
Abstract
Severe HIV-associated neurocognitive disorders (HAND), such as HIV-associated dementia, and opportunistic CNS infections are now rare complications of HIV infection due to comprehensive highly active antiretroviral therapy (HAART). By contrast, mild to moderate neurocognitive disorders remain prevalent, despite good viral control in peripheral compartments. HIV infection seems to provoke chronic CNS injury that may evade systemic HAART. Penetration of antiretroviral drugs across the blood–brain barrier might be crucial for the treatment of HAND. This review identifies and evaluates the available clinical evidence on CSF penetration properties of antiretroviral drugs, addressing methodological issues and discussing the clinical relevance of drug concentration assessment. Although a substantial number of studies examined CSF concentrations of antiretroviral drugs, there is a need for adequate, well designed trials to provide more valid drug distribution profiles. Neuropsychological benefits and neurotoxicity of potentially CNS-active drugs require further investigation before penetration characteristics will regularly influence therapeutic strategies and outcome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
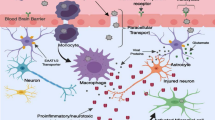
HIV-associated neurocognitive disorders (HAND) remain a challenge for the treatment of HIV infection. After the virus has penetrated the CNS in early stages of infection, both infected lymphocytes crossing the blood–brain barrier (BBB) and resident macrophages and microglia sustain HIV replication in the CNS [1], leading to neuronal damage and HAND [2]. As a result of highly active antiretroviral therapy (HAART), the incidence of HIV-associated dementia (HAD) and HIV-associated CNS opportunistic infections has declined, but mild to moderate neurocognitive impairment remains prevalent [3–6]. HAART can improve and often reverse neurocognitive dysfunction and suppress the viral burden in the CSF, a suggested surrogate marker for CNS infection [7, 8]. Benefits of therapy, however, vary from individual to individual. Even with suppression of HIV-RNA in the CSF to undetectable levels, milder forms of neurocognitive dysfunction may persist [9, 10] and markers of intrathecal immunoactivation regularly remain elevated [11–13]. While the viral load in the systemic compartment rapidly falls below the detection limit after the initiation of HAART, the antiviral response is often delayed in the CSF relative to the blood [14]. All these observations suggest that HAART is not as effective in the CNS as it is in peripheral compartments, raising the concern of insufficient penetration of antiretroviral drugs (ARVs) across the BBB. The ability of ARVs to reach therapeutic concentrations within the CNS is crucial in the face of the high-replication rates of CNS infection, as occurs in HAD [1], and might also reduce ongoing low-grade viral replication [15–17], possibly preventing the genetic compartmentalization of HIV infection, the development of a drug-resistant virus and irreversible damage within the CNS.
The CHARTER (CNS HIV Antiretroviral Therapy Effects Research) study group has devised a ranking scheme in order to quantify and compare the effectiveness of ARVs in the CNS. A revised version of this system was proposed in 2010 (see Table 1) [18]. On the basis of information from the literature on measured CSF concentrations, physiochemical drug characteristics and effectiveness in the CNS (reflected by suppression of CSF viral load and improved neurocognitive performance), the ranking system divides drugs into four categories according to penetration estimates. Individual ranking scores of the drugs included in a therapeutic regimen are summed up in the CNS penetration-effectiveness (CPE) rank [15, 18]. Altogether, the application of this ranking system has been successful. Higher CPE scores, consistent with higher penetration estimates, are associated with lower HIV-RNA levels in the CSF [15, 17, 19]. There has also been an association between higher CPE scores and neurocognitive improvement in HAND-affected patients [16, 19–21] and perinatally HIV-infected children [22], though results have not always been consistent [17].
Although at present the role of CNS penetration by ARVs for the treatment of various forms of HAND is controversial, the extent to which components of HAART can be detected in the CNS is of strong interest for two reasons. First, to provide extensive information for prospective trials to further investigate this question. Secondly, the fact that HIV is a neurotropic virus that penetrates the CNS early in the course of disease implies that the CNS must be one of the target sites for therapy. Healthcare providers who treat neurological manifestations of HIV infection should be aware of basic pharmacological properties of HAART components. The aim of this systematic review is to synthesize and evaluate the available clinical data on the penetration of ARVs into the CSF. The findings are discussed in the context of their clinical implications.
1.1 Transport of Drugs Across the Blood–Brain Barrier
Passive transport across the BBB is influenced by the chemical and physical properties of a drug. The main contributing factors are ionization, molecular weight, lipophilicity and protein binding. High molecular weight can potentially impair passive drug transport across biological membranes. In this context, the molecular weight of some components of HAART, for example of many protease inhibitors (PIs), might be critical [23]. In contrast, lipophilic properties enhance passive drug diffusion, being generally directly proportional to the transport rate of a drug across lipid membranes. However, highly lipophilic drugs may be ‘trapped’ inside the membrane, complicating partition into the opposite extracellular compartment [23, 24]. Furthermore, the affinity to plasma proteins limits penetration, as the passage of drugs across the BBB is restricted to the unbound fraction [25].
In addition to passive drug diffusion and facilitated transport, a variety of active transporters carry anti-HIV drugs across the BBB and the blood–CSF barrier. Transport occurs in both directions and is affected by interaction, inhibition and induction by concomitant drugs [26]. Among a number of potential and more or less characterized transporters localized at the barriers to the CNS, the efflux transporter P-glycoprotein (P-gp) from the family of multidrug resistance-associated proteins (MRPs) was investigated most extensively. Expressed on the luminal surface of brain capillary endothelium and in the choroid plexus’ epithelial cells [27], P-gp limits delivery of several ARVs to the CNS by active efflux, representing an efficient component of the BBB [23, 25].
1.2 Methods of Literature Review
We performed a systematic search for studies assessing drug concentrations of commonly used anti-HIV drugs in the CSF, which are zidovudine (AZT), stavudine (d4T), lamivudine (3TC), abacavir sulfate (ABC), tenofovir disoproxil fumarate (TDF), emtricitabine (FTC), nevirapine (NVP), efavirenz (EFV), etravirine (ETV), saquinavir (SQV), ritonavir (RTV), indinavir (IDV), nelfinavir (NFV), amprenavir (APV), lopinavir (LPV), atazanavir (ATV), fosamprenavir (FPV), darunavir (DRV), enfuvirtide (T-20), maraviroc (MVC) and raltegravir (RAL). PubMed was searched from 1980 to June 2012 for relevant studies. The following combinations of keywords were used: (‘highly active antiretroviral therapy’ OR HAART OR cART) AND (CSF OR ‘cerebrospinal fluid’); [drug name] AND (CSF OR ‘cerebrospinal fluid’); [drug name] AND (CNS OR ‘central nervous system’ OR brain). Additionally, reference lists of review articles were hand searched. Abstract data from the Conferences on Retroviruses and Opportunistic Infections (CROI) from 1997 to 2012 were searched. Reports on clinical studies were included when they provided concentration values of one or more of the above-mentioned ARVs in the CSF. Case reports and clinical trials considering less than four CSF samples per dose were excluded. Preliminary data from conference abstracts were included only if one or less published studies were available for a drug. Reports in languages other than English, French or German were excluded. From eligible reports, relevant information was extracted, including study design, study size, drug regimen, CSF post-dose sampling time, CSF drug concentrations, CSF-to-plasma concentration ratio, estimated antiviral activity in the CSF, neurological status of study subjects and neurological outcome measures.
2 Results
2405 records were identified through searching of PubMed. Sixty-six published studies met the eligibility criteria. Additionally, two unpublished conference abstracts were included in the review. The characteristics of eligible clinical studies are listed in Table 2, sorted by drug class and date of publication.
By now, CSF drug concentrations are available for all of the commonly used ARVs. Due to largely heterogeneous study designs and subject characteristics, we did not perform a quantitative meta-analysis in this review. Clinical data on CSF penetration of ARVs derive largely from observational trials with small study sizes. Generally, ARVs show limited penetration of the BBB, reflected by CSF-to-plasma concentrations ratios below 100 % in all studies included in this review. Still, drugs differ importantly in their ability to accumulate in the CSF.
2.1 Nucleoside and Nucleotide Reverse Transcriptase Inhibitors
Nucleoside and nucleotide reverse transcriptase inhibitors (NRTIs) such as zidovudine were the first drugs found to be effective against HIV-associated CNS disease. In vitro, NRTIs show remarkable activity against HIV replication in macrophages, the principal target cells for HIV in the CNS [96]. Clinical studies have demonstrated notable CSF penetration for zidovudine, stavudine, lamivudine, abacavir and emtricitabine (Table 2). In contrast, CSF concentrations of tenofovir have been relatively low with a median CSF-to-plasma concentration ratio of about 5 % [51, 52].
The degree of binding to plasma proteins is generally low for NRTIs, ranging from 0.7 % for tenofovir to 50 % for abacavir, and should not substantially affect the amount of drug available to be distributed into the CNS. Abacavir has the most marked lipophilic properties and the highest affinity to plasma proteins among this class of ARVs. About 50 % of systemic abacavir is bound to plasma proteins and thus not available for transport into the CNS; substantial lipophilicity, however, enhances its ability to cross cell membranes and to penetrate into body tissues, including the brain [97]. Indeed, measured CSF concentrations of abacavir suggest considerable penetration (see Table 2).
CSF-to-plasma concentration ratios of zidovudine, stavudine, lamivudine, abacavir and emtricitabine increase over time after dosing [32–34, 43, 44, 48, 50, 98]. Accumulation and elimination kinetics of these drugs are slower in the central compartment than in plasma, reflected by delayed peak concentrations and extended drug exposure in the CSF. Therefore, most of the values presented in Table 2 are influenced by the time span between drug intake and CSF sampling.
CSF concentrations of zidovudine, stavudine, lamivudine, abacavir and emtricitabine exceeded the 50 % inhibitory concentration (IC50), a measure of antiviral drug potency, in all studies evaluating this relationship and largely throughout the respective dosing interval. In contrast, tenofovir concentrations in the CSF exceeded IC50 in only a minority of samples [52]. In view of the remarkable efficacy of tenofovir in macrophages in vitro, it would be a promising agent for CNS HIV infection [96], activity in the CNS, however, seems to be limited by poor penetration [51, 52].
The exact entry route of NRTIs into the CNS is not clear. As CSF and plasma concentrations were not strongly associated with one another, processes other than simple passive diffusion are likely to play a role in the penetration of tenofovir into the CSF [52]. Several unspecific organic anion and cation transporters may contribute to brain uptake and efflux of NRTIs [26, 99].
2.2 Non-Nucleoside Reverse Transcriptase Inhibitors
Penetration of nevirapine into the CSF is generally good, likely due to the lipophilic properties of that drug [37, 38, 54]. Concentration values, however, have not been related to parameters of antiviral potency. In concordance with clinical results, Gibbs et al. [100] found the degree of accumulation in the brain to be greater for nevirapine than for zidovudine, stavudine, abacavir, lamivudine, ritonavir, amprenavir and tenofovir in a guinea pig brain perfusion model. Unlike nevirapine, CSF penetration of efavirenz has been reported to be less than 1 % of concomitant plasma concentrations [55, 56], though this cannot be taken to indicate pharmacological ineffectiveness or viral escape in the central compartment. The estimated unbound concentrations of efavirenz in the CSF approximate the free plasma fraction and exceed the 95 % inhibitory concentration (IC95) [55]. In addition, there is indirect evidence indicating that efavirenz does achieve relevant concentrations within the CNS, as this drug has widely recognized CNS adverse effects [101, 102]. Median CSF-to-plasma concentration ratios of etravirine have also been relatively low (1–4 %), but exceeded the IC50 [57, 58]. Extensive binding of etravirine to proteins, as observed in the blood (99.9 %), is not to be expected in the CSF, so that a contribution to viral control in the CNS is quite possible.
2.3 Protease Inhibitors
PIs have several physical and chemical characteristics that potentially impede passive diffusion into the central compartment [23]. A common property of this drug class is its extensive binding affinity to plasma proteins. Protein-bound fractions in the plasma range from 60 % for indinavir, 86 % for atazanavir and 90 % for fosamprenavir to more than 98 % for saquinavir, lopinavir, ritonavir and nelfinavir. Therefore, just a small fraction of the drug in the plasma is free to cross membranes. Molecular weights of PIs are high and might additionally limit penetration. PIs are highly lipophilic, a property generally favourable for passive transport, but penetration might be limited by ‘membrane trapping’ [23]. Lastly, P-gp-mediated efflux from the brain back to blood was demonstrated for PIs [103, 104].
Saquinavir, ritonavir, indinavir, nelfinavir, amprenavir, lopinavir and atazanavir have been detected in the CSF in a number of studies (see Table 2); for darunavir, two published studies were available [64, 88]. CSF concentrations of most PIs have been lower than expected from the estimated free plasma fraction, suggesting the influence of active transporters at the BBB and/or at the blood–CSF barrier. Indinavir, lopinavir, amprenavir and darunavir regularly exceeded inhibitory concentrations in the CSF, whereas saquinavir and ritonavir are not expected to achieve sufficient CSF concentrations.
A considerable number of studies investigated indinavir delivery to the CSF and found CSF-to-plasma ratios to be relatively high compared with other PIs. This high rate of permeation into the CSF can mainly be attributed to the only moderate affinity of indinavir to plasma proteins. Still, active retrograde transport of indinavir across the BBB is considered to limit accumulation in the CNS [66, 70]. Indinavir CSF-to-plasma ratios increase considerably within the dosing interval, reflecting delayed drug delivery from the plasma to the CSF. The CSF is supposed to act as a slowly equilibrating compartment, leading to stable concentrations and a longer elimination half-life of indinavir in the CSF relative to the systemic compartment [66, 69, 70, 72]. Lopinavir has demonstrated similar pharmacokinetic characteristics [81]. Under co-administration of ritonavir, CSF concentrations of indinavir increase more than 2-fold, paralleling minimum indinavir concentrations in plasma [72]. Ritonavir is a potent inhibitor of cytochrome P450 (CYP) 3A, thereby delaying systemic metabolism of indinavir and increasing the amount of drug available for transfer to the CNS [23, 68, 72]. CSF concentrations of other PIs might be similarly affected by co-administration of ritonavir [84, 87]. Other types of interaction between PIs have been detected, for example indinavir is suggested to exert an added inhibitory effect on lopinavir metabolism that might result in increased delivery of lopinavir to the CSF [76].
Although indinavir is predicted to exhibit better CSF penetration than other PIs, lower CSF-to-plasma ratios do not automatically mean reduced efficacy in the CNS. Limited penetration can be balanced by the potency of some PIs [105]. Compared with indinavir, the fraction of lopinavir penetrating into the CSF has been smaller, but closer in agreement with the reported free fraction in plasma [80]. Furthermore, CSF concentrations of lopinavir have been stable and still in the range of IC50 at the end of a 12-h dosing interval [81].
The interpretation of PI measurements in the CSF should account for the binding of drugs to CSF proteins. As a result of low protein levels in the CSF in unimpaired individuals, the estimated protein-bound fraction of drugs is generally lower in the CSF than in plasma, a fact that complicates the interpretation of CSF-to-plasma ratios of drugs with high affinity to proteins, such as PIs. The protein-bound fraction of indinavir in the CSF presumably ranges from 0 to 3 % and might be negligible [72]. In contrast, estimates for CSF protein binding of nelfinavir and lopinavir are relatively high (60–90 %), but still much lower than in plasma [78, 79]. Consequently, the CSF-adjusted IC95 of nelfinavir might be several times lower than in the plasma [78]. Under that condition, nelfinavir might contribute to inhibiting viral replication in the CNS, although absolute CSF concentrations have been low [78].
2.4 Other Antiretroviral Drugs
Enfuvirtide, an HIV fusion inhibitor, has not been detected in the CSF and might not contribute to viral suppression in the CNS [89]. A substantial fraction of the entry inhibitor maraviroc, in contrast, appears to reach the CSF, leading to concentrations beyond the mean 90 % effective concentration to inhibit viral replication (EC90) [90]. The low molecular weight of the drug and the relatively low plasma protein binding of about 76 % probably facilitate penetration to HIV sanctuary sites [91, 106]. Like PIs, maraviroc is a substrate of P-gp, a fact that might explain CSF concentrations being several-fold lower than the estimated free plasma fraction [92, 93]. Co-administration of ritonavir has been associated with higher CSF concentrations of maraviroc, presumably due to inhibition of maraviroc metabolism, thereby increasing systemic maraviroc exposure, leading to enhanced delivery to the CSF [90]. Very importantly, HIV strains infecting macrophages and microglia in the brain are predominantly C-C chemokine receptor type 5 (CCR5) tropic [107, 108], which is a precondition for virological efficacy of maraviroc.
Raltegravir, an inhibitor of HIV integrase, is also a suggested substrate for P-gp-mediated transport from the brain back into the systemic circulation. In a study by Yilmaz et al. [94] median CSF raltegravir concentrations have been nearly 4-fold lower than unbound drug concentrations in the plasma, exceeding the upper limit of the IC95 range in about half of the patients. Croteau et al. [95] found absolute CSF raltegravir concentrations comparable to these previous results, but drew different conclusions. Referencing IC50, which is lower than IC95, CSF concentrations are reported to exceed the concentration required to inhibit wild-type HIV in vitro in all individuals, suggesting that raltegravir is likely to contribute to the suppression of viral replication in the CNS.
3 Discussion
3.1 Considerations for the Assessment of CSF Penetration
Results from drug concentration assessment in the CSF are characterized by substantial intra- and inter-individual variability. Various factors might contribute to the variation among individuals and across clinical studies. In this context, baseline subject characteristics like dosing schedules, stage of disease, drug adherence and background HAART regimens deserve consideration. Some of these variables can be controlled by means of a comprehensive study design. Calculation and presentation of CSF-to-plasma ratios in addition to absolute CSF drug concentrations will address differential drug intake and systemic drug metabolism. Even so, this parameter has its limitations. On the one hand, CSF-to-plasma ratios are usually based on total drug concentrations and do not take protein-bound fractions into account. This is of particular concern for drugs with high affinity to plasma proteins such as most PIs. On the other hand, the ratios tend to change over time within the dosing interval. The ratio between the area under the concentration-time curve (AUC) in CSF and plasma might be a more accurate indicator for drug penetration than CSF-to-plasma ratios from samples obtained at single time points [50, 69, 70]. Ideally, intensive CSF sampling and simultaneous plasma sampling over the entire dosing interval or population-based pharmacokinetic modelling would provide information about the concentration time profile and address host genetic variability in CSF pharmacokinetics [109, 110]. For practical reasons, however, most studies included in this survey have simply assessed drug concentrations as a function of time. Of note, study subjects mostly received chronic oral dosing. CSF concentrations of ARVs can be significantly higher and are usually much more stable after long-term oral therapy than after a single dose [44], alleviating the problem of time dependency in drug measurement. Multiple dosing should therefore precede the assessment of CSF drug concentrations, particularly when drugs are known to accumulate in the CNS.
While absolute drug concentrations and CSF-to-plasma ratios provide pharmacokinetic information, antiviral drug potency parameters account for intracellular metabolism of drugs and allow estimation of antiviral effectiveness. Most of the reviewed studies compared the respective CSF drug concentrations with IC50 or IC95 in vitro. Assessment of the antiviral potency of a drug in vitro results in a concentration-effect curve that tends to be linear between 20 % and 80 % of maximum effect [111], therefore IC50 is generally less variable than IC95. IC50 is usually referenced in clinical resistance testing reports assessing the fold change in susceptibility of test virus as compared with wild-type virus. As long as the exact drug concentrations required to inhibit HIV strains in the central compartment are not defined, there are no recommendations on which of these reference standards to use in the context of CSF drug assessment. The majority of the studies included in our survey have referenced IC50.
Inhibitory concentrations have some limitations. First, reference inhibitory concentrations show marked variability depending on laboratory methods, viral strains and on whether they are derived from lymphocyte cell lines or from macrophages and monocytes. Compared with lymphocytes, in vitro inhibitory concentrations in macrophages are lower for NRTIs, similar for non-nucleotide reverse transcriptase inhibitors (NNRTIs) and higher for PIs [96]. Moreover, the IC50 is normally assessed in incubation media approximating conditions in the blood and thus containing more proteins than the CSF. Assessment of the IC50 in the presence of CSF would be desirable, but is not routinely performed due to methodological problems. Compared to standard in vitro conditions, the fraction of unbound, active drug is expected to be higher in the CSF, presumably leading to a lower IC50 in that compartment.
Importantly, in cells chronically infected by HIV, such as persistently infected macrophages in the CNS, the proviral DNA is firmly integrated within the host cell genome, and virus replication occurs independently of reverse transcriptase. Therefore, all reverse transcriptase inhibitors seem to be ineffective in these cells [96]. The inclusion of PIs in the therapeutic regimen allows for targeting of that cellular reservoir of HIV, however, the activity of PIs in chronically infected macrophages is several-fold lower than in lymphocytes [112]. As a consequence, even with PI concentrations in the CSF exceeding referenced in vitro inhibitory concentrations, CNS-standing-infected macrophages might escape from therapy.
Lastly, the effect of blood–CNS barrier disruption on ARV CNS penetration deserves consideration. Viral proteins and host inflammatory mediators affect the integrity of the BBB in the course of CNS-HIV infection, reflected by elevated CSF-to-plasma albumin ratios as a sign of abnormal BBB permeability soon after initial exposure [113]. and breakdown of tight junctions in patients with HIV encephalitis [114]. BBB disruption has been correlated with the severity of neurocognitive impairment [115], whereas in the majority of neurologically asymptomatic HIV-infected individuals, the BBB has been intact [116, 117]. These findings suggest that the delivery of ARVs to the CNS might be facilitated in patients with advanced HAND. Indeed, CSF concentrations of maraviroc have been higher in patients with neurological impairment than in neurologically asymptomatic individuals [82, 90, 91]. The CSF-to-plasma-to-albumin ratio mostly has not yet been associated with ARV concentrations in the CSF [33, 34, 66, 70, 72, 90], although evidence is not consistent [51, 94]. Penetration values derived from studies with neurologically asymptomatic subjects should not simply be extrapolated to patients with severe HAND until the effects of HIV infection on the BBB are better understood.
3.2 CSF as a Surrogate for CNS Drug Exposure
An important issue concerning CNS drug availability is the relevant sampling site. Clinical studies are generally bound to measure drug concentrations in the CSF as a surrogate for CNS drug exposure. Conversely, animal experiments can precisely quantify drug concentrations within the CNS and occasionally also point to the mechanisms and routes of drug entry. Providing information about drug concentrations in both the brain and the CSF, studies in animals have investigated the extent to which drug concentrations correlate in both compartments. Various experimental sampling and drug measurement techniques have been developed and were discussed in detail [118–120]. Table 3 presents reports on animal studies providing CSF-to-plasma or brain-to-plasma ratios of selected drugs with high penetration estimates, namely zidovudine, stavudine, abacavir, nevirapine, indinavir and maraviroc. Animal experiments have shown that drug concentrations in the CSF regularly differ from those in the brain. For example, brain-to-plasma ratios of saquinavir and nelfinavir have been found to be several-fold higher than CSF-to-plasma ratios in rodents and in nonhuman primates, respectively [140, 141]. Conversely, studies on animals consistently reported zidovudine and stavudine concentrations to be higher in the CSF than in brain samples, suggesting efflux mechanisms localized at the BBB [127, 131, 133, 142–144]. On one hand, brain levels are of direct interest: they indicate whether the BBB penetration is sufficient to inhibit the replication of virus residing in the brain. On the other hand, drug accumulation in the ventricular CSF itself could target infected perivascular and meningeal macrophages [24, 145]. In a comprehensive review, Shen et al. [25] assessed the applicability of CSF sampling for the assessment of CNS drug delivery in animals, concluding that CSF penetration studies remain a practical option for the assessment of drug availability in the CNS. Still, studies have to account for inherent physiochemical properties of drugs, such as lipophilicity, which determine the relationship between CSF and CNS concentrations [25]. However, in clinical studies CSF sampling is the most important way to get an idea of drug concentrations in the CNS. Comparative assessment of drug delivery in animal brain tissue and in human CSF might elucidate distribution kinetics and effective drug concentrations in the mammalian brain.
3.3 Widespread Neuropsychological Impairment Despite Highly Active Antiretroviral Therapy
Inadequate antiviral activity of ARVs in the CNS as a result of poor penetration is only one of several hypotheses that might explain persisting low-grade HIV replication in the CNS and persisting high prevalence of mild to moderate HAND under HAART. Other mutually non-exclusive explanations have been reviewed recently [146]. For example, in times of prolonged survival of HIV-infected individuals, age-associated disorders and complications of substance abuse gain more importance and might mimic, aggravate and interact with HIV-related neurobehavioural disorders, thereby posing challenges to diagnosis of HAND [5]. Evidence, however, did not confirm neither a significant interactive HIV and age effect on cognitive function in an early 2000 cohort [147] nor an association between substance abuse and neurocognitive disorders in the CHARTER cohort [148]. Another focus of interest is the role of sustained intrathecal immune activation in HAND pathogenesis. HAART does not appreciably suppress CNS inflammatory markers despite systemically effective treatment and undetectable CSF HIV-RNA, suggesting continuous brain damage by host mediators of inflammation and subsequent neurocognitive impairment [11, 13, 149]. Lastly, there are increasing concerns of whether ARVs may have CNS toxic effects that are related to continuing high rates of HAND. In a cohort study, discontinuation of HAART in patients under good peripheral virological control unexpectedly resulted in significant improvement in neurocognitive function over 96 weeks off therapy [150]. A potential explanation is HAART-induced neurotoxicity, however, in the absence of a control group, practice effects that might have resulted in better neuropsychological test performance cannot be ruled out, and there might have been patient selection bias. Increasing the CNS penetration of ARVs might increase the likelihood of drug-related toxicity in the brain, but so far the mechanisms that might lead to toxic neuronal damage by ARVs remain hypothetical. More longitudinal studies will be necessary to answer these questions [148].
A risk of poor CNS penetration might arise from the selection of HIV strains with resistance patterns different from those of plasma HIV strains, consistent with genetic compartmentalization of virus within the CNS. Indeed, in a substantial proportion of subjects, HIV strains in the CNS have genotypically diverged from strains in the blood [151]. Levels of compartmentalization have been highest in patients with chronic infection or HAD [152] or after long-time therapy [38]. At present, however, it is not absolutely clear whether discordant HIV drug resistance between both compartments is related to insufficient CNS exposure to ARVs.
While targeting HAART to the CNS, therapeutic strategies should ensure efficacy in the systemic compartment at the same time. Low nadir CD4+ cell counts in the blood have been a robust predictor of neurocognitive impairment in both the pre-HAART and the HAART eras, suggesting that CNS impairment that is at least partially irreversible begins during early severe immune suppression [148]. Consequently, early treatment initiation aimed at preventing systemic immunosuppression might reduce the risk of HAND irrespective of the regimen’s penetration effectiveness.
3.4 CNS-Active Drugs and Clinical Outcome
The pool of randomized controlled trials assessing the neuropsychological outcome under therapy with CNS-penetrating drugs is growing. In the pre-HAART era, study designs included single drug regimens based on NRTIs, providing evidence for CNS efficacy of single drugs. Since the introduction of PIs, NRTI monotherapy is expected to be inferior to combination therapy, so that patients in clinical studies are now predominantly being treated with multiple drugs. That might allow for the evaluation of the respective multidrug regimen, but the applicability for single drugs remains vague. As an alternative to standard HAART, i.e. combination triple therapy, ritonavir-boosted PI monotherapy has recently been considered for patients with intolerance to NRTIs or for treatment simplification. According to current recommendations, only patients under stable virological control and without any history of failure on prior PI-based therapy are eligible for PI monotherapy [153]. The poor availability of PIs in the CSF, however, gives rise to concerns over residual HIV replication in the CNS under nucleoside-sparing therapy. Large randomized cohort studies comparing standard triple HAART with lopinavir or darunavir monotherapy did not find nervous system adverse events to be more frequent in the monotherapy group after up to 96 weeks [154, 155]. Another study established neurological adverse events in only a small proportion of patients (2 %) under darunavir monotherapy, consistent with an elevated CSF viral load in these subjects [156]. Neuropsychological functioning, as assessed using a questionnaire, did not differ between patients randomized to darunavir monotherapy or to combination triple therapy [157]. Contrary to these findings, another study reported more patients experiencing therapeutic failure in the systemic compartment under lopinavir monotherapy (n = 29) than under triple HAART, consistent with both CSF HIV-RNA levels in the detectable range and neurological symptoms in most failing patients [158]. Moreover, 32 % of non-failing monotherapy patients had detectable HIV-RNA in the CSF at follow-up. Reintroduction of triple therapy in patients with therapeutic failure has been followed by improvement of neurological symptoms [156] and by decrease of the CSF viral load [105, 158]. The impact of PI monotherapy on virus levels in the CSF deserves further investigation.
Several studies addressed the question as to whether HAART including drugs that are more efficient in the CNS (neuroHAART) may be associated with better neurocognitive functioning. Evidence on this topic has recently been reviewed, providing qualitative and quantitative analysis [159]. Four longitudinal studies met the minimum quality criteria for inclusion in the meta-analysis, and all of them found a positive effect of neuroHAART (defined according to the CPE ranking in most studies) on neurocognitive functioning [159]. Despite the overall success of the CPE score as a tool for clinical practice, further validation will be necessary and some questions remain open. A large retrospective study confirmed survival benefit in patients with neurological AIDS-defining conditions to be associated with a CPE score of ≥ 1.5 in the early HAART era; however, the association was not maintained in the later HAART era, perhaps as a result of more powerful HAART regimens [160]. Critics see limitations of the CPE score in the insufficient reflection of pharmacodynamic aspects, genotypic resistance and drug-drug interactions [110, 161]. Furthermore, the question of whether the early initiation of HAART with targeted neuroactive drugs in neurologically asymptomatic patients can prevent HAND has not yet been resolved.
As the level of evidence on CNS effectiveness is increasing, estimates on CSF penetration are beginning to impact decisions about the therapy of HAND. According to the 2011 treatment guidelines by the European AIDS Clinical Society (EACS), inclusion of potentially CNS-active drugs should be considered in all patients with diagnosed HAND and is explicitly recommended in HAND-affected patients with a CSF viral load of >50 cells/mL in the absence of viremia [153]
4 Conclusion
Treatment of HAND requires viral load control both in the systemic and in the CNS compartments. While suppression of viral load is frequently obtained in the blood plasma as a result of potent HAART, drug penetration into the CNS is the focus of interest. The ability of ARVs to penetrate the BBB is believed to influence the extent of neurocognitive improvement and the decay of viral load in the CSF. Drug concentrations in the CSF are indicative for concentrations in the CNS and they can be assessed in the clinical context. Still, evidence on CSF distribution is sparse for several antiretroviral substances, including drugs introduced more recently, and is based on observational studies rather than on controlled clinical trials.
In the light of ongoing HAND and of the potential benefit of CNS-active drugs, clinical CSF penetration studies that respect relevant study design issues will be required. Early preclinical drug development should include assessment of CNS drug delivery in animals. CSF penetration studies and validated neuropsychological testing in a subgroup of patients in the course of new drug applications will lead to a better understanding of drug potency in the brain. Ultimately, large clinical cohort studies will be critical to provide guidelines for a well directed selection of HAART for patients with HAND.
References
Ellis RJ, Gamst AC, Capparelli E, et al. Cerebrospinal fluid HIV RNA originates from both local CNS and systemic sources. Neurology. 2000;54(4):927–36.
Ellis RJ, Moore DJ, Childers ME, et al. Progression to neuropsychological impairment in human immunodeficiency virus infection predicted by elevated cerebrospinal fluid levels of human immunodeficiency virus RNA. Arch Neurol. 2002;59(6):923–8.
d’Arminio Monforte A, Cinque P, Mocroft A, on behalf of the EuroSIDA Study Group, et al. Changing incidence of central nervous system diseases in the EuroSIDA cohort. Ann Neurol. 2004;55(3):320–8.
Clifford DB. HIV-associated neurocognitive disease continues in the antiretroviral era. Top HIV Med. 2008;16(2):94–8.
Heaton RK, Clifford DB, Franklin DR Jr, on behalf of the CHARTER Group, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER study. Neurology. 2010;75(23):2087–96.
Cysique LA, Brew BJ. Prevalence of non-confounded HIV-associated neurocognitive impairment in the context of plasma HIV RNA suppression. J Neurovirol. 2011;17(2):176–83.
Robertson KR, Robertson WT, Ford S, et al. Highly active antiretroviral therapy improves neurocognitive functioning. J Acquir Immune Defic Syndr. 2004;36(1):562–6.
Sacktor NC, Lyles RH, Skolasky RL, et al. Combination antiretroviral therapy improves psychomotor speed performance in HIV-seropositive homosexual men: Multicenter AIDS Cohort Study (MACS). Neurology. 1999;52(8):1640–7.
Clifford DB, McArthur JC, Schifitto G, on behalf of the Neurologic AIDS Research Consortium, et al. A randomized clinical trial of CPI-1189 for HIV-associated cognitive-motor impairment. Neurology. 2002;59(10):1568–73.
Letendre SL, McCutchan JA, Childers ME, on behalf of the HNRC Group, et al. Enhancing antiretroviral therapy for human immunodeficiency virus cognitive disorders. Ann Neurol. 2004;56(3):416–23.
Edén A, Price RW, Spudich S, et al. Immune activation of the central nervous system is still present after >4 years of effective highly active antiretroviral therapy. J Infect Dis. 2007;196(12):1779–83.
Yilmaz A, Price RW, Spudich S, et al. Persistent intrathecal immune activation in HIV-1-infected individuals on antiretroviral therapy. J Acquir Immune Defic Syndr. 2008;47(2):168–73.
Hagberg L, Cinque P, Gisslen M, et al. Cerebrospinal fluid neopterin: an informative biomarker of central nervous system immune activation in HIV-1 infection. AIDS Res Ther. 2010;3(7):15.
Eggers C, Hertogs K, Stürenburg HJ, et al. Delayed central nervous system virus suppression during highly active antiretroviral therapy is associated with HIV encephalopathy, but not with viral drug resistance or poor central nervous system drug penetration. AIDS. 2003;17(13):1897–906.
Letendre S, Marquie-Beck J, Capparelli E, on behalf of the CHARTER Group, et al. Validation of the CNS Penetration-Effectiveness rank for quantifying antiretroviral penetration into the central nervous system. Arch Neurol. 2008;65(1):65–70.
Cysique LA, Vaida F, Letendre S, et al. Dynamics of cognitive change in impaired HIV-positive patients initiating antiretroviral therapy. Neurology. 2009;73(5):342–8.
Marra CM, Zhao Y, Clifford DB, on behalf of the AIDS Clinical Trials Group 736 Study Team, et al. Impact of combination antiretroviral therapy on cerebrospinal fluid HIV RNA and neurocognitive performance. AIDS. 2009;23(11):1359–66.
Letendre SL, Ellis RJ, Ances BM, et al. Neurologic complications of HIV disease and their treatment. Top HIV Med. 2010;18(2):45–55.
Canestri A, Lescure FX, Jaureguiberry S, et al. Discordance between cerebral spinal fluid and plasma HIV replication in patients with neurological symptoms who are receiving suppressive antiretroviral therapy. Clin Infect Dis. 2010;50(5):773–8.
Tozzi V, Balestra P, Salvatori MF, et al. Changes in cognition during antiretroviral therapy: comparison of 2 different ranking systems to measure antiretroviral drug efficacy on HIV-associated neurocognitive disorders [published erratum appears in J Acquir Immune Defic Syndr 2009 Dec 1; 52 (4): 529]. J Acquir Immune Defic Syndr. 2009;52(1):56–63.
Smurzynski M, Wu K, Letendre S, et al. Effects of central nervous system antiretroviral penetration on cognitive functioning in the ALLRT cohort. AIDS. 2011;25(3):357–65.
Patel K, Ming X, Williams PL, on behalf of the International Maternal Pediatric Adolescent AIDS Clinical Trials 219/219C Study Team, et al. Impact of HAART and CNS-penetrating antiretroviral regimens on HIV encephalopathy among perinatally infected children and adolescents. AIDS. 2009;23(14):1893–901.
Gimenez F, Fernandez C, Mabondzo A. Transport of HIV protease inhibitors through the blood–brain barrier and interactions with the efflux proteins, P-glycoprotein and multidrug resistance proteins. J Acquir Immune Defic Syndr. 2004;36(2):649–58.
Strazielle N, Ghersi-Egea JF. Factors affecting delivery of antiviral drugs to the brain. Rev Med Virol. 2005;15(2):105–33.
Shen DD, Artru AA, Adkison KK. Principles and applicability of CSF sampling for the assessment of CNS drug delivery and pharmacodynamics. Adv Drug Deliv Rev. 2004;56(12):1825–57.
Varatharajan L, Thomas SA. The transport of anti-HIV drugs across blood–CNS interfaces: summary of current knowledge and recommendations for further research [published erratum appears in Antiviral Res 2009 Nov; 84 (2): 203]. Antiviral Res. 2009;82(2):A99–109.
Soontornmalai A, Vlaming ML, Fritschy JM. Differential, strain-specific cellular and subcellular distribution of multidrug transporters in murine choroid plexus and blood–brain barrier. Neuroscience. 2006;138(1):159–69.
Pizzo PA, Eddy J, Falloon J, et al. Effect of continuous intravenous infusion of zidovudine (AZT) in children with symptomatic HIV infection. N Engl J Med. 1988;319(14):889–96.
Balis FM, Pizzo PA, Murphy RF, et al. The pharmacokinetics of zidovudine administered by continuous infusion in children. Ann Intern Med. 1989;110(4):279–85.
Surbone A, Yarchoan R, McAtee N, et al. Treatment of the acquired immunodeficiency syndrome (AIDS) and AIDS-related complex with a regimen of 3′-azido-2′,3′-dideoxythymidine (azidothymidine or zidovudine) and acyclovir: a pilot study. Ann Intern Med. 1988;108(4):534–40.
Lane HC, Falloon J, Walker RE, et al. Zidovudine in patients with human immunodeficiency virus (HIV) infection and kaposi sarcoma: a phase II randomized, placebo-controlled trial [published erratum appears in Ann Intern Med 1990 Mar 1; 112 (5): 388]. Ann Intern Med. 1989;111(1):41–50.
Tartaglione TA, Collier AC, Coombs RW, et al. Acquired immunodeficiency syndrome: cerebrospinal fluid findings in patients before and during long-term oral zidovudine therapy [published erratum appears in Arch Neurol 1991 Dec; 48 (12): 1238]. Arch Neurol. 1991;48(7):695–9.
Burger DM, Kraaijeveld CL, Meenhorst PL, et al. Penetration of zidovudine into the cerebrospinal fluid of patients infected with HIV. AIDS. 1993;7(12):1581–7.
Rolinski B, Bogner JR, Sadri I, et al. Absorption and elimination kinetics of zidovudine in the cerebrospinal fluid in HIV-1-infected patients. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;15(3):192–7.
Hoetelmans RM, Kraaijeveld CL, Meenhorst PL, et al. Penetration of 3′-amino-3′-deoxythymidine, a cytotoxic metabolite of zidovudine, into the cerebrospinal fluid of HIV-1-infected patients. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;15(2):131–6.
Foudraine NA, Hoetelmans RM, Lange JM, et al. Cerebrospinal-fluid HIV-1 RNA and drug concentrations after treatment with lamivudine plus zidovudine or stavudine. Lancet. 1998;351(9115):1547–51.
van Praag RM, van Weert EC, van Heeswijk RP, et al. Stable concentrations of zidovudine, stavudine, lamivudine, abacavir, and nevirapine in serum and cerebrospinal fluid during 2 years of therapy. Antimicrob Agents Chemother. 2002;46(3):896–9.
Antinori A, Perno CF, Giancola ML, et al. Efficacy of cerebrospinal fluid (CSF)-penetrating antiretroviral drugs against HIV in the neurological compartment: different patterns of phenotypic resistance in CSF and plasma. Clin Infect Dis. 2005;41(12):1787–93.
Kline MW, Dunkle LM, Church JA, et al. A phase I/II evaluation of stavudine (d4T) in children with human immunodeficiency virus infection. Pediatrics. 1995;96(2 Pt 1):247–52.
Haworth SJ, Christofalo B, Anderson RD, et al. A single-dose study to assess the penetration of stavudine into human cerebrospinal fluid in adults. J Acquir Immune Defic Syndr Hum Retrovirol. 1998;17(3):235–8.
Fletcher CV, Brundage RC, Remmel RP, et al. Pharmacologic characteristics of indinavir, didanosine, and stavudine in human immunodeficiency virus-infected children receiving combination therapy. Antimicrob Agents Chemother. 2000;44(4):1029–34.
Gisolf EH, Enting RH, Jurriaans S, et al. Cerebrospinal fluid HIV-1 RNA during treatment with ritonavir/saquinavir or ritonavir/saquinavir/stavudine. AIDS. 2000;14(11):1583–9.
Haas DW, Clough LA, Johnson BW, et al. Evidence of a source of HIV type 1 within the central nervous system by ultraintensive sampling of cerebrospinal fluid and plasma. AIDS Res Hum Retroviruses. 2000;16(15):1491–502.
Brady KA, Boston RC, Aldrich JL, et al. Stavudine entry into cerebrospinal fluid after single and multiple doses in patients infected with human immunodeficiency virus. Pharmacotherapy. 2005;25(1):10–7.
van Leeuwen R, Katlama C, Kitchen V, et al. Evaluation of safety and efficacy of 3TC (lamivudine) in patients with asymptomatic or mildly symptomatic human immunodeficiency virus infection: a phase I/II study. J Infect Dis. 1995;171(5):1166–71.
Lewis LL, Venzon D, Church J, et al. Lamivudine in children with human immunodeficiency virus infection: a phase I/II study. The National Cancer Institute Pediatric Branch-Human Immunodeficiency Virus Working Group. J Infect Dis. 1996;174(1):16–25.
Mueller BU, Lewis LL, Yuen GJ, et al. Serum and cerebrospinal fluid pharmacokinetics of intravenous and oral lamivudine in human immunodeficiency virus-infected children. Antimicrob Agents Chemother. 1998;42(12):3187–92.
McDowell JA, Chittick GE, Ravitch JR, et al. Pharmacokinetics of [(14)C]abacavir, a human immunodeficiency virus type 1 (HIV-1) reverse transcriptase inhibitor, administered in a single oral dose to HIV-1-infected adults: a mass balance study. Antimicrob Agents Chemother. 1999;43(12):2855–61.
McDowell JA, Lou Y, Symonds WS, et al. Multiple-dose pharmacokinetics and pharmacodynamics of abacavir alone and in combination with zidovudine in human immunodeficiency virus-infected adults. Antimicrob Agents Chemother. 2000;44(8):2061–7.
Capparelli EV, Letendre SL, Ellis RJ, et al. Population pharmacokinetics of abacavir in plasma and cerebrospinal fluid. Antimicrob Agents Chemother. 2005;49(6):2504–6.
Calcagno A, Bonora S, Simiele M, et al. Tenofovir and emtricitabine cerebrospinal fluid-to-plasma ratios correlate to the extent of blood–brain barrier damage. AIDS. 2011;25(11):1437–9.
Best BM, Letendre SL, Koopmans P, on behalf of the CHARTER Study Group, et al. Low cerebrospinal fluid concentrations of the nucleotide HIV reverse transcriptase inhibitor, tenofovir. J Acquir Immune Defic Syndr. 2012;59(4):376–81.
Best BM, Letendre S, Capparelli EV, et al. Efavirenz and emtricitabine concentrations consistently exceed wild-type IC50 in cerebrospinal fluid: CHARTER findings [abstract no. 702]. 16th Conference on Retroviruses and Opportunistic Infections; 2009 Feb 8–11; Montréal.
Saitoh A, Sarles E, Capparelli E, et al. CYP2B6 genetic variants are associated with nevirapine pharmacokinetics and clinical response in HIV-1-infected children. AIDS. 2007;21(16):2191–9.
Tashima KT, Caliendo AM, Ahmad M, et al. Cerebrospinal fluid human immunodeficiency virus type 1 (HIV-1) suppression and efavirenz drug concentrations in HIV-1-infected patients receiving combination therapy. J Infect Dis. 1999;180(3):862–4.
Best BM, Koopmans PP, Letendre SL, on behalf of the CHARTER Group, et al. Efavirenz concentrations in CSF exceed IC50 for wild-type HIV. J Antimicrob Chemother. 2011;66(2):354–7.
Best B, Letendre S, Croteau D, et al. Therapeutic DRV and ETR concentrations in cerebrospinal fluid [abstract no. 643]. 18th Conference on Retroviruses an Opportunistic Infections; 2011 Feb 27–Mar 2; Boston (MA).
Tiraboschi JM, Niubo J, Vila A, et al. Etravirine concentrations in CSF in HIV-infected patients. J Antimicrob Chemother. Epub 2012 Feb 22.
Cameron DW, Japour AJ, Xu Y, et al. Ritonavir and saquinavir combination therapy for the treatment of HIV infection. AIDS. 1999;13(2):213–24.
Moyle GJ, Sadler M, Buss N. Plasma and cerebrospinal fluid saquinavir concentrations in patients receiving combination antiretroviral therapy. Clin Infect Dis. 1999;28(2):403–4.
Kravcik S, Gallicano K, Roth V, et al. Cerebrospinal fluid HIV RNA and drug levels with combination ritonavir and saquinavir. J Acquir Immune Defic Syndr. 1999;21(5):371–5.
Khaliq Y, Gallicano K, Venance S, et al. Effect of ketoconazole on ritonavir and saquinavir concentrations in plasma and cerebrospinal fluid from patients infected with human immunodeficiency virus. Clin Pharmacol Ther. 2000;68(6):637–46.
Yilmaz A, Fuchs D, Hagberg L, et al. Cerebrospinal fluid HIV-1 RNA, intrathecal immunoactivation, and drug concentrations after treatment with a combination of saquinavir, nelfinavir, and two nucleoside analogues: the M61022 study. BMC Infect Dis. 2006;27(6):63.
Calcagno A, Yilmaz A, Cusato J, et al. Determinants of darunavir cerebrospinal fluid concentrations: impact of once-daily dosing and pharmacogenetics. AIDS. 2012;26(12):1529–33.
Ståhle L, Martin C, Svensson JO, et al. Indinavir in cerebrospinal fluid of HIV-1-infected patients. Lancet. 1997;350(9094):1823.
Martin C, Sonnerborg A, Svensson JO, et al. Indinavir-based treatment of HIV-1 infected patients: efficacy in the central nervous system. AIDS. 1999;13(10):1227–32.
Letendre SL, Capparelli EV, Ellis RJ, et al. Indinavir population pharmacokinetics in plasma and cerebrospinal fluid: the HIV Neurobehavioral Research Center Group. Antimicrob Agents Chemother. 2000;44(8):2173–5.
van Praag RM, Weverling GJ, Portegies P, et al. Enhanced penetration of indinavir in cerebrospinal fluid and semen after the addition of low-dose ritonavir. AIDS. 2000;14(9):1187–94.
Zhou XJ, Havlir DV, Richman DD, on behalf of the AIDS Clinical Trials Study 343 Investigators, et al. Plasma population pharmacokinetics and penetration into cerebrospinal fluid of indinavir in combination with zidovudine and lamivudine in HIV-1-infected patients. AIDS. 2000;14(18):2869–76.
Haas DW, Stone J, Clough LA, et al. Steady-state pharmacokinetics of indinavir in cerebrospinal fluid and plasma among adults with human immunodeficiency virus type 1 infection. Clin Pharmacol Ther. 2000;68(4):367–74.
Foudraine NA, Jurriaans S, Weverling GJ, et al. Durable HIV-1 suppression with indinavir after failing lamivudine-containing double nucleoside therapy: a randomized controlled trial. Antivir Ther. 2001;6(1):55–62.
Haas DW, Johnson B, Nicotera J, et al. Effects of ritonavir on indinavir pharmacokinetics in cerebrospinal fluid and plasma. Antimicrob Agents Chemother. 2003;47(7):2131–7.
Marra CM, Lockhart D, Zunt JR, et al. Changes in CSF and plasma HIV-1 RNA and cognition after starting potent antiretroviral therapy. Neurology. 2003;60(8):1388–90.
Polis MA, Suzman DL, Yoder CP, et al. Suppression of cerebrospinal fluid HIV burden in antiretroviral naive patients on a potent four-drug antiretroviral regimen. AIDS. 2003;17(8):1167–72.
Solas C, Lafeuillade A, Halfon P, et al. Discrepancies between protease inhibitor concentrations and viral load in reservoirs and sanctuary sites in human immunodeficiency virus-infected patients. Antimicrob Agents Chemother. 2003;47(1):238–43.
Isaac A, Taylor S, Cane P, et al. Lopinavir/ritonavir combined with twice-daily 400 mg indinavir: pharmacokinetics and pharmacodynamics in blood, CSF and semen. J Antimicrob Chemother. 2004;54(2):498–502.
Aweeka F, Jayewardene A, Staprans S, et al. Failure to detect nelfinavir in the cerebrospinal fluid of HIV-1-infected patients with and without AIDS dementia complex. J Acquir Immune Defic Syndr Hum Retrovirol. 1999;20(1):39–43.
Karlström O, Ståhle L, Perrin L, et al. Efficacy of nelfinavir-based treatment in the central nervous system of HIV-1 infected patients. Scand J Infect Dis. 2006;38(5):371–4.
Yilmaz A, Ståhle L, Hagberg L, et al. Cerebrospinal fluid and plasma HIV-1 RNA levels and lopinavir concentrations following lopinavir/ritonavir regimen. Scand J Infect Dis. 2004;36(11–12):823–8.
Capparelli EV, Holland D, Okamoto C, on behalf of the HNRC Group, et al. Lopinavir concentrations in cerebrospinal fluid exceed the 50% inhibitory concentration for HIV. AIDS. 2005;19(9):949–52.
DiCenzo R, DiFrancesco R, Cruttenden K, et al. Lopinavir cerebrospinal fluid steady-state trough concentrations in HIV-infected adults. Ann Pharmacother. 2009;43(12):1972–7.
Garvey L, Nelson M, Latch N, et al. CNS effects of a CCR5 inhibitor in HIV-infected subjects: a pharmacokinetic and cerebral metabolite study. J Antimicrob Chemother. 2012;67(1):206–12.
Vernazza P, Daneel S, Schiffer V, et al. The role of compartment penetration in PI-monotherapy: the Atazanavir-Ritonavir Monomaintenance (ATARITMO) trial. AIDS. 2007;21(10):1309–15.
Best BM, Letendre SL, Brigid E, on behalf of the CHARTER Group, et al. Low atazanavir concentrations in cerebrospinal fluid. AIDS. 2009;23(1):83–7.
Sadler BM, Chittick GE, Polk RE, et al. Metabolic disposition and pharmacokinetics of [14C]-amprenavir, a human immunodeficiency virus type 1 (HIV-1) protease inhibitor, administered as a single oral dose to healthy male subjects. J Clin Pharmacol. 2001;41(4):386–96.
Saumoy M, Tiraboschi J, Gutierrez M, et al. Viral response in stable patients switching to fosamprenavir/ritonavir monotherapy (the FONT Study). HIV Med. 2011;12(7):438–41.
Croteau D, Letendre S, Best BM, on behalf of the CHARTER Group, et al. Therapeutic amprenavir concentrations in cerebrospinal fluid. Antimicrob Agents Chemother. 2012;56(4):1985–9.
Yilmaz A, Izadkhashti A, Price RW, et al. Darunavir concentrations in cerebrospinal fluid and blood in HIV-1-infected individuals. AIDS Res Hum Retroviruses. 2009;25(4):457–61.
Price RW, Parham R, Kroll JL, et al. Enfuvirtide cerebrospinal fluid (CSF) pharmacokinetics and potential use in defining CSF HIV-1 origin. Antivir Ther. 2008;13(3):369–74.
Yilmaz A, Watson V, Else L, et al. Cerebrospinal fluid maraviroc concentrations in HIV-1 infected patients. AIDS. 2009;23(18):2537–40.
Melica G, Canestri A, Peytavin G, et al. Maraviroc-containing regimen suppresses HIV replication in the cerebrospinal fluid of patients with neurological symptoms. AIDS. 2010;24(13):2130–3.
Tiraboschi JM, Niubo J, Curto J, et al. Maraviroc concentrations in cerebrospinal fluid in HIV-infected patients. J Acquir Immune Defic Syndr. 2010;55(5):606–9.
Croteau D, Best BM, Letendre S, on behalf of the CHARTER Group, et al. Lower than expected maraviroc concentrations in cerebrospinal fluid exceed the wild-type CC chemokine receptor 5-tropic HIV-1 50% inhibitory concentration. AIDS. 2012;26(7):890–3.
Yilmaz A, Gisslén M, Spudich S, et al. Raltegravir cerebrospinal fluid concentrations in HIV-1 infection. PLoS One. 2009;4(9):e6877.
Croteau D, Letendre S, Best BM, on behalf of the CHARTER Group, et al. Total raltegravir concentrations in cerebrospinal fluid exceed the 50-percent inhibitory concentration for wild-type HIV-1. Antimicrob Agents Chemother. 2010;54(12):5156–60.
Aquaro S, Caliò R, Balzarini J, et al. Macrophages and HIV infection: therapeutical approaches toward this strategic virus reservoir. Antiviral Res. 2002;55(2):209–25.
Yuen GJ, Weller S, Pakes GE. A review of the pharmacokinetics of abacavir. Clin Pharmacokinet. 2008;47(6):351–71.
Foudraine NA, Hoetelmans RM, Lange JM, et al. Cerebrospinal-fluid HIV-1 RNA and drug concentrations after treatment with lamivudine plus zidovudine or stavudine. Lancet. 1998;351(9115):1547–51.
Collins JM, Klecker RW Jr, Kelley JA, et al. Pyrimidine dideoxyribonucleosides: selectivity of penetration into cerebrospinal fluid. J Pharmacol Exp Ther. 1988;245(2):466–70.
Gibbs JE, Gaffen Z, Thomas SA. Nevirapine uptake into the central nervous system of the guinea pig: an in situ brain perfusion study. J Pharmacol Exp Ther. 2006;317(2):746–51.
Ciccarelli N, Fabbiani M, Di Giambenedetto S, et al. Efavirenz associated with cognitive disorders in otherwise asymptomatic HIV-infected patients. Neurology. 2011;76(16):1403–9.
Cespedes MS, Aberg JA. Neuropsychiatric complications of antiretroviral therapy. Drug Saf. 2006;29(10):865–74.
van der Sandt IC, Vos CM, Nabulsi L, et al. Assessment of active transport of HIV protease inhibitors in various cell lines and the in vitro blood–brain barrier. AIDS. 2001;15(4):483–91.
Kim RB, Fromm MF, Wandel C, et al. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J Clin Invest. 1998;101(2):289–94.
Letendre SL, van den Brande G, Hermes A, on behalf of the HIV Neurobehavioral Research Center Group, et al. Lopinavir with ritonavir reduces the HIV RNA level in cerebrospinal fluid. Clin Infect Dis. 2007;45(11):1511–7.
Walker DK, Bowers SJ, Mitchell RJ, et al. Preclinical assessment of the distribution of maraviroc to potential human immunodeficiency virus (HIV) sanctuary sites in the central nervous system (CNS) and gut-associated lymphoid tissue (GALT). Xenobiotica. 2008;38(10):1330–9.
He J, Chen Y, Farzan M, et al. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature. 1997;385(6617):645–9.
Li S, Juarez J, Alali M, et al. Persistent CCR5 utilization and enhanced macrophage tropism by primary blood human immunodeficiency virus type 1 isolates from advanced stages of disease and comparison to tissue-derived isolates. J Virol. 1999;73(12):9741–55.
Wynn HE, Brundage RC, Fletcher CV. Clinical implications of CNS penetration of antiretroviral drugs. CNS Drugs. 2002;16(9):595–609.
May S, Letendre S, Haubrich R, et al. Meeting practical challenges of a trial involving a multitude of treatment regimens: an example of a multi-center randomized controlled clinical trial in neuroAIDS. J Neuroimmune Pharmacol. 2007;2(1):97–104.
Morse GD, Catanzaro LM, Acosta EP. Clinical pharmacodynamics of HIV-1 protease inhibitors: use of inhibitory quotients to optimise pharmacotherapy. Lancet Infect Dis. 2006;6(4):215–25.
Perno CF, Newcomb FM, Davis DA, et al. Relative potency of protease inhibitors in monocytes/macrophages acutely and chronically infected with human immunodeficiency virus. J Infect Dis. 1998;178(2):413–22.
Spudich S, Gisslen M, Hagberg L, et al. Central nervous system immune activation characterizes primary human immunodeficiency virus 1 infection even in participants with minimal cerebrospinal fluid viral burden. J Infect Dis. 2011;204(5):753–60.
Dallasta LM, Pisarov LA, Esplen JE, et al. Blood–brain barrier tight junction disruption in human immunodeficiency virus-1 encephalitis. Am J Pathol. 1999;155(6):1915–27.
Avison MJ, Nath A, Greene-Avison R, et al. Neuroimaging correlates of HIV-associated BBB compromise. J Neuroimmunol. 2004;157(1–2):140–6.
Andersson LM, Hagberg L, Fuchs D, et al. Increased blood–brain barrier permeability in neuro-asymptomatic HIV-1-infected individuals: correlation with cerebrospinal fluid HIV-1 RNA and neopterin levels. J Neurovirol. 2001;7(6):542–7.
Abdulle S, Hagberg L, Gisslén M. Effects of antiretroviral treatment on blood–brain barrier integrity and intrathecal immunoglobulin production in neuroasymptomatic HIV-1-infected patients. HIV Med. 2005;6(3):164–9.
Enting RH, Hoetelmans RM, Lange JM, et al. Antiretroviral drugs and the central nervous system. AIDS. 1998;12(15):1941–55.
Sawchuk RJ, Yang Z. Investigation of distribution, transport and uptake of anti-HIV drugs to the central nervous system. Adv Drug Deliv Rev. 1999;39(1–3):5–31.
Sawchuk RJ, Elmquist WF. Microdialysis in the study of drug transporters in the CNS. Adv Drug Deliv Rev. 2000;45(2–3):295–307.
Hedaya MA, Sawchuk RJ. Effect of probenecid on the renal and nonrenal clearances of zidovudine and its distribution into cerebrospinal fluid in the rabbit. J Pharm Sci. 1989;78(9):716–22.
Sawchuk RJ, Hedaya MA. Modeling the enhanced uptake of zidovudine (AZT) into cerebrospinal fluid: 1. Effect of probenecid. Pharm Res. 1990;7(4):332–8.
Chu CK, Bhadti VS, Doshi KJ, et al. Brain targeting of anti-HIV nucleosides: synthesis and in vitro and in vivo studies of dihydropyridine derivatives of 3′-azido-2′,3′-dideoxyuridine and 3′-azido-3′-deoxythymidine. J Med Chem. 1990;33(8):2188–92.
Wong SL, Wang Y, Sawchuk RJ. Analysis of zidovudine distribution to specific regions in rabbit brain using microdialysis. Pharm Res. 1992;9(3):332–8.
Gallo JM, Sanzgiri Y, Howerth EW, et al. Zidovudine serum, cerebrospinal fluid, and brain concentrations following chronic administration of a new zidovudine formulation via an implantable pump in dogs [published erratum appears in J Pharm Sci 1992 Mar; 81 (3): 314]. J Pharm Sci. 1992;81(1):11–5.
Ståhle L, Guzenda E, Ljungdahl-Ståhle E. Pharmacokinetics and extracellular distribution to blood, brain, and muscle of alovudine (3′-fluorothymidine) and zidovudine in the rat studied by microdialysis. J Acquir Immune Defic Syndr. 1993;6(5):435–9.
Wong SL, Van Belle K, Sawchuk RJ. Distributional transport kinetics of zidovudine between plasma and brain extracellular fluid/cerebrospinal fluid in the rabbit: investigation of the inhibitory effect of probenecid utilizing microdialysis. J Pharmacol Exp Ther. 1993;264(2):899–909.
Lopez-Anaya A, Unadkat JD, Calkins DF, et al. Effect of age on distribution of zidovudine (azidothymidine) into the cerebrospinal fluid of macaca nemestrina. Pharm Res. 1993;10(9):1338–40.
Wang Y, Wong SL, Sawchuk RJ. Microdialysis calibration using retrodialysis and zero-net flux: application to a study of the distribution of zidovudine to rabbit cerebrospinal fluid and thalamus. Pharm Res. 1993;10(10):1411–9.
Tuntland T, Ravasco RJ, al-Habet S, et al. Efflux of zidovudine and 2′,3′-dideoxyinosine out of the cerebrospinal fluid when administered alone and in combination to macaca nemestrina. Pharm Res. 1994;11(2):312–7.
Masereeuw R, Jaehde U, Langemeijer MW, et al. In vitro and in vivo transport of zidovudine (AZT) across the blood–brain barrier and the effect of transport inhibitors. Pharm Res. 1994;11(2):324–30.
Wang Y, Sawchuk RJ. Zidovudine transport in the rabbit brain during intravenous and intracerebroventricular infusion. J Pharm Sci. 1995;84(7):871–6.
Yang Z, Brundage RC, Barbhaiya RH, et al. Microdialysis studies of the distribution of stavudine into the central nervous system in the freely-moving rat. Pharm Res. 1997;14(7):865–72.
Brewster ME, Anderson WR, Webb AI, et al. Evaluation of a brain-targeting zidovudine chemical delivery system in dogs. Antimicrob Agents Chemother. 1997;41(1):122–8.
Fox E, Bungay PM, Bacher J, et al. Zidovudine concentration in brain extracellular fluid measured by microdialysis: steady-state and transient results in rhesus monkey. J Pharmacol Exp Ther. 2002;301(3):1003–11.
Thomas SA, Segal MB. The transport of the anti-HIV drug, 2′,3′-didehydro-3′-deoxythymidine (D4T), across the blood–brain and blood–cerebrospinal fluid barriers. Br J Pharmacol. 1998;125(1):49–54.
Yang Z, Huang Y, Gan G, et al. Microdialysis evaluation of the brain distribution of stavudine following intranasal and intravenous administration to rats. J Pharm Sci. 2005;94(7):1577–88.
Thomas SA, Bye A, Segal MB. Transport characteristics of the anti-human immunodeficiency virus nucleoside analog, abacavir, into brain and cerebrospinal fluid. J Pharmacol Exp Ther. 2001;298(3):947–53.
Lin JH, Chiba M, Balani SK, et al. Species differences in the pharmacokinetics and metabolism of indinavir, a potent human immunodeficiency virus protease inhibitor. Drug Metab Dispos. 1996;24(10):1111–20.
Washington CB, Wiltshire HR, Man M, et al. The disposition of saquinavir in normal and P-glycoprotein deficient mice, rats, and in cultured cells. Drug Metab Dispos. 2000;28(9):1058–62.
Kaddoumi A, Choi SU, Kinman L, et al. Inhibition of P-glycoprotein activity at the primate blood–brain barrier increases the distribution of nelfinavir into the brain but not into the cerebrospinal fluid. Drug Metab Dispos. 2007;35(9):1459–62.
Galinsky RE, Hoesterey BL, Anderson BD. Brain and cerebrospinal fluid uptake of zidovudine (AZT) in rats after intravenous injection [published erratum appears in Life Sci 1990; 47 (23): 2171]. Life Sci. 1990;47(9):781–8.
Wang Y, Wei Y, Sawchuk RJ. Zidovudine transport within the rabbit brain during intracerebroventricular administration and the effect of probenecid. J Pharm Sci. 1997;86(12):1484–90.
Takasawa K, Terasaki T, Suzuki H, et al. In vivo evidence for carrier-mediated efflux transport of 3’-azido-3’-deoxythymidine and 2′,3′-dideoxyinosine across the blood–brain barrier via a probenecid-sensitive transport system. J Pharmacol Exp Ther. 1997;281(1):369–75.
Rennels ML, Gregory TF, Blaumanis OR, et al. Evidence for a ‘paravascular’ fluid circulation in the mammalian central nervous system, provided by the rapid distribution of tracer protein throughout the brain from the subarachnoid space. Brain Res. 1985;326(1):47–63.
Mothobi NZ, Brew BJ. Neurocognitive dysfunction in the highly active antiretroviral therapy era. Curr Opin Infect Dis. 2012;25(1):4–9.
Cysique LA, Maruff P, Bain MP, et al. HIV and age do not substantially interact in HIV-associated neurocognitive impairment. J Neuropsychiatry Clin Neurosci. 2011;23(1):83–9.
Heaton RK, Franklin DR, Ellis RJ, on behalf of the CHARTER Group and the HNCR Group, et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol. 2011;17(1):3–16.
Gisolf EH, van Praag RM, Jurriaans S, et al. Increasing cerebrospinal fluid chemokine concentrations despite undetectable cerebrospinal fluid HIV RNA in HIV-1-infected patients receiving antiretroviral therapy. J Acquir Immune Defic Syndr. 2000;25(5):426–33.
Robertson KR, Su Z, Margolis DM, on behalf of the A5170 Study Team, et al. Neurocognitive effects of treatment interruption in stable HIV-positive patients in an observational cohort. Neurology. 2010;74(16):1260–6.
Stingele K, Haas J, Zimmermann T, et al. Independent HIV replication in paired CSF and blood viral isolates during antiretroviral therapy. Neurology. 2001;56(3):355–61.
Harrington PR, Schnell G, Letendre SL, et al. Cross-sectional characterization of HIV-1 env compartmentalization in cerebrospinal fluid over the full disease course. AIDS. 2009;23(8):907–15.
European AIDS Clinical Society. Guidelines Version 6: October 2011 [online]. Available from URL: http://www.europeanaidsclinicalsociety.org/images/stories/EACS-Pdf/EACSGuidelines-v6.0-English.pdf [Assessed 2012 Jul 25].
Arribas JR, Delgado R, Arranz A, on behalf of the OK04 Study Group, et al. Lopinavir-ritonavir monotherapy versus lopinavir-ritonavir and 2 nucleosides for maintenance therapy of HIV: 96-week analysis. J Acquir Immune Defic Syndr. 2009;51(2):147–52.
Clumeck N, Rieger A, Banhegyi D, et al. 96 week results from the MONET trial: a randomized comparison of darunavir/ritonavir with versus without nucleoside analogues, for patients with HIV RNA <50 copies/mL at baseline. J Antimicrob Chemother. 2011;66(8):1878–85.
Katlama C, Valantin MA, Algarte-Genin M, et al. Efficacy of darunavir/ritonavir maintenance monotherapy in patients with HIV-1 viral suppression: a randomized open-label, noninferiority trial, MONOI-ANRS 136. AIDS. 2010;24(15):2365–74.
Winston A, Fätkenheuer G, Arribas J, et al. Neuropsychiatric adverse events with ritonavir-boosted darunavir monotherapy in HIV-infected individuals: a randomised prospective study. HIV Clin Trials. 2010;11(3):163–9.
Gutmann C, Cusini A, Günthard HF, on behalf of the Swiss HIV Cohort Study (SHCS), et al. Randomized controlled study demonstrating failure of LPV/r monotherapy in HIV: the role of compartment and CD4-nadir. AIDS. 2010;24(15):2347–54.
Cysique LA, Waters EK, Brew BJ. Central nervous system antiretroviral efficacy in HIV infection: a qualitative and quantitative review and implications for future research. BMC Neurol. 2011;11:148.
Lanoy E, Guiguet M, Bentata M, on behalf of the FHDH-ANRS CO4, et al. Survival after neuroAIDS: association with antiretroviral CNS Penetration-Effectiveness score. Neurology. 2011;76(7):644–51.
Brew BJ. HIV, the brain, children, HAART and ‘neuro-HAART’: a complex mix. AIDS. 2009;23(14):1909–10.
Acknowledgments
No funding was provided for the preparation of this article. There are no relevant conflicts of interest for any of the authors. All persons who made substantial contributions to the work met the criteria for authorship and are listed above.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Eisfeld, C., Reichelt, D., Evers, S. et al. CSF Penetration by Antiretroviral Drugs. CNS Drugs 27, 31–55 (2013). https://doi.org/10.1007/s40263-012-0018-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-012-0018-x