
Abstract
The “green” synthesis of galactomannan sulfates using a sulfamic acid–urea mixture has been studied for the first time. The effect of the time and temperature of the galactomannan sulfation process on the degree of substitution of galactomannan sulfates has been investigated. It is shown that, at a temperature of 70 °C with an increase in the process time up to 120 min, the degree of substitution increases up to 0.70. An increase in the process temperature up to 80 °C leads to the production of galactomannan sulfates with a degree of substitution of 1.67. With a further increase in the process temperature to 90 °C, the galactomannan structure is partially destructed, and the degree of substitution decreases. Embedding of the sulfate groups into the galactomannan structure has been confirmed by elemental analysis and Fourier-transform infrared spectroscopy. In addition, the initial and sulfated galactomannans have been characterized by X-ray diffraction, scanning electron microscopy, atomic force microscopy, and gel permeation chromatography. The thermal analysis shows that the initial galactomannan exhibits endothermic peaks at 254 and 294 °C and an exothermic peak at 315 °C, while sulfated galactomannan exhibits endothermic peaks at 209 and 275 °C and an exothermic peak at 281 °C. Using atomic force microscopy, it has been shown that the sulfated galactomannan film consists of spherical particles with an average diameter of 200–300 nm; according to the phase contrast data, it has the uniform composition without extraneous impurities.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Plant biomass is a renewable raw material and a sustainable resource for producing biologically active substances [1]. Recently, there has been growing interest in the use of phytogenic biologically active substances, which, as a rule, are safer than their analogues obtained synthetically or isolated from animal raw materials [2].
Plant polysaccharides are high-molecular-weight polycondensation products of monosaccharides linked to each other by the glycosidic bonds and forming linear or branched chains [3, 4]. Polysaccharides create the bulk of organic matter on Earth [5].
The roles of polysaccharides in human nutrition are diverse. They are harmless to the human body and digested by 69–95%, depending on the structure. Polysaccharides serve as a source of energy, affect lipid metabolism, play the role of enterosorbents, reduce cholesterol, and sorb microflora and salts of heavy metals [6,7,8]. Polysaccharides of higher plants, including galactomannans, exhibit the immunomodulating properties [9].
Galactomannans are plant polysaccharides consisting of β-(1–4)-D-mannose and α-(1–6)-D-galactose [10, 11]. The four major sources of seed galactomannans are locust bean (Ceratonia siliqua), guar gum (Cyamopsis tetragonoloba), tara gum (Caesalpinia spinosa Kuntze), and fenugreek gum (Trigonella foenum-graecum L.). Among them, only locust bean and guar gums are of considerable industrial and commercial importance. The galactose/mannose (G/M) ratio in galactomannans varies depending on their origin but conventionally ranges between 1.0:1.0–1.1 (Trigonella foenum-graecum L.), 1.0:1.6–1.8 (Cyamopsis tetragonoloba), 1.0:3.0 (Caesalpinia spinosa Kuntze), and 1.0:3.9–4.0 (Ceratonia siliqua). The conformation of the 1→4-linked backbone of β-D-mannan is similar to that of cellulose, which makes the former insoluble in water. However, the galactose side groups sterically break the interchain association and crystallization, thereby imparting a certain degree of solubility in water to galactomannans. As a result, the solubility of galactomannans increases with the degree of substitution of galactosyl: fenugreek and guar gum dissolve in cold water [11].
Galactomannans are non-toxic compounds widely used in the food and textile production, pharmaceutical industries, and medicine as food additives, stabilizers, flocculants, thickeners, and gelling agents [12,13,14,15]. The pharmacological studies showed that galactomannans and their derivatives exhibit the anticoagulant [16], hepatoprotective, and analgesic properties [17, 18]. It was established that the use of the partially hydrolyzed galactomannan positively affects the lipid and carbohydrate metabolism and normalizes the intestinal microflora [18].
A promising direction for modifying galactomannans is the preparation of their derivatives containing a sulfate group. Embedding of a sulfate group into a biopolymer macromolecule makes it water-soluble and increases its biodegradability [19, 20]. In addition, the sulfated derivatives of galactomannans not only can replace the widely used products of chemical modification of polymers but also find application in pharmaceuticals as potential antioxidants, antiviral drugs, and anticoagulants of a new class [21,22,23].
The SO3, H2SO4, and ClSO3H compounds currently used in sulfation of galactomannans are quite aggressive reagents and therefore require special equipment [21,22,23,24]. In addition, these processes need toxic organic solvents, e.g., pyridine, which limits their industrial use. In contrast to these reagents, sulfamic acid is a stable non-hygroscopic crystalline substance with the reactivity similar to that of the SO3–tertiary amine complex [24]. It is well-known that cellulose is destroyed upon heating with sulfamic acid, but, in the presence of urea, which plays a role of the main catalyst, its sulfation occurs [25].
The aim of this work was to synthesize galactomannan sulfates using the sulfamic acid–urea complex and study them by Fourier transform-infrared (FTIR) spectroscopy, X-ray diffraction (XRD), atomic force microscopy (AFM), thermogravimetric analysis (TGA), scanning electron microscopy (SEM), and gel permeation chromatography (GPC).
2 Experimental
The source of a raw material was peeled commercial galactomannan from Cyamopsis tetragonoloba (LLC “Mast-sl”). The monosaccharide composition was analyzed using the techniques reported by Wang et al. 2012 [26]. The galactose/mannose (G/M) ratio was 1.0:1.8.
2.1 Features of the galactomannan sulfation
2.1.1 Sulfation of galactomannan
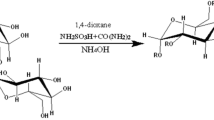
Sulfation of galactomannan (GM) was performed using the sulfamic acid–urea complex (SAA:U) according to the modified procedure from [27, 28]. To do this, the sulfating complex (SC) and galactomannan were triturated to obtain a homogeneous mass. The sulfating complex was obtained by preliminary mixing of 7.2 g (75 mmol) of sulfamic acid and 4.5 g (75 mmol) of urea. The ratio between galactomannan and the sulfating complex is 1:3 (mol/mol) (Fig. 1). The resulting reaction mixture was thermostated (at a temperature constant throughout the process) with continuous stirring at temperatures of 70, 80, and 90 °C within time frames of 30, 60, and 120 min. In this temperature range, the reaction mass melted.

Schematic of the galactomannan sulfation process with the use of the sulfamic acid–urea complex
2.1.2 Dissolution and neutralization
At the end of the thermostating process, the melt was cooled to room temperature. The obtained solid product was dissolved in 50 ml of water, and the unreacted sulfamic acid was neutralized with a 10% aqueous sodium hydroxide solution to pH 7–8. The resulting solution was evaporated in vacuum to a volume of 10–15 ml using a water jet pump.
2.1.3 Dialysis of sulfated galactomannan
The product was dialyzed against water in a MF-503-46 MFPI plastic bag (US) with a pore size of 3.5 kDa for 10 h to remove the excess reactants. Water is changed every hour (Fig. 1). In the dialysis process, the high-molecular-weight compounds were purified from the low-molecular-weight impurities, including inorganic compounds. The dissolved high-molecular-weight fraction remained in the dialysis bag. The low-molecular-weight products passed through the membrane of the dialysis bag to the environment (distilled water).
The degree of substitution (DS) is calculated as [26]:
where S% is the sulfur content determined by the elemental analysis.
To carry out the physicochemical study, the sample with a DS of 1.67 was taken.
2.2 Methods of the physicochemical analysis
2.2.1 Elemental analysis
The elemental analysis of sulfated galactomannan was performed on a ThermoQuest FlashEA-1112 elemental analyzer (Italy).
2.2.2 Fourier transform-infrared (FTIR) spectroscopy
The FTIR spectra of the initial and sulfated galactomannans were recorded using a Shimadzu IR Tracer-100 spectrometer (Japan) within the wavelength range of 400–4000 cm−1. The spectral data were analyzed using the OPUS program (version 5.0). Solid samples for the analysis were prepared in the form of tablets in a KBr matrix (2 mg sample/1000 mg KBr).
2.2.3 X-ray diffraction (XRD)
The X-ray diffraction study was carried out on a DRON-3 X-ray diffractometer (monochromatic CuKα radiation, λ = 0.154 nm) at a voltage of 30 kV and a current of 25 mA. The scanning step was 0.02 deg, and the intervals were 1 s per data point. The measurements were performed in the range of the Bragg angles 2Θ from 5.00 to 70.00Θ. The galactomannan and sulfated galactomannan crystallinity indices (CI) were taken from [29].
2.2.4 Scanning electron microscopy
Electron micrographs were obtained on a Hitachi TM-1000 scanning electron microscope (Japan) at an accelerating voltage of 15 kV and a magnification from × 100 to × 10,000 with a resolution of 30 nm. The electron micrographs were processed using the ImageJ software (version 1.8.0_112).
2.2.5 Atomic force microscopy
The sulfated galactomannan films were obtained as follows: The sulfated galactomannan (2 g) was dissolved in distilled water (30 ml) at room temperature. The obtained sulfated galactomannan solution was poured into a Petri dish and dried in an oven at a temperature of 45 °C to the constant weight. The obtained sulfated galactomannan films were separated from the Petri dish with tweezers and analyzed by atomic force microscopy. The semi-contact AFM study of the sulfated galactomannan films was carried out on an NT-MDT Solver P47 multimode scanning probe microscope (Moscow). Scanning was performed at no less than 3–4 points in several sites. The scanning rate was 1.5–2.0 Hz, and the image resolution was 256 × 256 pixels.
2.2.6 Thermogravimetric analysis
The thermogravimetric study and data analysis were performed using a NETZSCH STA 449 F1 Jupiter simultaneous thermal analysis instrument (Germany). The thermal degradation of the samples was analyzed in argon in the temperature range from 30 to 600 °C; the protective and purge gas flow rates were 20 and 50 ml/min, respectively. The samples were heated in a dynamic temperature regime (10 °C/min) in corundum crucibles. The measurement results were processed using the NETZSCH Proteus–Thermal Analysis.5.1.0 software supplied with the instrument.
2.2.7 Gel permeation chromatography
The average molecular weight Mw, average molecular mass Mn, and polydispersity of the sulfated galactomannan samples were determined by gel permeation chromatography using an Agilent 1260 Infinity II Multi-Detector GPC/SEC System chromatograph with two detections: by a refractometer (RI) and by a viscometer (VS). The separation was made on two Aquagel-OH columns using the solution 0.2MNaNO3 + 0,01 M NaH2PO4 in water (pH = 7) as a mobile phase. The column was calibrated by the polyethylene glycol standards (Agilent, USA). The eluent flow rate was 1 ml/min, and the sample volume was 100 μl. Before the analysis, the samples were dissolved in the mobile phase (1–5 mg/ml) and filtered through a 0.22-μm PES membrane filter (Agilent). Data collection and data processing were performed using the Agilent GPC/SEC MDS software.
3 Results and discussion
3.1 Synthesis of galactomannan sulfate
The mechanism of sulfation of polysaccharides with sulfamic acid is not well-understood. However, it was assumed [25, 30] that upon sulfation of alcohols, the reaction is first order for sulfamic acid and zero order for alcohol. This shows that the limiting stage is a kind of transformation in the acid molecule, specifically its decomposition into ammonia and sulfur trioxide.
We proposed to use the low-toxic non-corrosive sulfamic acid–urea mixture for sulfation of polymers [28, 31].
The rate of the direct interaction of alcohols with sulfamic acid is lower than the rate of catalyzed sulfation, because the S–N bond in sulfamic acid is stronger than in the donor–acceptor complex [25].
The reactivity of sulfamic acid increases in the presence of basic organic catalysts, including pyridine, urea, thiourea, acetamide, and picoline [25, 30]. Urea is the most effective agent. The increase in the reactivity of sulfamic acid in the presence of urea is explained by the formation of a donor–acceptor complex [24].
The galactomannan sulfation reaction with the sulfamic acid–urea complex and the subsequent isolation of galactomannan sulfate occurred according to the scheme shown in Fig. 2.

Scheme of the galactomannan sulfation reaction
In studying galactomannan sulfation with sulfamic acid, the time and temperature of the process were varied.
The data on the sulfur content in the galactomannan sulfate obtained under these experimental conditions are given in Table 1.
It was found that the DS of sulfated galactomannan can be controlled by varying the temperature and time of the sulfation process (Table 1).
A decrease in the process temperature to 70–90 °C instead of 150 °C reported by Sirvio et al. 2019 [27] is due to the properties of the initial GM, which, unlike cellulose, has an amorphous structure and is more exposed to the glycosidic bonds under the action of acids.
According to the gel permeation chromatography data for the sulfated GM samples obtained at different temperatures of the sulfation process, a decrease in the molecular weight of sulfated GM compared with the initial GM is observed (Table 2). Moreover, with increasing process temperature, the molecular weight of sulfated GM decreases. Obviously, along with the sulfation of the GM with sulfamic acid, depolymerization of the GM macromolecule occurs (Fig. 9). This is due to the fact that the glycosidic bonds connecting monosaccharide units to each other in hemicelluloses are sensitive to the action of acids; therefore, the treatment of galactomannan with acids causes its depolymerization. When sulfating galactomannan at a temperature of 70 °C, with an increase in the process time from 30 to 120 min, the DS increases from 0.39 to 0.70.

FTIR spectra: (1) galactomannan and (2) sodium salt of galactomannan sulfate

X-ray diffraction patterns of (1) galactomannan and (2) sodium salt of galactomannan sulfate

Typical AFM image of the sulfated galactomannan film. (a) Relief, (b) phase contrast, (c) 3D surface profile, and (d) particle size distribution

SEM images of (a) the initial galactomannan and (b) sulfated galactomannan samples

DSC (A) and DTG (B) analysis of (1) the initial galactomannan and (2) sulfated galactomannan samples

TGA analysis of (a) the initial galactomannan and (b) sulfated galactomannan samples

Molecular weight distribution for (1) galactomannan and sulfated galactomannan obtained at 80 °C for (2) 120 and (3) 300 min
Sulfated GM obtained at 80 °C had a higher DS than GM obtained at 70 °C with a maximum DS value of 1.67. However, an increase in the DS was observed with an increase in the process time to only 60 min. Upon further sulfation for a time of up to 300 min, the DS value decreased to 1.08. Sulfation of GM at a temperature of 90 °C showed that the DS of the sulfated polysaccharide decreases with time and attains a maximum value of 0.77 upon sulfation for 30 min.
The observed patterns of the change in the DS of GM with the process time and temperature can be explained as follows. An increase in the process temperature should lead to an increase in both the sulfation and depolymerization rate but to different extents. Obviously, the low-molecular-weight GM fractions not only have a high reactivity in the sulfation reaction but also undergo the faster depolymerization. Since the depolymerization rate increases with the process temperature, a great amount of the low-molecular-weight fraction of sulfated product with a sufficiently high DS is formed over time, which is removed during the dialysis purification. This leads to an apparent decrease in the DS of GM with time at a temperature of 90 °C. The high-molecular-weight fraction of GM, which has a lower reactivity in the sulfation reaction, leads to the formation of sulfated GM with a minor DS. Then, this sulfated GM remains and is identified after dialysis if the reaction occurs at high temperature for a long time. Thus, although the process of sulfation of GM at 70 °C occurs with some depolymerization, it is accompanied by a regular increase in the DS with time. A small (< 1) DS value suggests that the hydroxyl groups at the primary carbon atom (C6 in monosaccharide units) undergo sulfation [32, 33]. Sulfation at 80 °C proceeds at a faster rate; therefore, a product with a high DS value (1.26–1.67) is formed, increasing with a process time of up to 60 min. Then, in the course of the process, the amount of depolymerized (initial and sulfated) polysaccharides increases. This leads to the fact that, after purification of the reaction mass by dialysis, a low-molecular-weight sulfated GM with the high DS is removed, and the sulfated product with the lower DS remains, which is formed from the high-molecular-weight fractions with the low reactivity and weaker depolymerization. In this regard, in the range of 60–300 min, the DS noticeably decreases. A DS value within 1.26–1.67 is indicative not only of sulfation of the hydroxyl groups at C6 but also of partial sulfation of other hydroxyl groups of the monosaccharide units. An increase in the temperature of the sulfation process to 90 °C leads to an even higher degree of depolymerization, which increases during the process; therefore, after dialysis, only sulfated GM of the high-molecular-weight fractions of the initial GM, which have the lower reactivity for both sulfation and depolymerization, can be isolated. This causes a decrease in the DS from 0.77 to 0.23 during the process. Thus, in order to obtain sulfated GM with a high DS (1.67), the process should occur at a temperature of 80 °C for 60 min.
The observed regularity is consistent with the data reported by Wang et al. [26, 34] and probably related to the polysaccharide destruction at high temperature.
3.2 FTIR analysis
Embedding of the sulfate groups into the galactomannan structure is confirmed by the FTIR spectroscopy data (Fig. 3).
In the FTIR spectra of sulfated galactomannan, in contrast to the initial galactomannan, there is a high-intensity band at 1250 cm−1, which refers to the asymmetric stretching vibrations υas (O=S=O). The absorption bands in the range of 805–820 cm−1, which are not observed in the FTIR spectrum of the initial galactomannan, indicate the presence of the sulfate groups in the sodium salt of sulfated galactomannan (Fig. 3), which is consistent with the data reported by Wang et al. [26, 34].
In addition to the sulfate groups, the stretching vibrations of C=O groups at 1715 cm−1 are observed in the FTIR spectrum. The presence of a carbonyl group also confirms the formation of a carbamate group caused by an adverse reaction between polysaccharides and urea [27, 35]. The formation of esters of polysaccharide acids and carbamates is observed during phosphorylation of starch in the presence of urea [36], as well as using a deep eutectic solvent containing urea [27, 37].
3.3 X-ray diffraction (XRD)
The initial and sulfated galactomannan is analyzed by XRD (Fig. 4).
The galactomannan samples have an amorphous structure [38]. Comparison of XRD patterns of the samples of galactomannan and the sodium salt of galactomannan sulfate shows (Fig. 4) that further amorphization of the material structure occurs during sulfation. In the XRD pattern of the sample of the sodium salt of galactomannan sulfate, the peak smoothing was observed in the 2Θ angle range from 12 to 30°. The initial galactomannan has a crystallinity index of 0.34. The sulfated galactomannan has a crystallinity index of 0.13. Thus, the sulfation of galactomannan increases the amorphization by a factor of 2.
3.4 Atomic force microscopy
The synthesized sulfated galactomannan films are studied by the AFM technique (Fig. 5),which allows one to measure not only the lateral dimensions of nanoscale objects but also their height with the high (up to 0.1 nm) accuracy [39, 40].
According to the AFM data, the sulfated galactomannan film surface consists of homogeneous spherical particles with an average diameter from 200 to 300 nm. According to the phase-contrast images (Fig. 5b), the sulfated galactomannan film surface is quite homogeneous and contains no impurities.
3.5 Scanning electron microscopy
According to the scanning electron microscopy data, the initial galactomannan sample consists of rough asymmetric particles with average sizes from 200 to 800 μm (Fig. 6a). After sulfation, the samples have a morphology slightly different from the morphology of the initial galactomannan (Fig. 6b). The sodium salt of sulfated galactomannan consists of particles of different shapes with sizes of 50–1000 μm.
3.6 Thermal analysis
The differential scanning calorimetry (DSC) analysis (Fig. 7) shows the thermal behavior of the initial and sulfated galactomannans. For the initial galactomannan, endothermic peaks were detected at 254 and 294 °C, and an exothermic peak was detected at 315 °C. Sulfated galactomannan showed endothermic peaks at 209 and 275 °C and an exothermic peak at 281 °C.
Analysis of the DSC data showed that the early endothermic events are observed in the temperature range between 95 and 219 °C, which can be explained by the evaporation of water [41] due to the hydrophilic nature of the GM and SGM functional groups.
The GM and SGM calorimetric profiles contain endothermic and exothermic peaks. The first endothermic peak corresponds to the early removal of adsorbed water, while the second endothermic peak corresponds to the onset of thermal decomposition of organic matter (Fig. 7) [38, 42, 43]. It is well-known [37, 40,41,42] that the pyrolysis of polysaccharides is initiated by random destruction of glycosidic bonds with the subsequent decomposition. Thermograms contain peaks at 254 °C and 209 °C for the GM and SGM, respectively. This can be attributed to the elimination of galactose and mannose from the main chain of GM [41, 44,45,46]. The GM sulfation resulted in a decrease in the temperature of decomposition of the exothermic peak.
The thermogravimetric analysis of the initial and sulfated galactomannans is illustrated in Fig. 8. The temperatures of the broad peak of decomposition of GM and SGM obtained from the DSC curve are confirmed by the TGA thermograms. The thermal stability of the polymer is an important property, which can make the material suitable for use in food and pharmaceutical industries [38]. The TGA residual mass profiles demonstrated the stability of sulfated galactomannan over a narrower temperature range compared with the initial galactomannan.
3.7 Gel permeation chromatography
According to the galactomannan molecular mass distribution (Table 2), the initial sample is a low-molecular-weight type GM with MW ~ 660 kDa, which is consistent with the results reported in Ref. [47,48,49]. At the same time, GM has a bimodal particle mass distribution: the high-molecular-weight fraction with an MM of ~ 1500 kDa and the low-molecular-weight fraction with an MM of ~ 600 kDa. In this case, the degree of polydispersity was 2.55.
After the sulfation process occurring for 120 min at 80 °C, the destruction of the GM chains and the redistribution of molecular masses in the sample are probably observed; the peak of the low-molecular-weight fraction with MM ~ 600 kDa disappears; and a new peak corresponding to the reaction product with MM ~ 130 kDa arises. The proportion of the high-molecular-weight fraction in the sample also noticeably decreases (Fig. 9).
An increase in the sulfation process time to 300 min leads to further destruction of the GM polymer chains with a decrease in the molecular mass of the main fraction of the sulfated product to ~ 110 kDa; at the same time, a slight decrease in the total fraction of the low-molecular-weight part of the product is observed. Simultaneously, the proportion of the high-molecular-weight fraction, on the contrary, slightly increases, probably due to the higher resistance of the molecules against destruction and an increase in the degree of sulfation of GM. The molecular mass redistribution leads to an increase in polydispersity from 2.75 to 4.78.
4 Conclusions
A new method for sulfating galactomannan with the sulfamic acid–urea mixture was proposed. The high degree of substitution (1.67) was obtained at a temperature of 80 °C and a process time of 60 min.
Embedding of the sulfate groups into the galactomannan structure was confirmed by the elemental analysis and FTIR spectroscopy. The FTIR spectra of sulfated galactomannan contain bands at 1250 cm−1 and 805–820 cm−1, which indicate the presence of the sulfate groups.
The initial and sulfated galactomannan samples were also investigated by the X-ray diffraction, scanning electron microscopy, atomic force microscopy, and gel permeation chromatography techniques. It was shown using scanning electron microscopy that the initial galactomannan consists of particles of different shapes with a size ranging from 200 to 800 μm and sulfated galactomannan consists of particles of different shapes with the size range of 50–1000 μm.
The gel permeation chromatography study showed that the sulfation of galactomannan reduces the average molecular weight of sulfated galactomannan. In the gel permeation chromatogram, the initial galactomannan has a bimodal particle mass distribution: the high-molecular-weight fraction with an MM value of ~ 1500 kDa and the low molecular weight fraction with an MM value of ~ 600 kDa. An increase in the sulfation process time from 120 to 300 min leads to the destruction of the galactomannan polymer chains with a decrease in the molecular weight of the main fraction of the sulfated product from 130 to 110 kDa.
References
Dorea CMPG, Alvesa MGCF, Willa LSEP, Costa TG, Sabry DA (2012) A sulfated polysaccharide, fucans, isolated from brown algae Sargassum vulgare with anticoagulant, antithrombotic, antioxidant and anti-inflammatory effects. Carbohydr Polym 91:467–475. https://doi.org/10.1016/j.carbpol.2012.07.075
Oliveira RCR, Almeida RR, Goncalves TA (2016) A review of plant sulfated polysaccharides and their relations with anticoagulant activities. J Dev Drugs 5:3. https://doi.org/10.4172/2329-6631.1000166
Kaith BS, Sharma R, Kalia S (2015) Guar gum based biodegradable, antibacterial and electrically conductive hydrogels. Int J Biol Macromol 75:266–275. https://doi.org/10.1016/j.ijbiomac.2015.01.046
Thombare N, Jha U, Mishra S, Siddiqui MZ (2016) Guar gum as a promising starting material for diverse applications: a review. Int J Biol Macromol 88:361–372. https://doi.org/10.1016/j.ijbiomac.2016.04.001
Da Silva BP, Parente JP (2002) Chemical properties and biological activity of a polysaccharide from Melocactus depressus. Planta Med 68(1):74–76. https://doi.org/10.1055/s-2002-20052
Gracher AHP, Santana AG, Cipriani TR, Lacomini M (2015) A procoagulant chemically sulfated mannan. Carbohydr Polym 136:177–186. https://doi.org/10.1016/j.carbpol.2015.09.022
Reis RL (2008) Natural-based polymers for biomedical applications / Reis R L, Neves N M , Mano J F, Gomes M E, Marques A P, Azevedo H S. – Lisboa: Elsevier 832 p
Goun EA, Petrichenko VM, Solodnikov SU, Suhinina TV (2002) Anti cancer and anti-thrombin activity of Russian plants. J Ethnopharmacol 81:337–342. https://doi.org/10.1016/s0378-8741(02)00116-2
Silveira JLM, Bresolin TMB (2011) Pharmaceutical use of galactomannans. Quim Nova 34(2):292–299
Cerqueira MA, Bourbon AI, Pinheiro AC, Martins JT, Souza BWS, Teixeira JA, Vicenta AA (2011) Galactomannans use in the development of edible films/coatings for food applications. Trends Food Sci Technol 22:662–671. https://doi.org/10.1016/j.tifs.2011.07.002
Prajapati VD, Jani GK, Moradiya NG, Randeria NP, Nagar BJ, Naikwadi NN, Variya BC (2013) Galactomannan: a versatile biodegradable seed polysaccharide. Int J Biol Macromol 60:83–92. https://doi.org/10.1016/j.ijbiomac.2013.05.017
Filatova AV, Azimova LB, Turaev AS (2020) Study of the process of gelation of galactomannan from the seeds of Styphnolobium japonicum (Fabaceae). Chem Plant Raw Mater 1:33–39. https://doi.org/10.14258/jcprm.2020015485
Mercier T, Guldentops E, Lagrou K, Maertens J (2018) Galactomannan, a surrogate marker for outcome in invasive Aspergillosis: finally coming of age. Front Microbiol 9:661. https://doi.org/10.3389/fmicb.2018.00661
Perera N, Yang FL, Chang CM, Lu YT, Zhan SH, Tsai YT, Hsieh JF, Li LH, Hua KF, Wu SH (2017) Galactomannan from Antrodia cinnamomea enhances the phagocytic activity of macrophages. Org Lett 19(13):3486–3489. https://doi.org/10.1021/acs.orglett.7b01468
Tolstenkov AC, Drozd HH, Lapikova EU, Makarov VA, Mestechkina NM, Bannikova GE, Ilyina AB, Varlamov VP (2007) Effect of galactomannan from seeds of Cyamopsis Tetragonoloba (L.) taub on anticoagulant activity of rat plasma with intravenous administration. Clin Hematol Hemorheol Cardiovasc Surg 7:242–243 (in Rus)
Mestechkina NM, Anulov OV, Scherbukhin VD (1998) Study of galactomannan seed Amorphafruticosa L. Appl Biochem Microbiol (Rus) 34(5):549–552
Mestechkina NM, Dovletmuradov K, Scherbukhin VD (1991) Galactomannan common licorice seeds (Glyzyrrhizaglabra). Appl Biochem Microbiol (Rus) 27(3):435–441
Krishtanova NA, Safonova MJ, Bolotova VTs (2005) Prospects for the use of plant polysaccharides as therapeutic and therapeutic agents. Proceed Voronezh St Univ Ser: Chem Biol Pharm 1:212–221. (in Russ)
Caputo HE, Strau JE, Grinstaff MW (2019) Design, synthesis, and biomedical applications of synthetic sulphated polysaccharides. Chem Soc Rev 48:2338–2365. https://doi.org/10.1039/c7cs00593h
Vasilyeva NY, Levdanskiy AV, Kazachenko AS, Djakovich L, Pinel K, Kuznetsov BN (2013) Sulfation of mechanoactivated arabinogalactan with sulfuric anhydride–pyridine complex in pyridine medium. J Sib Fed Univ Chem 6(2):158–169
Mestechkina NM, Egorov AV, Shcherbukhin VD (2006) Synthesis of galactomannan sulfates. Appl Biochem Microbiol 42:326–330. https://doi.org/10.1134/S0003683806030185
Wang X, Wang J, Zhang J, Zhao B, Yao J, Wang Y (2009) Structure-antioxidant relationships of sulfated galactomannan from guar gum. Int J Biol Macromol 46(1):59–66. https://doi.org/10.1016/j.ijbiomac.2009.10.004
Zhang Z, Wang H, Chen T, Zhang H, Liang J, Kong W, Yao J, Zhang J, Wang J (2019) Synthesis and structure characterization of sulfated galactomannan from fenugreek gum. Int J Biol Macromol 125:1184–1191. https://doi.org/10.1016/j.ijbiomac.2018.09.113
Kuznetsov BN, Levdansky VA, Kuznetsova SA, Garyntseva NV, Sudakova IG, Levdansky AV (2018) Integration of peroxide delignification and sulfamic acid sulfation methods for obtaining cellulose sulfates from aspen wood. Eur J Wood Prod 76(3):999–1007. https://doi.org/10.1007/s00107-017-1262-z
Al-Horani RA, Desai UR (2010) Chemical sulfation of small molecules – advances and challenges. Tetrahedron. 66(16):2907–2918. https://doi.org/10.1016/j.tet.2010.02.015
Wang J, Zhao B, Wang X, Yao J, Zhang J (2012) Structure and antioxidant activities of sulfated guar gum: homogeneous reaction using DMAP/DCC catalyst. Int J Biol Macromol 50(5):1201–1206. https://doi.org/10.1016/j.ijbiomac.2012.03.009
Sirvio JA, Ukkola J, Liimatainen H (2019) Direct sulfation of cellulose fibers using a reactive deep eutectic solvent to produce highly charged cellulose nanofibers. Cellulose 26(4):2303–2316. https://doi.org/10.1007/s10570-019-02257-8
Akman F, Kazachenko AS, Vasilyeva NY, Malyar YN (2020) Synthesis and characterization of starch sulfates obtained by the sulfamic acid-urea complex. J Mol Struct 1208:127899. https://doi.org/10.1016/j.molstruc.2020.127899
Park S, Baker JO, Himmel ME, Parilla PA, Jonson DK (2010) Cellulose crystallinity index: measurement techniques and their impact on integrating cellulose performance. Biotechnol Biofuels 3:10. https://doi.org/10.1186/1754-6834-3-10
Spillane W, Malaubier JB (2014) Sulfamic acid and its n- and o-substituted derivatives chemical reviews 114(4):2507–2586. https://doi.org/10.1021/cr400230c
Kuznetsov BN, Vasilyeva NY, Kazachenko AS, Skvortsova GP, Levdansky VA, Lutoshkin MA (2018) Development of the method of Abies wood ethanol lignin sulfation using sulfamic acid. J Sib Fed Univ Chem 1(11):122–130. https://doi.org/10.17516/1998-2836-0063
Muschin T, Yoshida T (2012) Structural analysis of galactomannans by NMR spectroscopy. Carbohydr Polym 87(3):1893–1898. https://doi.org/10.1016/j.carbpol.2011.08.059
Muschin T, Budragchaa D, Kanamoto T, Nakashima H, Ichiyama K, Yamamoto N, Shuqin H, Yoshida T (2016) Chemically sulfated natural galactomannans with specific antiviral and anticoagulant activities. Int J Biol Macromol 89:415–420. https://doi.org/10.1016/j.ijbiomac.2016.05.005
Wang J, Niu S, Zhao B, Wang X, Yao J, Zhang J, Zhao W, Zhao Y (2013) Regioselective synthesis of sulfated guar gum: comparative studies of structure and antioxidant activities. Int J Biol Macromol 62:734–740. https://doi.org/10.1016/j.ijbiomac.2013.10.005
Yin C, Shen X (2007) Synthesis of cellulose carbamate by supercritical CO2-assisted impregnation: structure and rheological properties. Eur Polym J 43:2111–2116. https://doi.org/10.1016/j.eurpolymj.2007.01.041
Heinze U, Klemm D, Unger E, Pieschel F (2003) New starch phosphate carbamides of high swelling ability: synthesis and characterization. Starch-Staerke 55:55–60. https://doi.org/10.1002/star.200390017
Willberg-Keyrilainen P, Hiltunen J, Ropponen J (2018) Production of cellulose carbamate using urea-based deep eutectic solvents. Cellulose 25:195–204. https://doi.org/10.1007/s10570-017-1465-9
Mudgil D, Barak S, Khatkar BS (2012) X-ray diffraction, IR spectroscopy and thermal characterization of partially hydrolyzed guar gum. Int J Biol Macromol 50(4):1035–1039. https://doi.org/10.1016/j.ijbiomac.2012.02.031
Bhushan B, Fuchs H, Hosaka S (2004) Applied scanning probe methods I. Springer-Verlag, New York
Kuznetsov BN, Vasilyeva NY, Levdansky AV, Karacharov AA, Krylov AS, Mazurova EV, Bondarenko GN, Levdansky VA, Kazachenko AS (2017) The Raman spectroscopy, XRD, SEM, and AFM study of Arabinogalactan sulfates obtained using sulfamic acid. Rus J Bioorg Chem 43(7):722–726. https://doi.org/10.1134/S106816201707010X
Cerqueira MA, Souza BWS, Simões J, Teixeira JA, Domingues MRM, Coimbra MA, Vicente AA (2011) Structural and thermal characterization of galactomannans from non-conventional sources. Carbohydr Polym 83(1):179–185. https://doi.org/10.1016/j.carbpol.2010.07.036
Ramos-Sanchez MC, Rey FJ, Rodriguez ML, Martin-Gil FJ, Martin-Gil J (1988) DTG and DTA studies on fungical polysaccharides. Thermochim Acta 134:55–60. https://doi.org/10.1016/0040-6031(88)85217-1
Rey FJ, Ramos-Sanchez MC, Rodriguez-Mendez ML, Martin-Gil J, Martin-Gil FJ (1988) DTG and DTA studies on typical sugars. Thermochim Acta 134:67–72. https://doi.org/10.1016/0040-6031(88)85216-X
Zohuriaan MJ, Shokrolahi F (2004) Thermal studies on natural and modified gums. Polym Test 23:575–579. https://doi.org/10.1016/j.polymertesting.2003.11.001
Chaires-Martínez L, Salazar-Montoya JA, Ramos-Ramírez EG (2008) Physicochemical and functional characterization of the galactomannan obtained from mesquite seeds (Prosopis pallida). Eur Food Res Technol 227:1669–1676. https://doi.org/10.1007/s00217-008-0892-0
Vendruscolo CW, Ferrero C, Pineda EAG, Silveira JLM, Freitas RA, Jiménez-Castellanos MR, Bresolin TMB (2009) Physicochemical and mechanical characterization of galactomannan from Mimosa scabrella: effect of drying method. Carbohydr Polym 76(1):86–93. https://doi.org/10.1016/j.carbpol.2008.09.028
Wang Q, Ellisa PR, Ross-Murphy SB (2003) Dissolution kinetics of guar gum powders—II. Effects of concentration and molecular weight. Carbohydr Polym 53:75–83. https://doi.org/10.1016/S0144-8617(03)00009-2
Hirata T, Nishimoto T (1991) DSC, DTA, and TG of cellulose untreated and treated with flame-retardants. Thermochim Acta 193:99–106. https://doi.org/10.1016/0040-6031(91)80177-K
Varma AJ, Kokane SP, Pathak G, Pradhan SD (1997) Thermal behavior of galactomannan guar gum and its periodate oxidation products. Carbohydr Polym 32(2):111–114. https://doi.org/10.1016/S0144-8617(96)00155-5
Acknowledgments
The experiments were conducted on the equipment of the Krasnoyarsk Territorial Center for Collective Use, Federal Research Center «Krasnoyarsk Science Center SB RAS».
Funding
The reported study was funded by RFBR, project number 20-33-70256.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kazachenko, A.S., Malyar, Y.N., Vasilyeva, N.Y. et al. «Green» synthesis and characterization of galactomannan sulfates obtained using sulfamic acid. Biomass Conv. Bioref. 12, 2705–2714 (2022). https://doi.org/10.1007/s13399-020-00855-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13399-020-00855-2