
Abstract
To investigate the independent and interactive effects of opiate addiction and HIV on neuroinflammation, we measured microglial/macrophage activation and astrogliosis in multiple regions of human brain. Samples of thalamus, frontal gray matter, and frontal white matter were obtained from 46 individuals categorized as: HIV negatives, HIV-negative opiate addicts, HIV positives, HIV-positive opiate addicts, HIV encephalitis (HIVE), and HIVE opiate addicts. Activated brain microglia/macrophages and astrocytosis were quantified by morphometric analysis of immunohistochemical stains for CD68, HLA-D, CD163, and GFAP. The effects of HIV grouping, opiate addiction, and their interaction on expression of the markers were examined in a series of two-way ANOVAs. In opiate addicts, there was generally higher baseline expression of CD68 and HLA-D in HIV negatives, and lower expression in HIV and HIVE, compared to individuals without opiate abuse. Thus, for these markers, and for GFAP in frontal gray, opiates were associated with attenuated HIV effect. In contrast, for CD163, opiates did not significantly alter responses to HIV, and HIV effects were variably absent in individuals without opiate abuse. The divergent impact that opiate addiction displays on these markers may suggest a generally immunosuppressive role in the CNS, with decreased HIV-associated activation of markers CD68 and HLA-D that potentially reflect neurotoxic pathways, and preservation of CD163, thought to be an indicator of neuroprotective scavenger systems. These results suggest a complex impact of opiates on neuroinflammation in baseline and virally stimulated states.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Significant comorbidity exists between HIV infection and substance use disorders, such that the two have been considered “interlinked epidemics” (Nath et al. 2002). Abuse of opiates is a major transmission route, while abuse of other illicit substances, such as cocaine and methamphetamine, has become a primary risk factor for HIV infection (CDC 2007). Literature documenting diverse immunomodulatory effects of substances of abuse (SOA) has evolved, with unclear applicability to the progression of HIV or its neurobiology. In part, this is because of the complexities of the SOA epidemic, where polysubstance use patterns and medical co-morbidities make it difficult to disentangle independent effects in HIV-positive individuals with SOA histories.
Heroin is a common substance of abuse for HIV-positive persons who reside in NYC, where it is estimated that over 110,000 people are living with HIV (New York City Department of Health and Mental Hygiene 2011). One of the mechanisms postulated for the deleterious interaction of HIV and SOA in the human brain is immunomodulation (Burdo et al. 2006). The immunomodulatory effects of opiates make them of particular interest to neuroAIDS investigations because of the brain's inflammatory response to HIV, the fact that these substances modulate immunity in part via CNS receptor-mediated pathways, and the significant potential for functional and behavioral impact on multiple levels (Friedman et al. 2006; Hauser et al. 2007). However, it is likely that the interaction of opiates and local neuroimmune responsiveness to HIV is a highly complex phenomenon, as these substances also influence characteristics of the blood–brain barrier and exert effects on the peripheral immune cells that traffic into the CNS on a daily basis (Dhillon et al. 2007; Friedman et al. 2006; Seelbach et al. 2007). Thus, it is not surprising that there are conflicting reports of immunomodulatory effects in the brain and its cellular constituents, with or without the presence of HIV. While opiates are largely considered immunosuppressive, the few human brain studies that have been performed on intravenous heroin users have suggested enhanced microglial activation in response to HIV (Anthony et al. 2005; Arango et al. 2004; Chao et al. 1997; Hu et al. 2002). The immunomodulatory effects of other substances are even less clear. For example, with regard to cocaine (generally considered pro-inflammatory), studies of human brain to date have emphasized its catastrophic alterations to CNS vasculature that result in intracerebral stroke and hemorrhage, but have not elucidated what effect it might have on intrinsic parenchymal immunomodulation (Aggarwal et al. 1996; Crawford et al. 2006; Dhillon et al. 2007; Hauser et al. 2007; Tyor and Middaugh 1999).
Despite potential CNS vulnerability to SOA-HIV-1 interactions, the present literature describing human brain consequences is limited by very few studies of human brain tissue with appropriate control groups of HIV negatives. Additionally, existing data has been compiled on primarily Caucasian cohorts, limiting generalizability to minorities who currently comprise the majority of HIV-infected adults in the USA (CDC 2007). In this regard, tissue available from the Manhattan HIV Brain Bank presents a unique opportunity to examine neuroinflammation and SOA in the context of HIV, as the cohort is predominantly composed of minority individuals with high rates of substance use disorders. The current study aims to add to the growing body of literature on SOA–HIV interactions, by examining microglial activation in HIV-infected and seronegative substance abusers and comparing the neuroinflammatory influence of opiate addiction in a well characterized, primarily American ethnic minority, urban cohort.
Methods
Patient population
Study subjects were selected from the Manhattan HIV Brain Bank (MHBB; U01MH083501) in New York City, NY. The MHBB is a longitudinal, observational study of both HIV-positive and HIV-negative adults. As part of the subject evaluations, psychiatric and substance use histories and basic laboratory data inclusive of CD4 counts and HIV plasma loads are recorded. Data obtained from this population were obtained in compliance with the Mount Sinai School of Medicine IRB. A total of 46 participants were classified according to positive or negative status for the following variables: HIV, HIV encephalitis (HIVE), and history of opiate (heroin) addiction (administration routes for all but one patient were intravenous). Histories of opiate addiction were ascertained upon interview or chart review, and in 25 patients, confirmed with a semi-structured psychiatric interview, the Psychiatric Research Interview for Substance and Mental Disorders (Hasin et al. 1996) and in 21, urine toxicology. Subjects for this study were a subset of the larger holdings of the MHBB, and were selected by the following criteria: appropriate clinical data available for accurate opiate characterization and immunological status (CD4 count) and absence of opportunistic infections, significant anoxic ischemic damage, and other neuropathologies in the regions of interest that were known to contribute to changes in the microglial/astroglial markers of interest. Once these criteria were met by a sample, we then attempted to match as closely as possible the demographic characteristics (age, gender, and race/ethnicity) of each group, so that there was no significant difference between the groups in mean age, gender composition, and race/ethnicity. This resulted in the following distribution:
-
Group 1
HIV negative; no opiate use syndrome (n = 7)
-
Group 2
HIV negative; opiate addict (n = 7)
-
Group 3
HIV positive; no opiate use syndrome (n = 10)
-
Group 4
HIV positive; opiate addict (n = 13)
-
Group 5
HIV encephalitis; no opiate use syndrome (n = 4)
-
Group 6
HIV encephalitis; opiate addict (n = 5)
Males accounted for 67 % of the total sample and average age was 47 years (8.6). For the participants with HIV, median CD4 count (cells/mm3) was 15.5 (range = 1 − 336) and mean viral load (log10) was 4.39 (1.69). Of note, the groups were composed almost exclusively of African Americans (43.5 %) and Hispanics (50 %). Finally, 44 % of the opiate addicts had documentation of being subsequently maintained on methadone and 36 % were taking or receiving non-methadone opiates.
Human brain processing
At the time of patient demise, brains were obtained and routinely processed as has been previously published (Morgello et al. 2001). A minimum of 57 routine hematoxylin and eosin-stained sections were examined for each brain. Neuropathologic analysis to diagnose the presence of HIVE was performed by a board-certified neuropathologist (SM). With only one exception, the brains used in this study were free of active opportunistic infection or tumor; the one exception was an opiate addict with HIVE (group 6), who had a circumscribed temporal lobe lymphoma which did not involve the brain regions sampled.
The three regions of interest selected for this study included thalamus, frontal gray matter, and frontal white matter. These regions were selected for their comparability to a prior study of brain microglial activation in polysubstance-using Caucasian patients with HIV, and because several of these regions are sites of predilection for HIV-associated neuropathologies (Arango et al. 2004).
Tissue microarray and immunohistochemical staining
Paraffin donor blocks were chosen in the regions of interest (frontal gray, frontal white, and thalamus), and used to create 12 tissue microarrays (TMAs) (three slides for each region) with cores of 1.0 mm diameter (Battifora 1986; Kononen et al. 1998). This redundancy was to ensure that all 46 patients had at least two adherent punches in each region. Microarrays were sectioned at 5 μm, and mounted on coated slides (Fisher superfrost plus, Fisher Corp) for immunohistochemical staining.
Immunohistochemical staining was performed with four antibodies. Details of the primary antibodies manufacturer, clone, and dilutions used are summarized in Table 1. After incubation with the primary antibody, slides were incubated for 30 min with anti-mouse or anti-rabbit Ig ImmPRESS reagent (Vector Laboratories, CA) prior to development with diaminobenzidene chromogen. To minimize run-to-run variability, all sections were stained with each antibody in one cycle. Hematoxylin counterstain was applied, to facilitate identification of the cells of interest.
Morphometric analysis
After immunohistochemical staining, slides were examined with a Nikon Labphot-2 light microscope. Six successive fields at ×40 magnification (total area of analysis, 0.03 mm2) were taken from each tissue core with a Coolpix 950 digital camera attached to the microscope by a Coolpix MDC lens. The percentage of tissue area occupied by antibody-positive cells was determined by an automated system developed at the Mount Sinai School of Medicine (Wu et al. 2008). Two cores were analyzed for each antibody in each anatomical region from each patient, for a total of 12 images. The raw percentage area measures for positive staining in the 12 images were recorded in a Microsoft Excel workbook and were averaged prior to the general analysis.
Validation of TMA as a representation of whole slide IHC
TMAs have the advantage of containing multiple specimens on a single slide, allowing for more uniformity of immunohistochemical staining. Unfortunately, TMAs also reduce the amount of tissue analyzed. It has been shown for several common antigen/antibody pairs that two needle cores adequately represent antigen expression on a whole tissue section with 95 % accuracy; we wished to validate this in our system.
Blocks of deep white matter were chosen from six MHBB cases. Whole 5-μM slices were taken from each block and used in immunohistochemistry using monoclonal anti-CD68 antibody. The slides were visualized with a light microscope and 30 random areas of deep white matter were photographed and used for morphometric analysis. After the whole sections were cut and stained, a TMA was constructed from the residual paraffin blocks. The CD68-stained slides were used as a guide to select five areas of deep white matter, which were punched from the blocks with a 1.0-mm-diameter needle. With six cases represented, this resulted in a TMA with a total of 30 tissue cores. Five-micron slices were cut from the TMA block and stained for CD68 as described. Six images were photographed from each core and analyzed for area of staining. The data were organized in Excel and statistical analysis performed.
Pearson's correlation tests were applied to analyze the number of punches in a TMA that were equivalent to a standard tissue section in morphometric analysis. Five Pearson correlation tests were performed: test 1 used the average of six area measures from punch 1 against six area measures generated from the first six images of the slice; test 2 combined 12 area measures generated from punches 1 and 2 against the first 12 measures generated from the slice, and so on, until the final test was done on 30 images generated from all five punches against 30 measures generated from the 30 images of the slice.
Performing the five correlation tests of punch to slide staining allowed us to determine how many punches were equivalent to a tissue slice in our quantitative analysis. The tests revealed that there was an adequate and statistically significant correlation between two punches and the tissue slice (r = 0.983, p < 0.01).
Data analysis
Statistical analyses were performed using SPSS software (Chicago, IL) version 19.0 for Windows, and repeated using JMP version 9.0.0 on a Macintosh computer (SAS Institute). Two-way analysis of variance (ANOVA) was utilized for the primary analyses of the study. When interaction terms were significant, follow-up one-way ANOVAs were completed separately for the opiate and no opiate groups with HIV status as the independent variable, and post hoc analysis by Tukey's test. Chi-square analyses were applied to categorical variables. Independent variables in these analyses included HIV status (HIV negative, HIV positive, or HIVE) and opiate addiction history status. The dependent variable was average antibody staining area for each region of interest. An analysis of the relationship between microglial/astrocytic cell response and peripheral immunovirologic status in the HIV and opiate groups was completed using Spearman's bivariate correlations. Viral loads were log10 transformed prior to analyses.
Results
Effects of HIV status, opiate addiction, and their interaction, on neuroimmune and glial markers
To determine the effects of HIV grouping, opiate addiction, and their interaction on neuroimmune and glial markers, a series of two-way ANOVAs was performed in each brain region for each marker. The p values for the overall models and interaction terms from these analyses are presented in Table 2. For each opiate group, marker, and region, means and standard deviations are additionally detailed in Table 3, along with p values from simple tests of significance (one-way ANOVA) for HIV status when overall models indicated the presence of significant group differences. The effects of opiate addiction on marker expression in the HIV groups are depicted in Fig. 1.

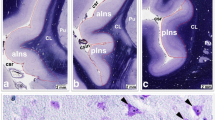
Immunohistochemical stains for CD68 (a–f) and CD163 (g–l) in frontal white matter of subjects with and without opiate addiction, with and without HIV and HIV encephalitis. (Diaminobenzidene chromogen, hematoxylin counterstain, original magnification ×40)
For CD68, the overall model (two-way ANOVA) demonstrated significant differences in frontal white matter (F (5,40) = 6.4903, p = 0.0002) and thalamus (F (5,39) = 2.6085, p = 0.0396), but not frontal gray matter (F (5,40) = 1.7877, p = 0.1375). Significant or trend level interactions between opiate status (addict, no abuse) and HIV grouping (negative, positive, and HIVE) were seen for CD68 in frontal white matter (p = 0.0323) and thalamus (p = 0.0906). In frontal white matter, for individuals with no opiate abuse, the HIVE group displayed significantly higher CD68 expression than HIV-positive and HIV-negative groups (p < 0.0001). Likewise, in thalamus, there was a stepwise effect of HIV status in that the HIVE group displayed significantly higher CD68 expression than HIV positive, who displayed higher levels than HIV negative (p = 0.0645). In contrast to the significant or trend level effects of HIV status on CD68 expression in individuals without history of opiate abuse, in subjects with opiate addiction, differences in CD68 expression were not significant in any brain region. For all brain regions, CD68 staining in HIV negatives was greater in individuals with opiate addiction than in those without; conversely, for all brain regions, CD68 staining in HIVE positives and HIVE was less in opiate addicts than in those without. Thus, in the presence of opiate addiction, baseline levels of CD68 in HIV negatives were elevated, and the increase of levels in association with HIV infection was decreased.
For HLA-D, the overall model demonstrated significant differences in the thalamus (F (5,39) = 3.4599, p = 0.0111) and trend level differences in frontal gray (F (5,40) = 2.2817, p = 0.0647), but not frontal white matter (F (5,40) = 1.3958, p = 0.2466). The interaction terms were significant or at trend level for HLA-D in frontal gray matter (p = 0.0716) and thalamus (p = 0.0300), wherein the effect of HIV status on expression showed a similar attenuation in opiate addicts as was seen with CD68. Follow-up one-way ANOVAs revealed that in the absence of opiate abuse, there was a trend for the HIV-positive group to demonstrate greater HLA-D staining than the HIV-negative group in frontal white matter (F (2,19) = 3.05, p = 0.0724). A significant HIV effect was also observed in the thalamus in the non-opiate group (F (2,18) = 4.30, p = 0.0308), where post hoc tests indicated that the HIV positive displayed higher HLA-D than both the HIV negative and HIVE groups. Differences in HLA-D expression in opiate addicts were not significant in any brain region; there was only a trend level effect in frontal gray matter (F (2, 23) – 2.87, p = 0.0783). In this region, post hoc tests revealed significantly greater HLA-D staining in the HIVE group than the HIV negatives and HIV positives.
In contrast to the patterns seen with CD68 and HLA-D, opiate addiction did not appear to attenuate the response of CD163 to HIV. For CD163, the overall model demonstrated significant differences in the frontal gray (F (5,40) = 3.8910, p = 0.0057) and frontal white matter (F (5,40) = 6.0320, p = 0.0003), but not thalamus (F (5,39) = 1.5207, p = 0.2058). No significant or trend level interactions were seen for CD163. In opiate addicts, the effect of HIV status on CD163 expression was significant in frontal white matter (F (2, 23) = 26.85, p < 0.0001) and frontal gray matter (F (2, 23) = 3.41, p = 0.0513). Post hoc tests revealed that the HIVE groups demonstrated significantly greater CD163 staining than both the HIV-positive and -negative groups in all regions tested. In contrast, in the absence of opiate abuse, CD163 did not show significant HIV effects in frontal white matter, but was significant in frontal gray matter (F (2, 19) = 6.14, p = 0.0093), where both the HIVE and HIV-positive groups demonstrated significantly greater CD163 staining than the HIV negatives.
The overall model for GFAP was significant only in frontal gray matter (F (5,40) = 3.1545, p = 0.0171), where no interaction effect was observed, but a simple main effect of HIV status was significant (F (2, 40) = 5.660, p = 0.007). In the absence of opiate abuse, a significant HIV effect was observed (F (2, 19) = 8.79, p = 0.0022). Post hoc analyses revealed that for these subjects without opiate abuse, both the HIVE and HIV-positive groups demonstrated significantly greater GFAP staining than the HIV negatives. Analyses for subjects with opiate addiction did not reach statistical significance, but qualitative examination of the mean staining values demonstrate that the HIVE group evidenced higher GFAP levels than HIV positive and HIV negatives (F (2, 23) = 2.23, p = 0.1312).
Thus, in general, individuals with opiate addiction showed higher mean levels of CD68 and HLA-D staining in the HIV-negative state, lower CD68 levels in the HIV positive and HIVE conditions, and lower HLA-D in the HIV-positive conditions, when contrasted to individuals without opiate abuse (Table 3). This effect was not seen for CD163, where opiate addicts had generally higher peak levels in HIVE and variable attenuation in HIV-negative states. Thus, opiate addiction appeared to attenuate the significant HIV-associated rise in CD68 and HLA-D, and enhance CD163 responsiveness.
Effects of CD4 count and plasma viral load on neuroimmune and glial markers
To determine the relationship between HIV patient (combined HIV and HIVE group) immunovirologic status and expression of the brain markers of interest, an analysis of the relationship between CD4 count and plasma viral load to CD68, HLA-D, CD163, and GFAP was completed within each opiate grouping (Table 4). HIV-positive opiate addicts did not have a significantly different mean CD4 count than HIV-positive individuals without opiate abuse (mean (SEM) CD4 for opiate addicts = 76 (21); for no opiate abuse = 46 (24); p = 0.3643); nor did they differ significantly in plasma HIV load (mean log10 HIV load for opiate addicts = 4.18 (0.42); for no opiate abuse = 4.65 (0.45); p = 0.4450).
We examined the correlation within each opiate group of immunovirologic indices and expression of brain markers. In the opiate addicts, the correlation between increasing CD4 and decreasing expression of CD68 was stronger in gray matter regions than in the non-abusing population. Additionally, within the opiate group, there was a negative correlation with CD163 that was not present in non-abusers. However, correlation of decreasing GFAP with increasing CD4 was not present in addicts, and present in gray matter regions of non abusers. Thus, these correlative analyses generally showed tighter relationship between attenuation of microglial markers and increasing CD4 count in the opiate abusers than non-opiate group, but a reversal of this phenomenon with regard to astrocyte marker GFAP. With regard to viral load, no correlations were seen in the absence of opiate addiction, whereas addicts had significant correlations of gray matter CD68 and white matter CD163 with plasma viral load.
Discussion
Documentation and analysis of the immunomodulatory impact of SOA has been undertaken for several decades, with the fundamental clinical observation that a spectrum of drug users show increased susceptibility to microbial infections (Cabral 2006; Friedman et al. 2006). With the onset of the HIV epidemic, concerns arose that SOA would modulate the natural history of infection, although a fully realized observation of this phenomenon and its underlying pathogenetic mechanism have remained problematic. This may largely be due to the significant behavioral and medical co-morbidities in HIV-infected addicts, and in part because of the polysubstance-using habits of the individuals under study (Burdo et al. 2006; Cabral 2006).
The complex immunomodulatory effects of SOA have been well documented in animal models and in vitro systems, and may be mediated at multiple levels, including receptor-initiated pathways in the CNS and peripheral immune effector cells (Burdo et al. 2006). Both innate and adaptive immunity can be significantly altered, and in some paradigms, SOA bias T helper cell divergence from a pro-inflammatory, anti-microbial Th1 pathway to the humoral Th2 (Friedman et al. 2006). Significant interactions between these effects and the immunosuppressive impact of HIV raise concerns of harmful synergies, and recently, focus on neuroAIDS disorders as an important location for these synergies has arisen (Berman et al. 2006). Thus, a nascent literature examining the impact of SOA on brain inflammation has evolved, largely in experimental systems, and to a lesser degree in primary human CNS materials. Access to and interpretation of primary human materials remain problematic, as substance users often display multiple confounds, have polysubstance habits, and CNS tissues and fluids are not easily acquired. In this regard, the MHBB represents a unique resource, as it follows a large number of HIV-infected individuals with substance use disorders, is targeted to their nervous system characterization, and the program banks CSF and brain tissue when available.
In general, exogenous opiates are considered immunosuppressive, although it has been noted that most experimental paradigms investigating this phenomenon have utilized acute administration, and more conflicting results may be obtained in models with subacute or chronic administration (Eisenstein et al. 2006). With regard to studies examining the impact of opiates on CNS cells, it has been demonstrated that activation of mu opioid receptors inhibits microglial cell chemotaxis and promotes their apoptosis, supporting the notion of an anti-inflammatory role within the brain (Chao et al. 1997; Hu et al. 2002). However, in the presence of HIV tat, astrocytes treated with morphine increase their release of MCP-1, RANTES, and IL-6, potentially contributing to a pro-inflammatory environment, although it should be noted that not all chemokines are pro-inflammatory and some may contribute to diminished responsiveness (El-Hage et al. 2005; Hauser et al. 2007). It has also been demonstrated that morphine enhances HIV replication in peripheral blood mononuclear cells, and has been shown to potentiate TNF alpha production from human microglial cell cultures (Peterson et al. 1998). The effects of morphine are mediated through mu opioid receptors and are thought to result from enhanced reactivity to other stimuli (such as HIV or its proteins) (Peterson et al. 1998). In contrast, human microglia exposed to the kappa opioid peptide dynorphin demonstrate direct upregulation of TNF alpha and IL-6 in the absence of other immune stimuli (Peterson et al. 1998).
Studies of opioids in animal models of HIV infection have arrived at contradictory conclusions regarding immunomodulation. Mice given systemic morphine showed increased numbers of macrophages/microglia at intracerebral sites of tat injection (El-Hage et al. 2006). However, simian models have shown conflicting results regarding enhanced viral virulence, with some studies showing beneficial, and others, detrimental effects of chronic morphine administration (Burdo et al. 2006). For SIV neuropathogenesis, one study has documented enhanced CNS replication in the context of chronic morphine exposures (Kumar et al. 2006).
In the context of the contradictory findings of animal and in vitro analyses, there have been few human brain studies published to examine the role of opiates in the neuroimmune response to HIV. All have been conducted in the Caucasian Edinburgh cohort, utilizing the same basic technique (immunohistochemistry) as the current study (Anthony et al. 2005; Arango et al. 2004; Tomlinson et al. 1999). One of these studies observed an upregulation of microglia in the brains of HIV-negative intravenous drug users (IVDU) when contrasted with HIV-negative individuals without drug addiction, and failed to find a significant difference in the density of microglial cells between HIV-negative IVDU and HIV-positive, pre-symptomatic IVDU (Tomlinson et al. 1999). Two studies examining the response to HIV encephalitis demonstrated trends for increased staining of microglia in IVDU when compared to non-IVDU individuals with HIV and HIVE; however, significance at p < 0.05 was not achieved in any region (Anthony et al. 2005; Arango et al. 2004).
In the context of these prior studies, our analysis of a predominantly minority cohort grouped into HIV negative, HIV positive, and HIVE, shows that with opiate addiction, there is an attenuation of macrophage/microglial activation, with tendencies for enhanced expression of CD68 and HLA-D in the absence of HIV, and diminution of an HIV-associated increase. Our results suggest that there may be a bi-phasic effect of chronic opiates in the CNS with regard to these stimulatory molecules: in the absence of other CNS immune stimuli, chronic opiate exposure may actuate glial-stimulatory effects, resulting in enhanced baseline immune responsiveness. When challenged with a pathogen such as HIV, the chronically opiate exposed brain may conversely show a decreased ability to respond, with impaired microglial function. Of interest, when plasma samples derived from members of the MHBB cohort were examined for levels of lipopolysaccharide (LPS), individuals with intravenous heroin abuse were found to have higher levels than those without, suggesting that decreased immune responsiveness to intestinal bacterial pathogens may be operant in this cohort (Ancuta et al. 2008). When injected systemically, LPS is capable of activating microglia and stimulating brain cytokine response in both the absence and presence of intrinsic brain pathologies (Combrinck et al. 2002; Perry 2004; Teeling et al. 2007). Our findings, taken together with the literature on LPS, may lead to the following hypothesis: with chronic opiates, attenuation of response to gut pathogens may result in basal CNS stimulation on the basis of circulating factors such as LPS. When infection enters the brain, as with HIVE, there may then be continued impedance to normal responsiveness in the context of the nervous system.
Of interest in the present study was the differential effect of opiates on CD163 expression, when contrasted with CD68 and HLA-D. CD163 is a hemoglobin scavenger receptor, and marks perivascular macrophages in the normal human CNS (Fabriek et al. 2005; Kim et al. 2006). These cells are critical to antigen presentation and co-stimulation, and are important targets of HIV infection (Fabriek et al. 2005; Kim et al. 2006). They are important in the transmission of systemic inflammatory stimuli to the CNS and are constitutively activated (Galea et al. 2008). There is a suggestion in the literature that these cells are important in the anti-inflammatory response; in HIV neuropathology, it has already been noted that the pattern of CD163 expression is different and distinguishable from the pattern of other markers of activated microglia, including HLA-DR (Roberts et al. 2004). Thus, the divergent effects noted in the present study build on these initial observations: while opiates abrogate the effect of HIV on stimulatory molecules CD68 and HLA-D, their apparent enhancement of HIV effects on CD163 suggests that this may be a critical component of suppressing CNS immune responsiveness. Further examination of this potential pathway is indicated.
Interesting divergence was also seen with regard to microglial and astrocytic markers and their correlation with immunovirologic indices in opiate and non-opiate groups. While HIV-opiate interactions were present in microglia in multiple regions of brain in this study, a similar effect was not seen for astrocytes. Furthermore, non-opiate groups showed correlations of GFAP with CD4, but not opiate addicts. This may suggest that mechanisms of chronic opiate action on the CNS may vary between cell compartments. Alternatively, it may be that the one marker we chose to study, GFAP, may not reflect the full extent of opiate interactions in this cell compartment. Further study may be warranted.
Finally, the apparent contradiction between the present study and prior studies of the predominantly Caucasian Edinburgh cohort needs to be addressed. While our study found evidence of opiate-associated immunosuppression, the opposite effect was seen in subjects from Edinburgh (Anthony et al. 2005; Arango et al. 2004; Tomlinson et al. 1999). There are many potential reasons for this discrepancy. First, it has been noted that the Edinburgh group is largely polysubstance, and it is unclear how the neurobiological impact of their SOA utilization was determined to be a result of the opiate component. In our cohort, most individuals were maintained on methadone or other medical opiates. This raises the possibility, or even probability, that the effects witnessed in brains from the MHBB reflect chronic utilization of non-heroin opiates, and not other SOA. Another factor that was not reported in the Edinburgh studies was the comparative CD4 and HIV load in analytic groups, which may have biased reactivities. In MHBB, the significant differences that were observed were not due to systemic immunovirologic parameters. It is also possible that because of the dramatically different demographic compositions of the two samples, differences may reflect a disparity based on some unidentified racial characteristic. Future studies are indicated, using larger sample sizes, with more attention to the temporal and quantitative aspects of the SOA, and any potential demographic variability. Additionally, it will be essential to correlate the results of this study with behavioral and cognitive phenotypes, as neuroinflammation is critical to the clinical manifestations of neuroAIDS disorders.
References
Aggarwal SK, Williams V, Levine SR, Cassin BJ, Garcia JH (1996) Cocaine-associated intracranial hemorrhage: absence of vasculitis in 14 cases. Neurology 46:1741–1743
Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, Zaman T, Stone D, Mefford M, Morgello S, Singer EJ, Wolinsky SM, Gabuzda D (2008) Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS One 3:e2516
Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE (2005) Does drug abuse alter the microglial phenotype and cell turnover in the context of advancing HIV infection? Neuropathol Appl Neurobiol 31:325–338
Arango JC, Simmonds P, Brettle RP, Bell JE (2004) Does drug abuse influence the microglial response in AIDS and HIV encephalitis? AIDS 18:S69–S74
Battifora H (1986) The multitumor (sausage) tissue block: novel method for immunohistochemical antibody testing. Lab Invest 55:244–248
Berman JW, Carson MJ, Chang L, Cox BM, Fox HS, Gonzalez RG, Hanson GR, Hauser KF, Ho WZ, Hong JS, Major EO, Maragos WF, Masliah E, McArthur JC, Miller DB, Nath A, O'Callaghan JP, Persidsky Y, Power C, Rogers TJ, Royal W 3rd (2006) NeuroAIDS, drug abuse, and inflammation: building collaborative research activities. J Neuroimmune Pharmacol 1:351–399
Burdo T, Katner S, Taffe M, Fox H (2006) Neuroimmunity, drugs of abuse, and neuroAIDS. J Neuroimmune Pharmacol 1:41–49
Cabral GA (2006) Drugs of abuse, immune modulation, and AIDS. J Neuroimmune Pharmacol 1:280–295
CDC (2007) HIV/AIDS Fact sheet: a glance at the HIV/AIDS epidemic. Center for Disease Control and Prevention (CDC)
Chao CC, Hu S, Shark KB, Sheng WS, Gekker G, Peterson PK (1997) Activation of mu opioid receptors inhibits microglial cell chemotaxis. J Pharmacol Exp Ther 281:998–1004
Combrinck MI, Perry VH, Cunningham C (2002) Peripheral infection evokes exaggerated sickness behaviour in pre-clinical murine prior disease. Neuroscience 112(1):7–11
Crawford FC, Wood ML, Wilson SE, Mathura VS, Hollen TR, Geall F, Kolippakkam DN, Mullan MJ (2006) Cocaine induced inflammatory response in human neuronal progenitor cells. J Neurochem 97:662–674
Dhillon NK, Peng F, Bokhari S, Callen S, Shin SH, Zhu X, Kim KJ, Buch SJ (2007) Cocaine-mediated alteration in tight junction protein expression and modulation of CCL2/CCR2 axis across the blood-brain barrier: implications for HIV-dementia. J Neuroimmune Pharmacol 3(1):52–56
Eisenstein TK, Rahim RT, Feng P, Thingalaya NK, Meissler JJ (2006) Effects of opioid tolerance and withdrawal on the immune system. J Neuroimmune Pharmacol 1:237–249
El-Hage N, Gurwell JA, Singh IN, Knapp PE, Nath A, Hauser KF (2005) Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia 50:91–106
El-Hage N, Wu G, Wang J, Ambati J, Knapp PE, Reed JL, Bruce-Keller AJ, Hauser KF (2006) HIV-1 tat and opiate-induced changes in astrocytes promote chemotaxis of microglia through the expression of MCP-1 and alternative chemokines. Glia 53:132–146
Fabriek BO, van Haastert ES, Galea I, Polfliet MMJ, Dopp ED, van den Heuvel MM, van den Berg TK, de Groot CJA, der Valk PV, Dijkstra CD (2005) CD163-positive pervascular macrophages in the human CNS express molecules for antigen recognition and presentation. Glia 51:297–305
Friedman H, Pross S, Klein TW (2006) Addictive drugs and their relationship with infectious diseases. FEMS Immunol Med Microbiol 47:330–342
Galea I, Felton LM, Waters S, van Rooijen N, Perry VH, Newman TA (2008) Immune-to-brain signaling: the role of cerebral CD163-positive macrophages. Neurosci Lett 448:41–46
Hasin D, Trautman K, Miele G, Samet S, Smith M, Endicott J (1996) Psychiatric Research Interview for Substance and Mental Disorders (PRISM): reliability for substance abusers. Am J Psychiatry 153:1195–1201
Hauser KF, El-Hage N, Stiene-Martin A, Maragos WF, Nath A, Persidsky Y, Volsky DJ, Knapp PE (2007) HIV-1 neuropathogenesis: glial mechanisms revealed through substance abuse. J Neurochem 100:567–586
Hu S, Sheng WS, Lokensgard JR, Peterson PK (2002) Morphine induces apoptosis of human microglia and neurons. Neuropharmacology 42:829–836
Kim WK, Alvarez X, Fisher J, Bronfin B, Westmoreland S, McLaurin J, Williams K (2006) CD163 identifies perivascular macrophages in normal and viral encephalitic brains and potential precursors to perivascular macrophages in blood. Am J Pathol 168(3):822–834
Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton S, Torhorst J, Mihatsch MJ, Sauter G, Kallioniemi OP (1998) Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med 4:844–847
Kumar R, Orsoni S, Norman L, Verma AS, Tirado G, Giavedoni LD, Staprans S, Miller GM, Buch SJ, Kumar A (2006) Chronic morphine exposure causes pronounced virus replication in cerebral compartment and accelerated onset of AIDS in SIV/SHIV-infected Indian rhesus macaques. Virology 354:192–206
Morgello S, Gelman BB, Kozlowski PB, Vinters HV, Masliah E, Cornford M, Cavert W, Marra C, Grant I, Singer EJ (2001) The National NeuroAIDS Tissue Consortium: a new paradigm in brain banking with an emphasis on infectious disease. Neuropathol Appl Neurobiol 27:326–335
Nath A, Hauser KF, Wojna V, Booze RM, Maragos W, Prendergast M, Cass W, Turchan JT (2002) Molecular basis for interactions of HIV and drugs of abuse. J Acquir Immune Defic Syndr 31(Suppl):S62–S69
New York City Department of Health and Mental Hygiene (2011) New York City HIV/AIDS annual surveillance statistics. New York City Department of Health and Mental Hygiene, New York
Perry VH (2004) The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav Immun 18:407–413
Peterson PK, Molitor TW, Chao CC (1998) The opioid-cytokine connection. J Neuroimmunol 83:63–69
Roberts ES, Masliah E, Fox HS (2004) CD163 identifies a unique population of ramified microglia in HIV encephalitis (HIVE). J Neuropathol Exp Neurol 63(12):1255–1264
Seelbach MJ, Brooks TA, Egleton RD, Davis TP (2007) Peripheral inflammatory hyperalgesia modulates morphine delivery to the brain: a role for P-glycoprotein. J Neurochem 102:1677–1690
Teeling JL, Felton LM, Deacon RMJ, Cunningham C, Rawlins JNP, Perry VH (2007) Sub-pyrogenic systemic inflammation impacts on brain and behavior, independent of cytokines. Brain Behav Immun 21:836–850
Tomlinson GS, Simmonds P, Busuttil A, Chiswick A, Bell JE (1999) Upregulation of microglia in drug users with and without pre-symptomatic HIV infection. Neuropathol Appl Neurobiol 25:369–379
Tyor WR, Middaugh LD (1999) Do alcohol and cocaine abuse alter the course of HIV-associated dementia complex? J Leukoc Biol 65:475–481
Wu HS, Murray J, Morgello S (2008) Segmentation of brain immunohistochemistry images using clustering of linear centroids and regional shapes. J Imaging Sci Technol 52:040502–040502-11
Acknowledgments
The authors thank the participants and staff of the Manhattan HIV Brain Bank. This work is supported by grants U01MH083501 from the National Institutes of Mental Health (NIMH) and Neurological Disorders and Stroke (NINDS) (to SM) and UL1RR029887 from the National Center for Research Resources (NCRR) (to the Mount Sinai School of Medicine).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Byrd, D., Murray, J., Safdieh, G. et al. Impact of opiate addiction on neuroinflammation in HIV. J. Neurovirol. 18, 364–373 (2012). https://doi.org/10.1007/s13365-012-0118-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-012-0118-x