. Gjertsen, K. Petersen, O. Bruserud, Expression profile of heat shock proteins in acute myeloid leukaemia patients reveals a distinct signature strongly associated with FLT3 mutation status--consequences and potentials for pharmacological intervention. Br. J. Haematol. 156, 468–480 (2012). doi:10.1111/j.1365-2141.2011.08960.x
M. Zackova, D. Mouckova, T. Lopotova, Z. Ondrackova, H. Klamova, J. Moravcova, Hsp90 - a potential prognostic marker in CML. Blood Cells Mol. Dis. 50, 184–189 (2013). doi:10.1016/j.bcmd.2012.11.002
W. Yu, Q. Rao, M. Wang, Z. Tian, D. Lin, X. Liu, J. Wang, The Hsp90 inhibitor 17-allylamide-17-demethoxygeldanamycin induces apoptosis and differentiation of Kasumi-1 harboring the Asn822Lys KIT mutation and down-regulates KIT protein level. Leuk. Res. 30, 575–582 (2006). doi:10.1016/j.leukres.2005.08.028
J.E. Lancet, I. Gojo, M. Burton, M. Quinn, S.M. Tighe, K. Kersey, Z. Zhong, M.X. Albitar, K. Bhalla, A.L. Hannah, M.R. Baer, Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia 24, 699–705 (2010). doi:10.1038/leu.2009.292
C. Gallerne, A. Prola, C. Lemaire, Hsp90 inhibition by PU-H71 induces apoptosis through endoplasmic reticulum stress and mitochondrial pathway in cancer cells and overcomes the resistance conferred by Bcl-2. Biochim. Biophys. Acta 1833, 1356–1366 (2013). doi:10.1016/j.bbamcr.2013.02.014
S.R. Ambati, E.C. Lopes, K. Kosugi, U. Mony, A. Zehir, S.K. Shah, T. Taldone, A.L. Moreira, P.A. Meyers, G. Chiosis, M.A. Moore, Pre-clinical efficacy of PU-H71, a novel HSP90 inhibitor, alone and in combination with bortezomib in Ewing sarcoma. Mol. Oncol. 8, 323–336 (2014). doi:10.1016/j.molonc.2013.12.005
H. Zong, A. Gozman, E. Caldas-Lopes, T. Taldone, E. Sturgill, S. Brennan, S.O. Ochiana, E.M. Gomes-DaGama, S. Sen, A. Rodina, J. Koren 3rd, M.W. Becker, C.M. Rudin, A. Melnick, R.L. Levine, G.J. Roboz, S.D. Nimer, G. Chiosis, M.L. Guzman, A hyperactive signalosome in acute myeloid leukemia drives addiction to a tumor-specific Hsp90 species. Cell Rep. 13, 2159–2173 (2015). doi:10.1016/j.celrep.2015.10.073
C. Hurtz, K. Hatzi, L. Cerchietti, M. Braig, E. Park, Y.M. Kim, S. Herzog, P. Ramezani-Rad, H. Jumaa, M.C. Muller, W.K. Hofmann, A. Hochhaus, B.H. Ye, A. Agarwal, B.J. Druker, N.P. Shah, A.M. Melnick, M. Muschen, BCL6-mediated repression of p53 is critical for leukemia stem cell survival in chronic myeloid leukemia. J. Exp. Med. 208, 2163–2174 (2011). doi:10.1084/jem.20110304
J.M. Polo, T. Dell’Oso, S.M. Ranuncolo, L. Cerchietti, D. Beck, G.F. Da Silva, G.G. Prive, J.D. Licht, A. Melnick, Specific peptide interference reveals BCL6 transcriptional and oncogenic mechanisms in B-cell lymphoma cells. Nat. Med. 10, 1329–1335 (2004). doi:10.1038/nm1134
C. Duy, J.J. Yu, R. Nahar, S. Swaminathan, S.M. Kweon, J.M. Polo, E. Valls, L. Klemm, S. Shojaee, L. Cerchietti, W. Schuh, H.M. Jack, C. Hurtz, P. Ramezani-Rad, S. Herzog, H. Jumaa, H.P. Koeffler, I.M. de Alboran, A.M. Melnick, B.H. Ye, M. Muschen, BCL6 is critical for the development of a diverse primary B cell repertoire. J. Exp. Med. 207, 1209–1221 (2010). doi:10.1084/jem.20091299
B. Sumithra, U. Saxena, A.B. Das, Alternative splicing within the Wnt signaling pathway: role in cancer development. Cell. Oncol. (Dordr) 39, 1–13 (2016). doi:10.1007/s13402-015-0266-0
A.E. Abrahamsson, I. Geron, J. Gotlib, K.H. Dao, C.F. Barroga, I.G. Newton, F.J. Giles, J. Durocher, R.S. Creusot, M. Karimi, C. Jones, J.L. Zehnder, A. Keating, R.S. Negrin, I.L. Weissman, C.H. Jamieson, Glycogen synthase kinase 3beta missplicing contributes to leukemia stem cell generation. Proc. Natl. Acad. Sci. U. S. A. 106, 3925–3929 (2009). doi:10.1073/pnas.0900189106
N.M. Ghahhari, S. Babashah, Interplay between microRNAs and WNT/beta-catenin signalling pathway regulates epithelial-mesenchymal transition in cancer. Eur. J. Cancer 51, 1638–1649 (2015). doi:10.1016/j.ejca.2015.04.021
J.P. Radich, H. Dai, M. Mao, V. Oehler, J. Schelter, B. Druker, C. Sawyers, N. Shah, W. Stock, C.L. Willman, S. Friend, P.S. Linsley, Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc. Natl. Acad. Sci. U. S. A. 103, 2794–2799 (2006). doi:10.1073/pnas.0510423103
A.M. Coluccia, A. Vacca, M. Dunach, L. Mologni, S. Redaelli, V.H. Bustos, D. Benati, L.A. Pinna, C. Gambacorti-Passerini, Bcr-Abl stabilizes beta-catenin in chronic myeloid leukemia through its tyrosine phosphorylation. EMBO J. 26, 1456–1466 (2007). doi:10.1038/sj.emboj.7601485
L. Wang, L.S. You, W.M. Ni, Q.L. Ma, Y. Tong, L.P. Mao, J.J. Qian, J. Jin, beta-Catenin and AKT are promising targets for combination therapy in acute myeloid leukemia. Leuk. Res. 37, 1329–1340 (2013). doi:10.1016/j.leukres.2013.06.023
A. Hamad, Z. Sahli, M. El Sabban, M. Mouteirik, R. Nasr, Emerging therapeutic strategies for targeting chronic myeloid leukemia stem cells. Stem Cells Int. 2013, 724360 (2013). doi:10.1155/2013/724360
J. Yeung, M.T. Esposito, A. Gandillet, B.B. Zeisig, E. Griessinger, D. Bonnet, C.W. So, beta-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 18, 606–618 (2010). doi:10.1016/j.ccr.2010.10.032
S. Song, T. Christova, S. Perusini, S. Alizadeh, R.Y. Bao, B.W. Miller, R. Hurren, Y. Jitkova, M. Gronda, M. Isaac, B. Joseph, R. Subramaniam, A. Aman, A. Chau, D.E. Hogge, S.J. Weir, J. Kasper, A.D. Schimmer, R. Al-awar, J.L. Wrana, L. Attisano, Wnt inhibitor screen reveals iron dependence of beta-catenin signaling in cancers. Cancer Res. 71, 7628–7639 (2011). doi:10.1158/0008-5472.CAN-11-2745
A. Gandillet, S. Park, F. Lassailly, E. Griessinger, J. Vargaftig, A. Filby, T.A. Lister, D. Bonnet, Heterogeneous sensitivity of human acute myeloid leukemia to beta-catenin down-modulation. Leukemia 25, 770–780 (2011). doi:10.1038/leu.2011.17
E.C. Hales, J.W. Taub, L.H. Matherly, New insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling axis: targeted therapy of gamma-secretase inhibitor resistant T-cell acute lymphoblastic leukemia. Cell. Signal. 26, 149–161 (2014). doi:10.1016/j.cellsig.2013.09.021
F. Damm, B. Markus, F. Thol, M. Morgan, G. Gohring, B. Schlegelberger, J. Krauter, M. Heuser, O.A. Bernard, A. Ganser, TET2 mutations in cytogenetically normal acute myeloid leukemia: clinical implications and evolutionary patterns. Genes Chromosomes Cancer 53, 824–832 (2014). doi:10.1002/gcc.22191
C. Lobry, P. Ntziachristos, D. Ndiaye-Lobry, P. Oh, L. Cimmino, N. Zhu, E. Araldi, W. Hu, J. Freund, O. Abdel-Wahab, S. Ibrahim, D. Skokos, S.A. Armstrong, R.L. Levine, C.Y. Park, I. Aifantis, Notch pathway activation targets AML-initiating cell homeostasis and differentiation. J. Exp. Med. 210, 301–319 (2013). doi:10.1084/jem.20121484
C. Dierks, R. Beigi, G.R. Guo, K. Zirlik, M.R. Stegert, P. Manley, C. Trussell, A. Schmitt-Graeff, K. Landwerlin, H. Veelken, M. Warmuth, Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 14, 238–249 (2008). doi:10.1016/j.ccr.2008.08.003
M.Y. Konopleva, C.T. Jordan, Leukemia stem cells and microenvironment: biology and therapeutic targeting. J. Clin. Oncol. 29, 591–599 (2011). doi:10.1200/JCO.2010.31.0904
L. Jin, Y. Tabe, S. Konoplev, Y. Xu, C.E. Leysath, H. Lu, S. Kimura, A. Ohsaka, M.B. Rios, L. Calvert, H. Kantarjian, M. Andreeff, M. Konopleva, CXCR4 up-regulation by imatinib induces chronic myelogenous leukemia (CML) cell migration to bone marrow stroma and promotes survival of quiescent CML cells. Mol. Cancer Ther. 7, 48–58 (2008). doi:10.1158/1535-7163.MCT-07-0042
L. Brault, A. Rovo, S. Decker, C. Dierks, A. Tzankov, J. Schwaller, CXCR4-SERINE339 regulates cellular adhesion, retention and mobilization, and is a marker for poor prognosis in acute myeloid leukemia. Leukemia 28, 566–576 (2014). doi:10.1038/leu.2013.201
E. Weisberg, A.K. Azab, P.W. Manley, A.L. Kung, A.L. Christie, R. Bronson, I.M. Ghobrial, J.D. Griffin, Inhibition of CXCR4 in CML cells disrupts their interaction with the bone marrow microenvironment and sensitizes them to nilotinib. Leukemia 26, 985–990 (2012). doi:10.1038/leu.2011.360
S. Tavor, I. Petit, S. Porozov, A. Avigdor, A. Dar, L. Leider-Trejo, N. Shemtov, V. Deutsch, E. Naparstek, A. Nagler, T. Lapidot, CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 64, 2817–2824 (2004)
Y. Wang, Y. Liu, S.N. Malek, P. Zheng, Y. Liu, Targeting HIF1alpha eliminates cancer stem cells in hematological malignancies. Cell Stem Cell 8, 399–411 (2011). doi:10.1016/j.stem.2011.02.006
M. Shahjahani, J. Mohammadiasl, F. Noroozi, M. Seghatoleslami, S. Shahrabi, F. Saba, N. Saki, Molecular basis of chronic lymphocytic leukemia diagnosis and prognosis. Cell. Oncol. 38, 93–109 (2015). doi:10.1007/s13402-014-0215-3
A. Chávez-González, S. Avilés-Vázquez, D. Moreno-Lorenzana, H. Mayani, in Stem cell biology in normal life and diseases, ed. by K. Alimoghaddam (InTech, 2013)
J.M. Goldman, How I treat chronic myeloid leukemia in the imatinib era. Blood 110, 2828–2837 (2007). doi:10.1182/blood-2007-04-038943
R. Bhatia, M. Holtz, N. Niu, R. Gray, D.S. Snyder, C.L. Sawyers, D.A. Arber, M.L. Slovak, S.J. Forman, Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood 101, 4701–4707 (2003). doi:10.1182/blood-2002-09-2780
M. Copland, A. Hamilton, L.J. Elrick, J.W. Baird, E.K. Allan, N. Jordanides, M. Barow, J.C. Mountford, T.L. Holyoake, Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood 107, 4532–4539 (2006). doi:10.1182/blood-2005-07-2947
H.G. Jorgensen, E.K. Allan, N.E. Jordanides, J.C. Mountford, T.L. Holyoake, Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood 109, 4016–4019 (2007). doi:10.1182/blood-2006-11-057521
A. Hamilton, G.V. Helgason, M. Schemionek, B. Zhang, S. Myssina, E.K. Allan, F.E. Nicolini, C. Muller-Tidow, R. Bhatia, V.G. Brunton, S. Koschmieder, T.L. Holyoake, Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood 119, 1501–1510 (2012). doi:10.1182/blood-2010-12-326843
J. Bertacchini, N. Ketabchi, L. Mediani, S. Capitani, S. Marmiroli, N. Saki, Inhibition of Ras-mediated signaling pathways in CML stem cells. Cell. Oncol. 38, 407–418 (2015). doi:10.1007/s13402-015-0248-2
J.H. Black, J.A. McCubrey, M.C. Willingham, J. Ramage, D.E. Hogge, A.E. Frankel, Diphtheria toxin-interleukin-3 fusion protein (DT(388)IL3) prolongs disease-free survival of leukemic immunocompromised mice. Leukemia 17, 155–159 (2003). doi:10.1038/sj.leu.2402744
T. Cierpicki, J. Grembecka, Challenges and opportunities in targeting the menin-MLL interaction. Future Med. Chem. 6, 447–462 (2014). doi:10.4155/fmc.13.214
Acknowledgments
We would like to thank all our colleagues for the insights that we have attempted to summarize. We apologize to the colleagues whose work could not be cited, owing to space limitations. SB is funded by the Iranian Council for Stem Cell Research and Technology Development (11/77230) and Tarbiat Modares University, Tehran, Iran. MLG is funded by the Irma T. Hirschl/Monique Weill-Caulier Trust, the US National Institutes of Health (NIH) (R01 CA172546; 5R01CA102031), and the Leukemia and Lymphoma Foundation. ACG is funded by the Instituto de Bebidas para la Salud y el Bienestar, México and the Fundación IMSS, México.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
None declared.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
(PPTX 55 kb)
Supplementary Fig. 2
(PPTX 286 kb)
Supplementary Fig. 3
(PPTX 342 kb)
Rights and permissions
About this article

Cite this article
Chavez-Gonzalez, A., Bakhshinejad, B., Pakravan, K. et al. Novel strategies for targeting leukemia stem cells: sounding the death knell for blood cancer. Cell Oncol. 40, 1–20 (2017). https://doi.org/10.1007/s13402-016-0297-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-016-0297-1