
Abstract
The long-term impact of chronic human immunodeficiency virus (HIV) infection on brain status in injecting drug users (IDU) treated with highly active antiretroviral therapy (HAART) is unknown. Viral persistence in the brain with ongoing neuroinflammation may predispose to Alzheimer-like neurodegeneration. In this study, we investigated the brains of ten HAART-treated individuals (six IDU and four non-DU), compared with ten HIV negative controls (six IDU and four non-DU). HIV DNA levels in brain tissue were correlated with plasma and lymphoid tissue viral loads, cognitive status, microglial activation and Tau protein and amyloid deposition. Brain HIV proviral DNA levels were low in most cases but higher in HIV encephalitis (n = 2) and correlated significantly with levels in lymphoid tissue (p = 0.0075), but not with those in plasma. HIV positive subjects expressed more Tau protein and amyloid than HIV negative controls (highest in a 58 year old), as did IDU, but brain viral loads showed no relation to Tau and amyloid. Microglial activation linked significantly to HIV positivity (p = 0.001) and opiate abuse accentuated these microglial changes (p = 0.05). This study confirms that HIV DNA persists in brains despite HAART and that opiate abuse adds to the risk of brain damage in HIV positive subjects. Novel findings in this study show that (1) plasma levels are not a good surrogate indicator of brain status, (2) viral burden in brain and lymphoid tissues is related, and (3) while Tau and amyloid deposition is increased in HIV positive IDU, this is not specifically related to increased HIV burden within the brain.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Opportunities to investigate the neuropathology and brain viral characteristics in human immunodeficiency virus (HIV) positive subjects have been comparatively limited since highly effective antiretroviral therapy (HAART) was introduced more than 15 years ago. HAART is effective in controlling HIV replication with the result that RNA viraemia may become virtually undetectable in well-maintained subjects. However, viral persistence in so-called sanctuary sites, including lymphoid tissues and the central nervous system, is considered to be a significant problem (Langford et al. 2006; Kaul 2009; Marra et al. 2009) and many subjects with chronic HIV disease exhibit subtle cognitive impairment (Letendre et al. 2009; Zhao et al. 2009; Heaton et al. 2011; Schouten et al. 2011). There is debate in the literature as to whether the incidence of the more severe forms of cognitive deficit, including HIV-associated dementia, has actually increased despite the benefits of HAART (Kaul 2009; Schouten et al. 2011; Eisfeld et al. 2013). In addition, concern has been expressed that individuals with chronic HIV infection may be at risk of premature onset of Alzheimer-like neurodegenerative disorders (Brew et al. 2009; Xu and Ikezu 2009).
Illicit drug use is a significant co-morbid factor for HIV infection, not only in exposure to primary infection and simultaneous exposure to the risk of hepatitis C (HCV) transmission, but also in drug-associated psychological and cognitive disorders that contribute to non-compliance with therapy (Ersche et al. 2006; Robbins et al. 2008; Reddy et al. 2012)
Despite the importance of cognitive decline in chronic HIV infection, relatively few studies have investigated the viral burden in brain tissue from HAART-treated individuals (Langford et al. 2006; Zhao et al. 2009; Lamers et al. 2010, 2011; Borjabad et al. 2011; Gelman et al. 2012, 2013). There has been no investigation that compared, on a case by case basis, the viral loads in blood, lymphoid tissue and the brain with the level of neuroinflammation and the expression of neurodegenerative protein markers, all of which are pertinent to the debate regarding premature onset of Alzheimer-like conditions. The influence of opiate misuse on the viral burden in the brains of HAART-treated individuals is unknown. The aim of this study was to examine the hypothesis that microglial activation, signalling a chronic inflammatory state in the brain, and the accumulation of neurodegenerative proteins are related to the brain viral burden in HAART-treated individuals and that abuse of opiates is synergistic in this setting.
Materials and methods
Study subjects
This study included ten HIV positive subjects (six injecting drug users (IDU) aged 35–58 years, mean 45.8 years, and four who did not use drugs (non-DU) aged 28–40 years, mean 38.3 years) and ten HIV negative subjects (six IDU aged 35–52 years, mean 42.2 years, and four non-DU aged 34–40 years, mean 37 years) (Table 1). Three of the four non-DU acquired HIV by heterosexual intercourse and one was a homosexual male. Six of the HIV positive subjects were in late-stage HIV infection (CDC C3); the remaining four were still at CDC stages A or B. All had been treated with HAART for up to 7.9 years and most had received Zidovudine before the start of HAART. Cases with opportunistic infections or tumours in the brain were excluded from this study.
The IDU all used heroin at the time of HIV infection and were subsequently maintained on oral methadone, but they also misused a variety of other drugs including dihydrocodeine, benzodiazepines and temazepam, with cannabis and excess alcohol in some cases. No subjects in this study used either amphetamines or cocaine. All six HIV positive IDU, but only one HIV positive non-DU, were infected with HCV. The HIV negative IDU and non-DU were all HCV-negative.
Neuropsychological assessment of intellectual function, memory and mood was undertaken in the HIV positive cohort as previously described (Bell et al. 1996). For the HIV negative controls, cognitive status was estimated from medical records and reporting by next of kin and close contacts.
For subjects in this study who died in hospital, the post mortem examination was authorized by next of kin. Some subjects died suddenly and examination to establish the cause of death was then instructed by the appropriate legal authorities. In all 20 cases, including the medico-legal autopsies, the deceased's next of kin authorized the use of brains and tissue samples for research. Three HIV positive subjects had specifically requested that their bodies be used for this purpose. Brain and tissue samples were retained in, and selected from, the UK Medical Research Council HIV brain bank in Edinburgh (www.edinburghbrainbanks.ed.ac.uk). Selection was on the basis of a history of HAART intake for a significant period in the HIV positive group, with absent cerebral opportunistic conditions, and the ten HIV negative control subjects were then matched as closely as possible. Ethical approval for this study was obtained from the Lothian Research Ethics Committee.
Immunohistochemical examination and image analysis
Following fixation in buffered formalin, autopsy tissues were processed through a routine 41-h programme in a vacuum infiltration processor (Tissue Tek) and embedded in paraffin wax using a Tissue Tek embedding console. Brains were fixed for 2 weeks; fixation time for smaller tissue samples including spleen and lymph nodes was shortened to 2–4 days. All tissues were examined and reported initially as part of the routine post mortem examination. Sections stained with haematoxylin and eosin, and with special stains as necessary, were used for this purpose. In the present study, immunohistochemical staining was performed using the antibodies and protocols listed in Table 2. Standard avidin-biotin complex techniques were not deemed sufficiently sensitive for some antibodies and tyramide signal amplification was used in these circumstances, as described previously (Ramage et al. 2005). Diaminobenzidine was used as the visualising agent for all antibodies. Brain tissue selected for immunohistochemistry included blocks from the frontal and occipital lobes as close as possible to the origin of frozen samples, hippocampus and basal ganglia. Spleen and lymph node sections were examined only with haematoxylin and eosin and with HIV p24 antibody.
All sections were assessed blind to the case origin and clinical status and decoded only when all the measurements and counts had been completed. Sections of brain tissue stained for CD68 and MHCII (microglial/macrophage markers) were quantified for percentage area positive staining using a computerized image analysis system, Image Pro Plus (Media Cybernetics), because the numerous and irregular outline of positive cells precluded manual counts, and this method provided an objective assessment (Anthony et al. 2010). For CD68 and MHCII stained sections, 24 contiguous images were assessed in grey (GM) and white matter (WM) separately, using an ×10 objective and averaging the percentage immunopositive area across the 24 GM and 24 WM images. Sections stained for Tau protein (AT8) were assessed manually by screening the entire slide with a ×20 objective for the presence and total number of neuropil threads and neurofibrillary tangles within the grey matter. The total area of grey matter in each section was then calculated allowing the number of Tau-positive features/square centimetre to be determined. Sections stained for amyloid (4G8 and AB4) were assessed manually for the presence of immunopositive plaques in grey matter (present or absent), and for neuronal staining. Similarly, the sections stained for HIV p24 were assessed manually for the presence of any positive cells and each section scored positive or negative.
DNA extraction and purification
At the time of post mortem examination, fresh samples of brain and other organs were frozen for storage at −80 °C. Frozen tissue samples used in this study for estimation of viral load included all those still available as follows: frontal lobe in seven cases, basal ganglia in one case and occipital lobe in two cases. Lymphoid tissues included spleen in seven cases and lymph node in three cases. Cellular DNA was extracted from ~10 mg frozen lymphoid tissue using the QIAGEN AllPrep DNA/RNA mini kit. DNA was extracted from neural tissue by the phenol chloroform method as described previously (Wang et al. 2001). Nested PCR amplification of the gag and envV3 regions was carried out using 1 μg DNA and primers as previously described (Hughes et al.1997), except for envV3 which used primers 5′-CCAATYCCYATACATTATTGT-3′ (outer sense), 5′-TAGAAAAAYTCYCCYCYACAA-3′ (outer anti-sense), 5′-CAGTACAATGYACACATGGAA-3′ (inner sense) and (5′-AAYTTCTRGRTCCCCTCCTG-3′ (inner anti-sense). The number of proviral copies/106 cells was calculated from limiting dilution of DNA from the number of PCR positive reactions using gag primers and applying the Poisson equation to estimate the number of templates per microgram cellular DNA, making the assumption that 1 μg cellular DNA equates to 150,000 cells. Nucleotide sequencing of both gag and env V3 PCR products was used to confirm that, for each individual, closely related HIV sequences were detected in both lymphoid and neural tissues.
Results
Neuropathology, neuroinflammation and neurodegeneration
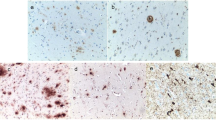
No cases of HIV encephalitis were identified on routine staining of the brains from the HIV positive groups and no giant cells were seen (Table 1). Abnormal blood vessels with thickened homogenized walls were present in the white matter of almost all the IDU (Fig. 1a) and lymphocytic infiltrates were present in the meninges and around small parenchymal vessels in some HIV positive brains (Fig. 1b). Alzheimer II astrocytes were present in the grey matter of individuals with liver dysfunction and occasional small infarcts were present in some cases (Table 1). Immunohistochemistry revealed the presence of HIV p24 positive microglial cells (Fig. 1c) in the white matter of two individuals, confirming the presence of HIV encephalitis (Table 3). While one of these (HIV 10) also showed occasional microglial nodules (Fig. 1c), the presence of HIV encephalitis was entirely unsuspected on routine neuropathological examination in the other (HIV 9) (Table 1). HIV p24 was positive in only two lymphoid tissues (Fig. 1d), and these results bore little relation to the viral burden in lymphoid tissue (Table 3), potentially the result of sampling different areas in a tissue type showing considerable heterogeneity in infection frequency.

a Abnormal thick-walled blood vessels (arrow) in the white matter of a 43-year-old HIV-infected IDU (subject 5); haematoxylin and eosin. b Perivascular lymphocytic infiltrate (arrow) in the meninges of an HIV-infected IDU (subject 5); negative control immunohistochemical section. c Microglial nodule positive for HIVp24 in the white matter of a 28-year-old HIV positive man infected heterosexually (subject 10), confirming the presence of HIV encephalitis. d Positivity for HIV p24 (arrow) in a hilar lymph node from a 38-year-old HIV positive man infected heterosexually (subject 9). Carbon deposition is present to the left of the figure. e Little evidence of CD68 positive microglia/macrophages in the white matter of a 37-year-old HIV negative, non-IDU control (subject C7); CD68 antibody. f Upregulation of CD68 positive microglia/macrophages in the white matter of a 47-year-old HIV positive IDU (subject 4); CD68 antibody. g Little evidence of MHCII positive microglia/macrophages in the white matter of an HIV negative non-IDU control (subject C7); MHCII antibody. h Upregulation of MHCII positive microglia/macrophages in the white matter of an HIV positive IDU (subject 4); MHCII antibody. i Tau positive neurofibrillary tangle in the cortex of a 47-year-old HIV positive IDU (subject 4); immunohistochemistry AT8 antibody. j Tau positive pre-tangle (fine arrow) and neuropil threads (thicker arrows) in the cortex of a 58-year-old HIV positive IDU (subject 2); immunohistochemistry AT8 antibody. k Positivity for amyloid protein within neurons and in large shadow plaques in the grey matter of an HIV positive IDU (subject 2); immunopositivity for 4G8 antibody. l Positivity for insoluble amyloid protein in shadow plaques in the grey matter of an HIV positive IDU (subject 2). Neurons are BA4 negative. Immunopositivity for BA4 antibody
Microglial markers were positive in all cases. CD68 positivity was observed in parenchymal and perivascular microglia, more in white matter than in grey (Fig. 1e, f), and was at its lowest level in the non-DU controls (Fig. 1e). CD68 staining was much accentuated in HIV positive subjects in whom CD68 positive macrophages and occasional microglial nodules were also present (Fig. 1f). The levels of CD68 in the four groups are compared in Fig. 2. Similar results were obtained with MHCII staining (Figs. 1g, h and 3). As expected, microglial activation is higher in HIV positive subjects than in negative (p = 0.001, 0.008 in white and gray matter, respectively, for CD68, p = 0.008, 0.04 for MHCII in white and gray matter using the Kruskal-Wallace non-parametric test). The presence of the only two HIV encephalitis cases in the non-DU group and associated prominent microglial/macrophage activation in at least one of these two cases has an effect on the mean level of neuroinflammation such that there appears to be no difference between HIV positive IDU and non-DU (p > 0.05); whereas in the HIV negative subjects, there is a significant difference between the two, being least in the non-DU (p = 0.05 and 0.01 for CD68 in white and gray matter, respectively).

CD68 immunopositive microglia/macrophages in grey and white matter of HIV positive and negative IDU and non-DU groups of subjects. The y-axis shows a log scale of percentage area of stained slide that is positive for CD68, as described in Materials and methods

MHCII immunopositive microglia/macrophages in grey and white matter of HIV positive and negative IDU and non-DU groups of subjects. The y-axis shows a log scale of percentage area of stained slide that is positive for MHCII, as described in Materials and methods
Tau positivity in the form of neuropil threads and neurofibrillary tangles was confined to gray matter, with the exception of a very occasional Tau positive thread in the white matter close to the grey/white matter junction. Tau positive neuropil threads were not distributed uniformly and tended to cluster, generally in the middle layers of the cortex, especially if tangles were also present. Well-formed neurofibrillary tangles (Fig. 1i) and pre-tangles (Fig. 1g) were present in those cases with a higher number of Tau positive threads (Fig. 1j). There were two outlying cases with respect to the levels of Tau positivity. The oldest HIV positive IDU (subject HIV 2, aged 58) had the highest levels of Tau positivity (30 neurites and 1 tangle/cm2) among the HIV positive subjects, in keeping with his age. The other Tau positive outlier was an HIV negative non-DU (C8, aged 40), who was found to have 20 neuropil threads and 6 tangles/cm2, but without any evidence of amyloid plaque positivity. Other Tau positive structures were observed in the cortex of subject C8, and it is likely that the changes represented the subclinical stages of a tauopathy in this individual. Both outliers were excluded from the group comparisons shown in Fig. 4. Declining positivity for Tau positivity is seen across the four groups, with IDU having more Tau than the non-DU (mean Tau scores 6.5 and 4.9, respectively) and HIV positive subjects more than HIV negative (mean Tau scores 7.5 and 4.3, respectively), again excluding the outliers, but these differences did not achieve statistical significance.

Tau-positive neuropil threads and tangles in HIV positive and negative IDU and non-DU groups of subjects. The two outlying individuals (one HIV + IDU and one HIV non-DU) are excluded. The y-axis shows the total number of Tau positive neuropil threads and tangles/cm2 in the grey matter, as described in Materials and methods
Staining for amyloid (4G8 and BA4) showed 4G8-positive neurons (Fig. 1k) in all cases and no discernible difference was noted in the intensity of neuronal staining between the groups. However, 4G8 positive shadow plaques (Fig. 1l) were confined to three HIV + ve IDU (HIV 1, HIV 2 and HIV 5) and were numerous only in subject HIV 2. This 58-year-old HIV positive IDU was the only subject to also show AB4 positive plaques (Fig. 1l). Taken together with his excess Tau positivity and with similar changes elsewhere in this brain, these changes are consistent with the onset of Alzheimer's disease in this subject. All the other subjects, both HIV positive and negative, were negative for amyloid plaques.
It is worth noting that having excluded the two outliers displaying the highest levels of Tau protein (HIV 2 and C8) from the Tau group comparisons in Fig. 4 on account of suspected advancing neurodegenerative conditions, the comparisons shown in Figs. 2 and 3 were re-examined in order to exclude the same two cases from the microglial data (results not shown). However, these exclusions made no difference to the microglial outcome. There was no significant correlation, on a case by case basis, between the neurodegenerative marker Tau protein (numerical score) or the presence of amyloid (plaques positive or negative) and the degree of neuroinflammation (CD68/MHCII) (p values comparing Tau scores with CD68 or MHCII in either white or gray matter were >0.05 by Spearman rank correlation test).
Viral load
Of the HIV positive subjects, six were in late-stage HIV infection (CDC C3), with commensurate low CD4 counts but variable plasma loads (Table 3). Five of these six had been treated with HAART for 3 years or more. Of the six CDC C3 subjects, three had virtually undetectable brain viral loads (<2 proviral copies/106 cells), one had moderate levels (14 copies/106 cells), and two had higher levels (up to 332 copies/106 cells) (Table 3). These last two subjects (HIV 9 and HIV10) displayed HIV p24 positivity in the brain, confirming the presence of HIV encephalitis, despite treatment with HAART for nearly 8 years in one case. The absence of HIV encephalitis in all other cases was confirmed by re-evaluation of the full neuropathological examination that formed part of the routine post mortem examination. Among the four subjects in stages CDC A3, B2 and B3, one (HIV 5) in CDC B2 had a moderate brain viral load (14 copies/106 cells) but the remainder had virtually undetectable proviral DNA in brain tissue.
Viral loads were always higher in lymphoid tissue than in brain (geometric mean values 1,500 DNA copies/106 cells in lymphoid compared to 13/106 cells in brain). Despite this difference, there was a significant association in viral loads in the two tissues (Table 3, Spearman's rank correlation test 0.68; p = 0.0075), but not with plasma loads (p = 0.59 and 0.75 for lymphoid tissue and brain, respectively). The variable plasma viral loads suggest that in some cases, HAART was not successful in containing disease or that compliance with therapy was incomplete. In this regard, there was no difference between IDU and non-DU. There was some association between cognitive impairment and plasma viral load but this did not achieve statistical significance (p = 0.15 by Spearman's rank correlation test), but none for brain viral load. Despite the one case (HIV 10) displaying HIV encephalitis, high brain viral load and moderately severe dementia, HCV-associated dysfunction and hepatic encephalopathy may be confounding factors that obscure any such association. There was no significant association between brain viral load and deposition of Tau and/or amyloid. The duration of HIV infection overall, and in CDC stage C3, appeared to have little impact on the development of HIV encephalitis.
Discussion
This study confirms that HIV is present in the brains of HIV positive subjects treated with HAART, but shows that even in advanced disease, a majority of brains in this series have very low viral loads. The possibility in such cases of blood contamination of the brain tissue samples cannot be ruled out, although the wide variation in HIV plasma levels and lack of correlation with brain viral loads in these cases argues against this. An early study of HIV RNA levels in the brains of HAART-treated individuals compared with those from the pre-HAART era showed that HAART conferred significant benefit in significantly reducing, while not eliminating, virus in the brain (Langford et al. 2006). Cerebrospinal fluid (CSF) is sometimes regarded as a surrogate marker for processes evolving in the brain, including brain viral load but CSF was not available in our cases. Cysique et al. (2011) reviewed studies of the effect of HAART on cognitive status and CSF viral loads and found that regimes with high central nervous system (CNS) penetrance were effective in maintaining cognition and reducing CSF load. Marra et al. (2009) showed that HAART, particularly combinations that crossed the blood–brain barrier successfully, controlled viral replication in CSF but, surprisingly, was associated with poorer cognitive function. Lamers et al. (2011) sequenced HIV gp120 from multiple brain regions and peripheral tissues in five subjects and found a high degree of compartmentalization with particularly high viral load in the meninges. In a larger study, Gelman et al. (2013) investigated the relationship between cognitive disorders and HIV gag/pol RNA and DNA in the brains of 148 HAART-treated subjects and found a positive relationship with cognitive impairment, the correlation being stronger for RNA than for DNA. In 2012, Gelman et al. demonstrated a strong association between HIV RNA level in the brain, and to a lesser extent HIV DNA, and the presence of HIV encephalitis in 24 subjects from the same resource. In contrast, Zhao et al. (2009) found lower levels of HIV DNA in the brains of subjects with dementia compared with those who had not had cognitive impairment, but the study subjects showed multiple brain pathologies. Lamers et al. (2010) also investigated HIV DNA levels in multiple brain tissues from 11 subjects with a range of brain pathology and showed a greater degree of viral evolution in gp41 and gp120 in cognitively impaired versus non-impaired individuals.
Imaging studies can also provide relevant data. Harezlak et al. (2011) reported that 268 subjects on stable HAART all displayed evidence of neuroinflammation, but that evidence of neuronal injury was confined to those with cognitive impairment. Findings in the present study with respect to microglial activation quantified in brain tissue sections are in accord with these imaging results. The lack of correlation between neurodegeneration and cognitive impairment in our study probably relates to the small number of subjects who displayed cognitive decline and to the Alzheimer-related end-stage degenerative proteins investigated here. Of the two subjects with a relatively high Tau protein burden, the one who also displayed amyloid plaques (HIV 2) had mild cognitive impairment but the other (C8), who did not have increased amyloid, did not.
In this study, we confirm the expected link between high brain levels of HIV DNA and the presence of HIV encephalitis. We also confirm that high levels of microglial activation (augmented CD68 and MHCII positivity) correlate with high viral loads and with HIV encephalitis. Drug use, largely opioids in this cohort, appears to be an additive factor in terms of microglial activation in that the levels for the latter in HIV positive IDU without HIV encephalitis overlapped with, or exceeded, the two non-DU with HIV encephalitis. While HCV is a possible confounding difference between the two HIV positive groups, the significantly higher levels of microglial activation in HIV negative IDU compared with HIV negative non-DU favour drug use as the cause, since both groups are HCV-negative. The finding that the microglial response is accentuated in drug users' brains is in line with previous findings from the Edinburgh cohort (Tomlinson et al. 1999; Anthony et al. 2005). In contrast, Byrd et al. (2012) concluded that opiates exert an immunosuppressive effect in the brain, in that microglial activation was less marked in opiate users than in non drug users, including those who were HIV infected.
SIV macaque models have also proved useful in investigating these responses. In this model, Clements et al. (2011) found that HAART was effective in reducing virus replication and inflammation in the brain when treatment was initiated early in infection, even when the drugs did not cross the blood–brain barrier. Zink et al. (2010) had reported similar results and showed that SIV replication was reduced in the macaque brain but that DNA persisted. Weed et al. (2012) reported that cocaine had no effect on the expression of neuroinflammatory and neurodegeneration markers in a similar model, or on SIV levels in the brain.
The presence of low-level viral DNA, and associated reactive and innate inflammatory responses in the brain, has been postulated to underpin neuronal injury observed in HIV-infected brains (Kaul 2009). While this has been compared to an Alzheimer-like process, Gisslen et al. (2009) found that abnormal levels of amyloid and Tau in the CSF of HIV positive and negative individuals did not adhere to the pattern of derangement seen in Alzheimer cases. In a report of 589 brains gathered in the US National NeuroAIDS Tissue Consortium, Everall et al. (2009) found no relation between cognitive disorder and HIV brain pathology despite the large number of cases. However, there did appear to be an association between cognitive impairment and the presence of Alzheimer II gliosis, consistent with hepatic encephalopathy, in a small subset of cases, underlining the potential importance of co-morbid factors such as HCV or alcohol-associated liver dysfunction. We have shown previously that Tau positivity is higher in IDU than in non-DU (Ramage et al. 2005; Anthony et al. 2010) and in HIV positive versus HIV negative subjects, particularly those treated with HAART, although the levels of Tau fall far short of the levels characteristic of Alzheimer's disease (Anthony et al. 2006). The present findings are in accord with the previous studies. Patrick et al. (2011) also reported increased levels of CDK5 and abnormal Tau phosphorylation in association with HIV encephalitis. Amyloid plaques were rare in the present study but were confined to HIV positive subjects, in line with other reports of an increase in amyloid deposition in association with HIV infection (Green et al. 2005; Andras and Toborek 2013). All three amyloid plaque-positive cases in the present study were HIV positive IDU, but one of these three was the oldest subject in the study by some margin. No relationship could be discerned between HIV brain viral load and the presence of amyloid plaques or the number of Tau positive features. In this study, cognitive impairment did not relate exclusively to high viral load or to HIV encephalitis. HCV is possibly neurotoxic (Paulino et al. 2011), which may be a confounding factor in HIV positive IDU. Even the drugs included in HAART, particularly protease inhibitors, have come under suspicion of contributing to neuronal injury, possibly mediated by a rise in drug-induced cerebrovascular disease (Gupta et al. 2012). The debate concerning varying levels of neuroprotection offered by different HAART regimes with different CNS penetrance is not yet resolved (Eisfeld et al. 2013). Opiates enhance neuronal damage by means of synaptodendritic loss (Hauser et al. 2012) in conjunction with activation or infection of microglial cells. Overall, the evidence is that opiates contribute to the development of neurodegenerative disorders in the context of HIV infection (Dutta and Roy 2012) and probably through a variety of mechanisms.
In summary, this study confirms a relationship between brain HIV burden (DNA), HIV encephalitis and microglial activation in HAART-treated subjects. We find that IDU is additive to HIV in promoting neuroinflammation. We did not find significant links between the degree of microglial activation, or the quantity of HIV DNA in the brain, with the expression of the neurodegenerative markers Tau and amyloid, although IDU accentuated Tau deposition, as we have shown previously. While this may be perceived as reassuring with respect to the risk of developing Alzheimer-like disorders in HIV positive subjects, it may be that in comparatively young individuals, any evidence of HIV-associated neuronal injury is more subtle, and at the synaptic level, rather than in the end-stage markers of neuronal senescence and death. However, the findings are different for HIV positive and negative IDU, and this study confirms that opiate IDU are at increased risk of developing neuronal and cerebrovascular disorders. A further novel finding in this study is the relationship between tissue viral levels (brain and lymphoid), suggesting a degree of coordinated tissue sequestration in infected individuals, whereas the unrelated plasma levels are more likely to reflect current therapy compliance. Plasma viral levels should not be relied upon to provide information regarding brain HIV status.
References
Andras IE, Toborek M (2013) Amyloid beta accumulation in HIV-1-infected brain: the role of the blood–brain barrier. IUBMB Life 65:43–49. doi:10.1002/iub.1106
Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE (2005) Does drug abuse alter microglial phenotype and cell turnover in the context of advancing HIV infection. Neuropathol Appl Neurobiol 31:325–338. doi:10.1111/j.1365-2990.00648.x
Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE (2006) Accelerated Tau deposition in the brains of individuals infected with human immunodeficiency virus-1 before and after the advent of highly active anti-retroviral therapy. Acta Neuropathol 111:529–538. doi:10.1007/s00401-006-0037-0
Anthony IC, Norrby KE, Dingwall T, Carnie FW, Millar T, Arango JC, Robertson R, Bell JE (2010) Predisposition to accelerated Alzheimer-related changes in the brains of human immunodeficiency virus negative opiate abusers. Brain 133:3685–3698. doi:10.1093/brain/awq263
Bell JE, Donaldson YK, Lowrie S, McKenzie CA, Elton RA, Chiswick A, Brettle RP, Ironside JW, Simmonds P (1996) Influence of risk group and zidovudine therapy on the development of HIV encephalitis and cognitive impairment in AIDS patients. AIDS 10:493–499
Borjabad A, Morgello S, Chao W, Kim SY, Brooks AI, Murray J, Potash MJ, Volsky DJ (2011) Significant effects of antiretroviral therapy on global gene expression in brain tissues of patients with HIV-1-associated neurocognitive disorders. PLoS Pathog. doi:10.1371/journal.ppat. 1002213
Brew BJ, Crowe SM, Landay A, Cysique LA, Guillemin G (2009) Neurodegeneration and ageing in the HAART era. J Neuroimmune Pharmacol 4:163–174. doi:10.1007/s11481-008-9143-1
Byrd D, Murray J, Safdieh G, Morgello S (2012) Impact of opiate addiction on neuroinflammation in HIV. J Neurovirol 18:364–373. doi:10.1007/s13365-012-0118-x
Clements JE, Gama L, Graham DR, Mankowski JL, Zink MC (2011) An SIV macaque model of HAART: viral latency in the periphery and the central nervous system. Curr Opin HIV AIDS 6:37–42. doi:10.1097/COH.0b03e3283412413
Cysique LA, Waters EK, Brew BJ (2011) Central nervous system antiretroviral efficacy in HIV infection: a qualitative and quantitative review and implications for future research. BMC Neurol 11:148. doi:10.1186/1471-2377-11-148
Dutta R, Roy S (2012) Mechanism(s) involved in opioid drug abuse modulation of HAND. Curr HIV Res 10:469–477. doi:10.2174/157016212802138805
Eisfeld C, Reichelt D, Evers S, Husstedt I (2013) CSF penetration by antiretroviral drugs. CNS Drugs 27:31–55. doi:10.1007/s40263-012-0018-x
Ersche KD, Clark L, London M, Robbins TW, Sahakian BJ (2006) Profile of executive and memory function associated with amphetamine and opiate dependence. Neuropsychopharmacology 31:1036–1047. doi:10.1038/si.nnp.1300889
Everall I, Vaida F, Khanlou N, Lazzaretto D, Achim C, Letendre S, Moore D, Ellis R, Cherner M, Gelman B, Morgello S, Singer E, Grant I, Masliah E, National NeuroAIDS Tissue Consortium (NNTC) (2009) Cliniconeuropathologic correlates of human immunodeficiency virus in the era of antiretroviral therapy. J Neurovirol 15:360–370. doi:10.3109/13550280903131915
Gelman BB, Chen T, Lisinicchia JG, Soukup VM, Carmical JR, Starkey JM, Masliah E, Commins DL, Brandt D, Grant I, Singer EJ, Levine AJ, Miller J, Winkler JM, Fox HS, Luxon BA, Morgello S, National NeuroAIDS Tissue Consortium (2012) The National NeuroAIDS Tissue Consortium brain gene array: two types of HIV-associated neurocognitive impairment. PLoS One. doi:10.1371/journal pone. 0046178
Gelman BB, Lisinicchia JG, Morgello S, Masliah E, Commins D, Achim CL, Fox HS, Kolson DL, Grant I, Singer E, Yiannoutsos CT, Sherman S, Gensler G, Moore DJ, Chen T, Soukup VM (2013) Neurovirological correlation with HIV-associated neurocognitive disorders and encephalitis in a HAART-era cohort. J Acquir Immune Defic Syndr 62:487–495. doi:10.1097/QAI.0b013e31827f1bdb
Gisslen M, Krut J, Andreasson U, Blennow K, Cinque P, Brew BJ, Spudich S, Hagberg L, Rosengren L, Price RW, Zetterberg H (2009) Amyloid and tau cerebrospinal fluid biomarkers in HIV infection. BMC Neurol 9:63. doi:10.1186/1471-2377-9-63
Green DA, Masliah E, Vinters HV, Beizai P, Moore DJ, Achim CL (2005) Brain deposition of beta-amyloid is a common feature in HIV positive patients. AIDS 19:407–411
Gupta S, Knight AG, Losso BY, Ingram DK, Keller JN, Bruce-Keller AJ (2012) Brain injury caused by HIV protease inhibitors: role of lipodystrophy and insulin resistance. Antiviral Res 95:19–29. doi:10.1016/j.antiviral.2012.04.010
Harezlak J, Buchthal S, Taylor M, Schifitto G, Zhong J, Daar E, Alger J, Singer E, Campbell T, Yiannoutsos C, Cohen R, Navia B, HIV Neuroimaging Consortium (2011) Persistence of HIV-associated cognitive impairment, inflammation and neuronal injury in era of highly active antiretroviral treatment. AIDS 25:625–633. doi:10.1097/QAD.0b013e3283427da7
Hauser KF, Fitting S, Dever SM, Podhaizer EM, Knapp PE (2012) Opiate drug use and the pathophysiology of neuro-AIDS. Curr HIV Res 10:435–452. doi:10.2174/157016212802138779
Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I, CHARTER Group, HNRC Group (2011) HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature and predictors. J Neurovirol 17:3–16. doi:10.1007/s13365-010-0006-1
Hughes ES, Bell JE, Simmonds P (1997) Investigation of the dynamics of the spread of human immunodeficiency virus to brain and other tissues by evolutionary analysis of sequences from the p17gag and env genes. J Virol 71:1272–1280
Kaul M (2009) HIV-1 associated dementia: update on pathological mechanisms and therapeutic approaches. Curr Opin Neurol 22:315–320. doi:10.1097/WCO.0b013e328329cf3c
Lamers SL, Salemi M, Galligan DC, Morris A, Gray R, Fogel G, Zhao L, McGrath MS (2010) Human immunodeficiency virus-1 evolutionary patterns associated with pathogenic processes in the brain. J Neurovirol 16:230–241. doi:10.3109/13550281003735709
Lamers SL, Gray RR, Salemi M, Huysentruyt LC, McGrath MS (2011) HIV-1 phylogenetic analysis shows HIV-1 transits through the meninges to brain and peripheral tissues. Infect Genet Evol 11:31–37. doi:10.1016/j.meegid. 2010.10.016
Langford D, Marquie-Beck J, de Almeida S, Lazzaretto D, Letendre S, Grant I, McCutchan JA, Masliah E, Ellis RJ, HIV Neurobehavioural Research Centre (HNRC) group (2006) Relationship of antiretroviral treatment to post mortem brain tissue viral load in human immunodeficiency virus-infected patients. J Neurovirol 12:100–107. doi:10.1080/13550280600713932
Letendre SL, Ellis RJ, Everall I, Ances B, Bharti A, McCutchan JA (2009) Neurologic complications of HIV disease and their treatment. Top HIV Med 17:46–56
Marra CM, Zhao Y, Clifford DB, Letendre S, Evans S, Henry K, Ellis RJ, Rodriguez B, Coombs RW, Schifitto G, McArthur JC, Robertson K, AIDS Clinical Trials Group 736 Study Team (2009) Impact of combination antiretroviral therapy on cerebrospinal fluid HIV RNA and neurocognitive performance. AIDS 23:1359–1366. doi:10.1097/QAD.0b013e32832c4152
Patrick C, Crews L, Desplats P, Dumaop W, Rockenstein E, Achim CL, Everall I, Masliah E (2011) Increased CDK5 expression in HIV encephalitis contributes to neurodegeneration via Tau phosphorylation and is reversed with Roscovitine. Am J Pathol 178:1646–1661. doi:10.1016/j.ajpath.2010.12.033
Paulino AD, Ubhi K, Rockenstein E, Adame A, Crews L, Letendre S, Ellis R, Everall IP, Grant I, Masliah E (2011) Neurotoxic effects of the HCV core protein are mediated by sustained activation of ERK via TLR2 signalling. J Neurovirol 17:327–340. doi:10.1007/s13365-011-0039-0
Ramage SN, Anthony IC, Carnie FW, Busuttil A, Robertson R, Bell JE (2005) Hyperphosphorylated tau and amyloid precursor protein deposition is increased in the brains of young drug abusers. Neuropathol Appl Neurobiol 31:439–448. doi:10.1111/j.1365-2990.2005.007670.x
Reddy PV, Pilakka-Kanthikeel S, Saxena SK, Saiyed Z, Nair MP (2012) Interactive effects of morphine on HIV infection: role in HIV-associated neurocognitive disorder. AIDS Res Treat. doi:10.1155/2012/953678
Robbins TW, Ersche KD, Everitt BJ (2008) Drug addiction and the memory systems of the brain. Ann NY Acad Sci 1141:1–21. doi:10.1196/annals.1441.020
Schouten J, Cinque P, Gisslen M, Reiss P, Portegies P (2011) HIV-1 infection and cognitive impairment in the cART era: a review. AIDS 25:561–575. doi:10.1097/QAD.0b013e3283437f9a
Tomlinson GS, Simmonds P, Busuttil A, Chiswick A, Bell JE (1999) Upregulation of microglia in drug users with and without pre-symptomatic HIV infection. Neuropathol Appl Neurobiol 25:369–379. doi:10.1046/j.1365-2990.1999.00197.x
Wang TH, Donaldson YK, Brettle RP, Bell JE, Simmonds P (2001) Identification of shared populations of human immunodeficiency virus type 1 infecting microglia and tissue macrophages outside the central nervous system. J Virol 75:11686–11699. doi:10.1128/JVI.75.23.11686-11699.2001
Weed M, Adams RJ, Hienz RD, Meulendyke KA, Linde ME, Clements JE, Mankowski JL, Zink MC (2012) SIV/macaque model of HIV infection in cocaine users: minimal effects of cocaine on behaviour, virus replication and CNS inflammation. J Neuroimmune Pharmacol 7:401–411
Xu J, Ikezu T (2009) The comorbidity of HIV-associated neurocognitive disorders and Alzheimer's disease: a foreseeable medical challenge in post-HAART era. J Neuroimmune Pharmacol 4:200–212. doi:10.1007/s11481-008-9136-0
Zhao L, Galligan DC, Lamers SL, Yu S, Shagrun L, Salemi M, McGrath MS (2009) High level of HIV-1 DNA concentrations in brain tissues differentiate patients with post-HAART AIDS dementia complex or cardiovascular disease from those with AIDS. Sci China C Life Sci 52:651–656. doi:10.1007/s11427-009-0085-5
Zink MC, Brice AK, Kelly KM, Queen SE, Gama L, Li M, Adams RJ, Bartizal C, Varrone J, Rahi A, Graham DR, Tarwater PM, Mankowski JL, Clements JE (2010) Simian immunodeficiency virus-infected macaques treated with highly active antiretroviral therapy have reduced central nervous system viral replication and inflammation but persistence of viral DNA. J Inf Dis 202:161–170. doi:10.1086/653213
Acknowledgments
The Edinburgh HIV Brain Bank is funded by the UK Medical Research Council. We thank the clinicians who looked after these subjects including Drs R Brettle and R Robertson and Professor C Leen, the clinical database manager Alan Wilson, and staff members who have assisted with the investigation of the tissue samples including Frances Carnie and Dr I Anthony. Lastly, this study would not have been possible without the generous trust and support of the donors and their families.
Conflict of interest
All three authors, Dr Donald Smith and Professors Peter Simmonds and Jeanne Bell, declare that they have no conflict of interest with the UK Medical Research Council.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Smith, D.B., Simmonds, P. & Bell, J.E. Brain viral burden, neuroinflammation and neurodegeneration in HAART-treated HIV positive injecting drug users. J. Neurovirol. 20, 28–38 (2014). https://doi.org/10.1007/s13365-013-0225-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-013-0225-3