
Abstract
Waxy corn possessing high amylopectin is widely employed as an industrial product. Traditional corn contains ~ 70–75% amylopectin, whereas waxy corn with the mutant waxy1 (wx1) gene possesses ~ 95–100% amylopectin. Marker-assisted breeding can greatly hasten the transfer of the wx1 allele into normal corn. However, the available gene-based marker(s) for wx1 are not always polymorphic between recipient and donor parents, thereby causing a considerable delay in the molecular breeding program. Here, a 4800 bp sequence of the wx1 gene was analyzed among seven wild-type and seven mutant inbreds employing 16 overlapping primers. Three polymorphisms viz., 4 bp InDel (at position 2406 bp) in intron-7 and two SNPs (C to A at position 3325 bp in exon-10 and G to T at position 4310 bp in exon-13) differentiated the dominant (Wx1) and recessive (wx1) allele. Three breeder-friendly PCR markers (WxDel4, SNP3325_CT1, and SNP4310_GT2) specific to InDel and SNPs were developed. WxDel4 amplified 94 bp among mutant-type inbreds, while 90 bp was amplified among wild-type inbreds. SNP3325_CT1 and SNP4310_GT2 revealed the presence-absence polymorphisms with an amplification of 185 bp and 189 bp of amplicon, respectively. These newly developed markers showed 1:1 segregation in both BC1F1 and BC2F1 generations, while 1:2:1 segregation was observed in BC2F2. The recessive homozygotes (wx1wx1) of BC2F2 identified by the markers possessed significantly higher amylopectin (97.7%) compared to the original inbreds (Wx1Wx1: 72.7% amylopectin). This is the first report of novel wx1 gene-based markers. The information generated here would help in accelerating the development of waxy maize hybrids.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Waxy corn, also known as “glutinous or sticky maize,” is a profitable, nutritious, and industrial crop (Talukder et al. 2022; Dong et al. 2019). The sticky quality of waxy corn is due to the endosperm starch, which contains ~ 95–100% amylopectin (Reddappa et al. 2022). In China and the majority of South-East Asian nations, it is generally eaten as food (Devi et al. 2017; Hossain et al. 2019). Waxy maize is widely employed in the paper, glue, and textile industries due to its unique properties (Bao et al. 2012; Yang et al. 2013). Several studies have reported that mutation in Waxy1 (Wx1) gene was first evidenced in the South-West province of China, specifically Yunnan and the surrounding areas (Wu et al. 2019; Liu et al. 2016). Traditional maize possesses ~ 25% amylose and ~ 75% amylopectin; however, the mutant wx1 gene enhances amylopectin to a level of ~ 40% (Zhang et al. 2013, Talukder et al. 2022). The Wx1 gene encodes granule-bound starch synthase-I (GBSSI) (Macdonald and Preiss 1985; Shure et al. 1983; Hossain et al. 2019). It is located on chromosome-9 and is composed of a 4.8 kb-long region with 14 exons (Mason-Gamer et al. 1998). The Wx1 gene has more than fifty mutants, having insertions and deletions (Huang et al. 2010). Two alleles with deletions viz., wx-D7 allele having a 30 bp deletion in the 7th exon and wx-D10 allele possessing a 15 bp deletion in the 10th exon, are the primary alleles in Chinese waxy corn germplasm (Bao et al. 2012). Besides, wx-B3, wx-B4, wx-m1, wx-m5, wx-m6, wx-m7, and wx-m9 are insertional mutations having Ac/Ds transposons, while wx-m8 allele has an insertion of a dSpm transposon, and wx-844 allele possesses an En/Spm transposable element (Huang et al. 2010).
In comparison to field corn hybrids, limited numbers of waxy corn hybrids are available worldwide (Talukder et al. 2022). Therefore, it is important to develop high amylopectin-based maize hybrids to meet the demand. The wx1 recessive mutation is responsible for the development of maize endosperm composed almost entirely of amylopectin (Qi et al. 2018). Marker-assisted selection (MAS) provides a great opportunity to introgress the wx1 gene into elite genotypes for the development of waxy corn hybrids with increased amylopectin. Molecular markers are considered a powerful tool for the identification of genomic regions controlling a trait of interest in genomics-assisted selection as these markers save a lot of time and resources by decreasing the number of breeding cycles needed to reconstitute the recurrent parent genome (Muthusamy et al. 2014; Hossain et al. 2018; Sarika et al. 2018; Singh and Singh 2015 and Zunjare et al. 2018). Three simple sequence repeat (SSR) markers (phi022, phi027, and phi061) present in the genic region of Wx1 have been widely used in molecular breeding (Zhang et al. 2013). Shin et al. (2006) further reported a co-dominant marker (wx-2507F/RG) to differentiate between the wild and mutant genotypes. However, in particular germplasm, these markers might not be polymorphic between the wild-type and the mutant alleles (Devi et al. 2017; Hossain et al. 2019; Talukder et al. 2022). Thus, the availability of new gene-based markers that are breeder-friendly would increase the efficiency of MAS in waxy corn breeding programs (Devi et al. 2017; Hossain et al. 2019; Talukder et al. 2022). As a result, the current study’s objectives were to (i) develop novel wx1 gene-based markers that are acceptable to breeders and (ii) validate those markers in segregating populations.
Materials and methods
Genetic materials
Seven diverse wild-type (Wx1-Wild1 to Wx1-Wild7) inbreds with low amylopectin and seven mutant-type (wx1-Mutant1 to wx1-Mutant7) inbreds with high amylopectin content were selected to identify DNA and protein sequence variations among them. The seven wild-type inbreds (HKI323, HKI1105, HKI1128, HKI161, HKI163, HKII93-1, and HKI193-2) are subtropically adapted and developed in India. Of these, HKI323, HKI1105, and HKI1128 are field corn inbreds, while HKI161, HKI163, HKII93-1, and HKI193-2 are quality protein maize (QPM) inbreds possessing the recessive opaque2 allele. These seven inbreds are genetically diverse, and they contributed to nine released hybrids viz., HM4 (HKI1105 × HKI323), HM8 (HKI1105 × HKI161), HM9 (HKI1105 × HKI1128), HM10: (HKI193-2 × HKI1128), HM11 (HKI1128 × HKI163), HQPM1 (HKI193-1 × HKI163), HQPM5 (HKI163 × HKI161), HQPM4 (HKI193-2 × HKI161), and HQPM7 (HKI193-1 × HKI161) in India. Besides, the seven waxy inbreds possessing the recessive waxy1 gene were developed by crossing seven diverse waxy germplasm with normal elite lines in India, and they are diverse in nature (Devi et al. 2017). The origin and pedigree details depicting the diverse nature of the 14 inbreds are presented in Table S1.
Genomic DNA isolation, PCR primers, PCR standardization, and sequencing
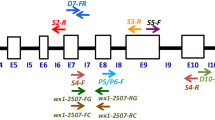
Genomic DNA was isolated from seeds of selected wild- and mutant-type waxy inbreds using the SDS-extraction method (Murray and Thompson 1980) and quantified in UV-spectrophotometer (BT-UVS-SBA-E. G-biosciences, India). With amplicon sizes between 400 and 600 bp, 16 overlapping primers covering the 4800 bp of the Wx1 gene (GenBank accession no. X03935) were designed using the Primer3 Online program. Using the Primer3 Online software and the technique described by Liu et al. (2012), single nucleotide polymorphism (SNP)-based primer sets were designed from two detected SNPs (Table 1). The names of the two primer sets are (i) SNP3325_CA1 F1/R and SNP3325_CA1 F2/R and (ii) SNP4310_GT2 F1 & R and SNP4310_GT2 F2 & R. WxDel4-F and WxDel4-R, a pair of InDel-specific primers, were also developed. Each primer was synthesized from M/s. Sequencher Pvt. Ltd.
Amplicons generated from overlapping primers in selected inbreds were sequenced from M/s. Sequencher Pvt. Ltd. For sequencing, a PCR mixture of 50 μl was prepared with 100 ng genomic DNA, 1 × OnePCR™ Mix (M/s. GeneDireX), and 0.25 μM each of the forward (F) and reverse (R) primers. The PCR assay was carried out on a Veriti 96-well thermal cycler (Applied Biosystems) with PCR conditions: initial denaturation at 95 °C: 5 min; 35 cycles consisting of denaturation at 95 °C: 45 s; primer annealing at 55–60 °C (optimized according to the primer Tm): 45 s; primer extension at 72 °C: 45 s; and final extension at 72 °C: 5 min. Ten microliters of PCR amplicon was checked on 2% agarose gel (Lonza, Rockland, ME, USA), and the remaining was processed for sequencing. The sequencing results were analyzed using the BioEdit software version 7.2.6 and the MEGA software version 7.0.26, with the ClustalW alignment tool to identify SNPs or InDels among selected wild and mutant genotypes. A 20 μl reaction mixture including 100 ng of template DNA, 1 × OnePCR™ Mix (M/s. GeneDireX), and 0.25 M of each of the forward and reverse primers was prepared for allele-specific and Indel-based PCR. The following PCR conditions were used for the Veriti 96-well thermal cycler (M/s. Applied Biosystems) during the PCR process: 35 cycles of initial denaturation at 95 °C: 5 min; primer annealing at 60 °C: 45 s; primer extension at 72 °C: 45 s; and final extension at 72 °C: 5 min. The efficiency of allele-specific PCRs was increased using a gradient PCR of different annealing temperatures (Table 1).
The PCR products of allele-specific and InDel-based PCRs were separated on 3% agarose gel (Lonza, Rockland, ME, USA) and 4% metaphor agarose (Lonza, Rockland, ME, USA), respectively, which were visualized on a UV-gel documentation system (M/s. Alpha Innotech, California, USA).
Validation of new wx1-specific marker in segregating generations
Seven inbreds (HKI161, HKI163, HKI323, HKI1105, HKI1128, HKI193-1, and HKI193-2) low in amylopectin were considered as recurrent parents, while MGU-102-wx1 was used as a donor line for the waxy trait. The recurrent (female) and donor parents (male) were crossed during the rainy season of July–November, 2016 at ICAR-Indian Agricultural Research Institute (IARI), New Delhi (28° 09′ N, 77° 13′ E, 229 MSL). F1s were grown and backcrossed at ICAR-Indian Institute of Maize Research (IIMR)-Winter Nursery Centre (WNC), Hyderabad (17° 19′ N, 78° 25′ E, 542.6 MSL) during the winter season of December 2016–April 2017. During the rainy season (2017), BC1F1 progenies were raised in Delhi, and foreground selection was performed by deploying newly developed wx1-specific markers. During the winter season (2017–18), selected BC2F1 progenies were raised and selfed in Hyderabad and subjected to foreground selection. During the rainy season (2018), the selected BC2F2 progenies were grown in New Delhi. Altogether, seven backcross populations, viz., cross-I (HKI323 × MGU-102-wx1), cross-II (HKI1105 × MGU-102-wx1), cross-III, (HKI1128 × MGU-102-wx1), cross-IV (HKI161 × MGU-102-wx1), cross-V (HKI163 × MGU-102-wx1), cross-VI (HKI193-1 × MGU-102-wx1), and cross-VII (HKI193-2 × MGU-102-wx1) each in BC1F1, BC2F1 and BC2F2 generations, were genotyped for the presence of wx1 gene. The details of all the backcrosses and selfed generations raised at different locations in different seasons are described in Table S2.
Estimation of amylopectin content
Seven randomly-selected wx1wx1 homozygotes in BC2F2 from each of the seven genetic backgrounds, along with the recurrent parents and waxy inbreds used in sequencing, were used for the estimation of amylopectin in triplicates. Total starch and amylose in the grains were estimated as described by Reddappa et al. (2022). The average of three technical replicates was used to compute the percentage of starch and amylose. Finally, amylopectin was calculated by subtracting the amount of amylose from the total starch content.
Statistical analysis
The chi-square analysis was used to evaluate the observed segregation pattern of the wx1 gene in seven segregating populations (BC1F1, BC2F1, and BC2F2) (Hossain et al. 2018). Using Microsoft Excel, the amount of amylopectin in each genotype was graphically depicted (2013).
Results
Sequence variation in Wx1 gene and development of Wx1-based markers
The generated sequences of the Wx1 gene in selected wild- and mutant-type inbreds were aligned along with wild-type Wx1 reference sequence (GenBank accession no. X03935; Klosgen et al. (1986)). A total of 220 SNPs and 185 InDels were observed among all the inbreds across the entire gene. However, a 4 bp InDel at position 2406 bp in intron-7 and two SNPs at position 3325 (“C” in the wild and “A” in the mutant) and 4310 (“G” in the wild and “T” in the mutant) differentiated wild and waxy genotypes (Fig. 1).

Sequence alignment for Wx1 gene showing polymorphisms differentiating wild and mutant wx1 alleles
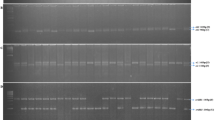
Inbreds of the mutant-type amplified 94 bp and those of the wild-type amplified 90 bp using a codominant marker (WxDel4) that encompassed 4 bp InDel (Fig. 2). In addition, a co-dominant marker (SNP3325_CA1) was developed, amplifying a 185 bp amplicon with primers SNP3325_CA1_F1 & R (wild-specific) and SNP3325_CA1_F2 & R (mutant-specific). Another co-dominant marker, SNP4310_GT2, was created with a primer set consisting of SNP4310_GT2_F1 & R (for wild-type DNA) and SNP4310_GT2_F2 & R (for mutant DNA). This primer combination generated an amplicon of 189 bp. While the mutant-specific primer set was only amplified in waxy inbreds, the wild-specific primer set was only amplified in wild-type inbreds (Figs. 3, 4).

Representative gel picture of segregation of WxDel4 marker in BC2F2 population; L, 50 bp ladder; R, dominant homozygote; D, recessive homozygote ad H, heterozygote; Yellow star indicates recessive homozygotes (waxy genotype)

Representative gel picture of segregation of SNP3325_CA marker in BC2F2 population; L, 50 bp ladder; R, dominant homozygote; D, recessive homozygote ad H, heterozygote; Yellow star indicates recessive homozygotes (waxy genotype). The order of the genotypes is same as Fig. 2

Representative gel picture of segregation of SNP4310_GT marker in BC2F2 population; L, 50 bp ladder; R, dominant homozygote; D, recessive homozygote ad H, heterozygote; Yellow star indicates recessive homozygotes (waxy genotype). (The order of the genotypes is same as Fig. 2)
Genotyping of backcross progenies
The InDel and SNP-based allele-specific markers developed under this study were used for genotyping seven populations of BC1F1, BC2F1, and BC2F2 generations. In BC1F1, WxDel4, SNP3325_CA1, and SNP4310_GT2 identified 53, 51, 38, 39, 39, 41, and 51 heterozygous plants in HKI323 × MGU-102-wx1, HKI1105 × MGU-102-wx1, HKI1128 × MGU-102-wx1, HKI161 × MGU-102-wx1, HKI163 × MGU-102-wx1, HKI193-1 × MGU-102-wx1, and HKI193-2 × MGU-102-wx1 populations, respectively (Table 2). WxDel4-FR showed a single amplicon in dominant homozygotes (Wx1/Wx1), and two amplicons in heterozygous plants (Wx1/wx1). Wild-specific SNP3325_CA1 and SNP4310_GT2 primer-set amplified bands in both homozygous dominant (Wx1/Wx1) and heterozygous plants (Wx1/wx1). While mutant specific SNP3325_CA1 and SNP4310_GT2 primer-set amplified in heterozygous plants (Wx1/wx1) only. All seven crosses had the recessive wx1 gene segregate according to the Mendelian inheritance pattern of 1 (Wx1Wx1):1 (Wx1wx1) (Table 2 and Fig. 2).
In case of BC2F1, using all the three markers (WxDel4, SNP3325_CA1, and SNP4310_GT2) and 51 heterozygous plants (Wx1/wx1) in HKI323 × MGU-102-wx1, 48 in HKI1105 × MGU-102-wx1, 42 in HKI1128 × MGU-102-wx1, 50 plants in HKI161 × MGU-102-wx1, 39 in HKI163 × MGU-102-wx1, 43 in HKI193-1 × MGU-102-wx1, and 41 in HKI193-2 × MGU-102-wx1 were identified (Table 2). WxDel4 separated both dominant homozygotes (Wx1Wx1) from heterozygotes (Wx1wx1). Wild-specific primer-set of SNP3325_CA1 and SNP4310_GT2 amplified in both dominant homozygotes (Wx1/Wx1) and heterozygotes (Wx1/wx1), whereas only heterozygous (Wx1/wx1) plants were amplified using mutant specific primer-set. The Mendelian inheritance pattern of 1 (Wx1wx1):1 (wx1wx1) was observed across seven crosses (Table 2).
In BC2F2 generation, WxDel4, SNP3325_CA1, and SNP4310_GT2 identified 20, 26, 19, 23, 19, 26, and 24 homozygous recessive (wx1wx1) plants in HKI323 × MGU-102-wx1, HKI1105 × MGU-102-wx1, HKI1128 × MGU-102-wx1, HKI161 × MGU-102-wx1, HKI163 × MGU-102-wx1, HKI193-1 × MGU-102-wx1, and HKI193-2 × MGU-102-wx1 populations, respectively (Table 2). The dominant homozygotes (Wx1Wx1), heterozygotes (Wx1wx1), and recessive homozygotes (wx1wx1) were identified using a WxDel4 marker (Fig. 2). Wild specific primer sets of SNP3325_CA1 and SNP4310_GT2 amplified expected fragments in dominant homozygotes (Wx1Wx1) and heterozygotes (Wx1/wx1) (Figs. 3, 4). On the other hand, mutant-specific primer sets of SNP3325 and SNP4310 amplified products in recessive homozygotes (wx1/wx1) and heterozygous plants (Wx1/wx1) (Figs. 3, 4). All seven crosses were in accordance with the Mendelian segregation ratio of 1 (Wx1Wx1):2 (Wx1wx1):1 (wx1wx1) (Table 2).
Amylopectin in selected wild and mutant genotypes, and among backcross-derived progenies
Amylopectin among the selected waxy inbreds (wx1-Mutant1 to wx1-Mutant7) was higher (mean 96.9%, range 96.1–97.5%) compared to the selected normal inbreds (Wx1-Wild1 to Wx1-Wild7) (mean 71.8%, range 68.1–74.9%) (Fig. S1). Among waxy inbreds, the wx1-Mutant1 genotype possessed the highest amylopectin (97.5%). The other six waxy genotypes, wx1-Mutant2, wx1-Mutant3, wx1-Mutant4, wx1-Mutant5, wx1-Mutant6, and wx1-Mutant7 had 96.5%, 97.3%, 97.1%, 96.4%, 96.1%, and 97.4% amylopectin, respectively. On the other hand, normal or wild type genotypes viz., Wx1-Wild1, Wx1-Wild2, Wx1-Wild3, Wx1-Wild4, Wx1-Wild5, Wx1-Wild6, and Wx1-Wild7 possessed 69.4%, 70.3%, 68.1, 73.5%, 72.2%, 74.2%, and 74.9% amylopectin, respectively. Approximately, a 1.4-fold increase in amylopectin was recorded among the waxy inbreds.
Amylopectin among the backcross-derived progenies (BC2F3 seeds harvested from BC2F2 plants) of HKI161, HKI163, HKI323, HKI1105, HKI1128, HKI193-1, and HKI193-2 showed a significant increase (mean 97.7%, range 95.4–99.6%) as compared to respective recurrent parents (mean 72.7%, range 68.2–75.8%) (Table S3). HKI323 had an average of 69.8% amylopectin, and its waxy versions possessed an average of 96.1%. HKI1105 possessed an average of 71.3% amylopectin, and its introgressed versions had 96.7% amylopectin. Waxy versions of HKI1128 had 97.4% amylopectin compared to 72.1% in HKI128. HKI161 had 74.1% amylopectin as compared to 98.5% amylopectin in its waxy versions (Fig. 5). HKI163 had 72.6% amylopectin, and its backcross-derived versions possessed 97.8% amylopectin. Waxy versions of HKI193-1 and HKI193-2 had 98.7% and 98.9% amylopectin compared to 74.2% and 74.7% amylopectin among the original versions, respectively. A similar fold increase in amylopectin was recorded among introgressed progenies.

Amylopectin content in MAS-derived progenies and their original versions
Discussion
Waxy maize rich in amylopectin has gained popularity throughout the world (Xiaoyang et al. 2017). It is generally consumed in the daily diet in the south-east region of Asia, and it helps poultry birds grow faster and better (Talukder et al. 2022). Waxy maize grains possess a wide range of industrial applications due to their chemical characteristics (Devi et al. 2017). Despite the widespread cultivation of waxy landraces, waxy hybrids with excellent grain yields are hard to come by. The primary method for introducing the mutant wx1 gene into field corn hybrids is molecular breeding. Few markers, including SSRs associated with wx1 genes, have been reported earlier and are being used in the MAS program. However, these markers are not universal as the markers are not polymorphic between the wild-type (Wx1) and mutant-type (wx1) germplasm across the globe, thereby limiting their application in MAS (Devi et al. 2017; Hossain et al. 2019). Here, we developed new gene-based InDels and SNP (allele-specific) markers for the selection of the wx1 gene in the molecular breeding program.
Gene-based markers specific to the wx1 gene
In the present study, one InDel marker (WxDel4) and two allele-specific SNP (SNP3325_CA1 and SNP4310_GT2) markers were developed. WxDel4 was developed from a 4 bp insertion-deletion polymorphism present in intron-7. Being present in an intron, the 4 bp InDel does not change the function of the wx1 gene. Okagaki et al. (1991) also discovered a 30 bp deletion in the Wx1 gene and created the mutant phenotype. A 5.6 kb retrotransposon insertion in the intron of the Wx1 gene also resulted in a stable waxy mutation (Marillonnet and Wessler 1997). The InDel marker developed here was codominant in nature and was able to distinguish heterozygotes from homozygotes (Chhabra et al. 2019). Sequence alignment of selected mutant and wild-type sequences of Wx1 alleles, considering X03935 as well as the Wx1-Reference gene sequence, revealed a C-to-A transition between the wild-type and mutant at 3325 bp position. The nucleotide polymorphism did not change amino acid as both nucleotide codons code for glycine. The seven mutant and seven wild-type alleles, as well as the Wx1-Reference genome sequence, also revealed a G to T transition at 4310 bp position, which also resulted in silent mutation. SNP markers are increasingly becoming popular due to their abundance, consistency, efficiency, less cost, and automation friendliness (Tong et al. 2016). A base change at the 3′ end of SNP primers typically correlates to a particular SNP site. According to the Liu et al. stated technique, an additional nucleotide alteration (A/T) has been made inside the three penultimate bases closest to the SNP site to increase the specificity of allele-specific SNP primers (2012). The newly created allele-specific SNP markers were reliable, affordable, and strong. The cost of SNP markers is lower than that of CAPS and dCAPS markers (Abhijith et al. 2020). To separate PCR amplicons, a straightforward 3% agarose gel can be employed. The markers generated in the current work are breeder-friendly since SNP-specific markers are based on straightforward PCR and agarose gel electrophoresis procedures rather than expensive and complex assays. Though any of these markers did not possess function, the polymorphisms could successfully differentiate the wild-type and mutant alleles of the Wx1 gene. These non-functional polymorphisms may be associated with the fitness of the Wx1 allele or strong linkage disequilibrium (LD) with the key functional polymorphisms. Thus, three markers developed in the present study possess a very high possibility of differentiating wild-type (Wx1) and mutant (wx1) alleles in various genetic background. However, the position and type of mutation within the Wx1 gene may vary in some waxy germplasm adapted to different regions of the world, and in such cases, new marker(s) would be required to be developed and validated.
Utilization of markers in molecular breeding
Marker-assisted backcross breeding (MABB) is preferable over traditional breeding particularly for the introgression of recessive alleles as heterozygous genotypes can be easily identified from the dominant homozygotes (Gupta et al. 2013). Additionally, progeny testing that takes a long time is avoided after each backcross (Hossain et al. 2018). The wx-2507F/RG InDel marker was found to be polymorphic between the wild-type and mutant alleles of Wx1 by Talukder et al. (2022). Three gene-based SSRs, phi027, phi061, and phi022, were employed by Zhang et al. (2013) to detect variation in the Wx1 gene in recurrent and donor parents. While Talukder et al. (2022) discovered phi022 as the polymorphism marker between the Wx1 and wx1 alleles, Yang et al. (2013) revealed phi022 and phi027 as polymorphic markers among the recurrent and donor parents. Three markers developed under the study were efficiently deployed for selecting foreground positive plants in BC1F1, BC2F1, and BC2F2 segregating generations. Among these, WxDel4 was an InDel marker with a codominance nature that was used in the current investigation to discriminate between the Wx1Wx1, wx1wx1, and Wx1wx1 genotypes. These markers are the best option for MAS. The research also showed that the Wx1 gene was segregated using the WxDel4 marker in both backcross and selfed generations in accordance with the Mendelian pattern. The other two markers (SNP3325_CA1 and SNP4310_GT2) were also co-dominant in nature. By comparing the amplicon pattern of different forward primers of a marker, heterozygotes were identified. For example, SNP3325_CA1_F1/R generated bands in Wx1Wx1 and Wx1wx1, while SNP3325_CA1_F2/R developed bands in Wx1wx1 and wx1wx1. Thus, individuals having an expression of bands by both the primer sets were considered heterozygotes, while individuals having bands only with one primer set were considered homozygotes depending upon the amplification by a specific type of the primer set. These two allele-specific SNP markers were successfully used in BC1F1, BC2F1, and BC2F2. Chhabra et al. (2020) developed four SNP-based markers for the sh2 gene in sweet corn. Abhijith et al. (2020) developed a breeder-friendly gene-based SNP (allele-specific) marker for the lpa1-1 gene responsible for low phytic acid in maize kernel. SNPs are successfully being used in marker-assisted breeding as they are more appropriate for genotyping than AFLP, RFLP, and SSR markers (Flint-Garcia et al. 2003). Next generation-based SNP genotyping though is high-throughput, but, costly and requires dedicated laboratory and special equipment (Shen et al. 2005). These SNP markers are easy to use as they simply require a PCR machine and gel electrophoretic system, thus involving low cost, and can be easily used by the breeders, especially in under-developed as well as developing countries (Chhabra et al. 2020).
Enhancement of amylopectin among wx1-based progenies
Amylose and amylopectin components make up maize starch (Hossain et al. 2019). Amylopectin is a homopolymer of glucopyranose with both α-(1 → 4) linkage and branching with α-(1 → 6), whereas amylose is a linear polymer of glucopyranose units joined by α-(1 → 4) linkage (Lin et al. 2019). According to the current study, waxy maize inbreds had about 40% more amylopectin than regular inbreds. According to Qi et al. (2020), field corn had a mean amylopectin concentration of 76.9%, but waxy maize had a mean amylopectin level of 94.9%. According to Yang et al. (2013), BC2F4 seeds had an average amylopectin concentration of 98.15%.
Stamp et al. (2016) also found > 97% amylopectin in waxy landraces compared to ~ 70–75% amylopectin in wild types of landraces. In the endosperm of maize, the wild-type Wx1 allele is involved in the synthesis of amylose and has been mapped at bin 9.03. (Brimhall et al. 1945; Nelson and Rines 1962). It has a 3718-bp-long coding sequence and 14 exons that range in size from 64 to 392 bp (Huang et al. 2010). A changed transcript is produced by a variety of mutations in the Wx1 gene, including premature stop codons, amino acid alterations in coding areas, and post-translational modifications. These mutations cause less amylose and more amylopectin to be present in grains (Wessler et al. 1986; Liu et al. 2007; Bao et al. 2012; Zhang et al. 2013). Thus, the higher amylopectin as observed in the BC2F3 seeds was due to the precise selection of recessive wx1 allele in the heterozygous condition in BC1F1 and BC2F1 populations, followed by the selection of homozygous wx1wx1 conditions in BC2F2 populations.
Conclusions
Comprehensive sequence analysis of the Wx1 gene in a set of wild and mutant inbreds revealed one 4 bp InDel and two SNPs. Three markers, viz., WxDel4, SNP3325_CA1, and SNP4310_GT2, were successfully developed and utilized in the genotyping of BC1F1, BC2F1, and BC2F2 populations segregating for the wx1 allele. All the markers differentiated the heterozygotes (Wx1wx1) from dominant homozygotes (Wx1Wx1) in BC1F1 and BC2F1 populations, while recessive homozygotes (wx1wx1) were identified in BC2F2 populations. The precise selection using markers was finally validated by an enhanced level of amylopectin among the selected wx1wx1-based progenies. These newly developed wx1-based markers would help in accelerating the pace of the waxy corn breeding program worldwide.
References
Abhijith KP, Muthusamy V, Chhabra R, Dosad S, Bhatt V, Chand G, Jaiswal SK, Zunjare RU, Vasudev S, Yadava DK, Hossain F (2020) Development and validation of breeder-friendly gene-based markers for lpa1–1 and lpa2–1 genes conferring low phytic acid in maize kernel. 3 Biotech 10:121. https://doi.org/10.1007/s13205-020-2113-x
Bao JD, Yao JQ, Zhu JQ (2012) Identification of glutinous maize landraces and inbred lines with altered transcription of waxy gene. Mol Breed 30:1707–1714
Brimhall B, Sprague GF, Sass JE (1945) A new waxy allele in corn and its effect on the properties of the endosperm starch. J Agronomy 37:937–944
Chhabra R, Hossain F, Muthusamy V, Baveja A, Mehta BK, Zunjare RU (2019) Development and validation of breeder-friendly functional markers of sugary1 gene encoding starch-debranching enzyme affecting kernel sweetness in maize (Zea mays). Crop Pasture Sci 70(10):868–875. https://doi.org/10.1071/CP19298
Chhabra R, Hossain F, Muthusamy V, Baveja A, Mehta BK, Zunjare RU (2020) Development and validation of gene-based markers for shrunken2-Reference allele and their utilization in marker-assisted sweet corn (Zea mays Sachharata) breeding programme. Plant Breed :1–10. https://doi.org/10.1111/pbr.12872
Devi EL, Chhabra R, Jaiswal SK, Hossain F, Zunjare RU, Goswami R, Muthusamy V, Baveja A, Dosad S (2017) Microsatellite marker-based characterization of waxy maize inbreds for their utilization in hybrid breeding. 3 Biotech 7:316
Dong L, Qi X, Zhu J, Liu C, Zhang X, Cheng B, Mao L, Xie C (2019) Supersweet and waxy: meeting the diverse demands for specialty maize by genome editing. Plant Biotechnol J 17(10):1853–1855. https://doi.org/10.1111/pbi.13144
Flint-Garcia SA, Thornsberry JM, Buckler ES (2003) Structure of linkage disequilibrium in plants. Ann Rev Plant Biol 54:357–374. https://doi.org/10.1146/annurev.arplant.54.031902.134907
Gupta HS, Babu R, Agrawal PK, Mahajan V, Hossain F, Nepolean T (2013) Accelerated development of quality protein maize hybrid through marker-assisted introgression of opaque-2 allele. Plant Breed 132:77–82
Hossain F, Muthusamy V, Pandey N, Vishwakarma AK, Baveja A, Zunjare RU, Thirunavukkarasu N, Saha S, Manjaiah KM, Prasanna BM, Gupta HS (2018) Marker-assisted introgression of opaque2 allele for rapid conversion of elite hybrids into quality protein maize. J Genet 97:287–298
Hossain F, Chhabra R, Devi EL, Zunjare RU, Jaiswal SK, Muthusamy V (2019) Molecular analysis of mutant granule-bound starch synthase-I (waxy1) gene in diverse waxy maize inbreds. 3 Biotech 9:3. https://doi.org/10.1007/s13205-018-1530-6
Huang BQ, Tian ML, Zhang JJ, Huang YB (2010) Waxy locus and its mutant types in maize (Zea mays L). J Integr Agr 9:1–10
Klosgen RB, Gierl A, Schwarz-Sommer Z, Saedler H (1986) Molecular analysis of the waxy locus of Zea mays. Mol Genet Genom 203:237–244
Lin F, Zhou L, He B, Zhang X, Dai H, Qian Y, Ruan L, Zhao H (2019) QTL mapping for maize starch content and candidate gene prediction combined with co-expression network analysis. Theor Appl Genet 132(7):1931–1941
Liu J, Rong T, Li W (2007) Mutation loci and intragenic selection marker of the granule-bound starch synthase gene in waxy maize. Mol Breed 20:93–102. https://doi.org/10.1007/s11032-006-9074-6
Liu J, Huang S, Sun M (2012) An improved allele-specific PCR primer design method for SNP marker analysis and its application. Plant Methods 8:34. https://doi.org/10.1186/1746-4811-8-34
Liu H, Wang X, Wei B, Wang Y, Liu Y, Zhang J, Hu Y, Yu G, Li J, Xu Z, Huang Y (2016) Characterization of genome-wide variation in Four-row Wax, a waxy maize landrace with a reduced kernel row phenotype. Front Plant Sci 7:667. https://doi.org/10.3389/fpls.2016.00667
Macdonald FD, Preiss J (1985) Partial purification and characterization of granule-bound starch synthases from normal and waxy maize. J Plant Physiol 78:849–852
Marillonnet S, Wessler SR (1997) Retrotransposon insertion into the maize waxy gene results in tissue-specific RNA processing. Plant Cell 9:967–978
Mason-Gamer RJ, Well CF, Kellogg EA (1998) Granule-bound starch synthase: structure, function, and phylogenetic utility. Mol Biol Evol 15:1658–1673
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4326. https://doi.org/10.1093/nar/8.19.4321
Muthusamy V, Hossain F, Thirunavukkarasu N, Choudhary M, Saha S, Bhat JS, Prasanna BM, Gupta HS (2014) Development of β-carotene rich maize hybrids through marker-assisted introgression of β-carotene hydroxylase allele. PLoS ONE 9:e113583
Nelson OE, Rines HW (1962) The enzymatic deficiency in the waxy mutant of maize. Biochem Biophy Res Commun 9:297–300
Okagaki RJ, Neuffer MG, Wessler SR (1991) A deletion common to two independently derived waxy mutations of maize. Genetics 128:425–431
Qi X, Dong L, Liu C, Mao L, Liu F, Zhang X, Cheng B, Xie C (2018) Systematic identification of endogenous RNA polymerase III promoters for efficient RNA guide-based genome editing technologies in maize. Crop J 6:314–320
Qi X, Wu H, Jiang H, Zhu J, Huang C, Zhang X, Liu C, Cheng B (2020) Conversion of a normal maize hybrid into a waxy version using in vivo CRISPR/Cas9 targeted mutation activity. Crop J. https://doi.org/10.1016/j.cj.2020.01.006
Reddappa SB, Chhabra R, Talukder ZA, Muthusamy V, Zunjare RU, Hossain F (2022) Development and validation of rapid and cost-effective protocol for estimation of amylose and amylopectin in maize kernels. 3 Biotech 12:62. https://doi.org/10.1007/s13205-022-03128-z
Sarika K, Hossain F, Muthusamy V, Zunjare RU, Baveja A, Goswami R, Bhat JS, Saha S, Gupta HS (2018) Marker-assisted pyramiding of opaque2 and novel opaque16 genes for further enrichment of lysine and tryptophan in sub-tropical maize. Plant Sci 272:142–152
Shen R, FanJB CD, Chang W, Chen J, Doucet D, Yeakley J, Bibikova M, Wickham GE, McBride C, Steemers F, Garcia F, Kermani BG, Gunderson K, Oliphant A (2005) High-throughput SNP genotyping on universal bead arrays. Mutat Res 573:70–82. https://doi.org/10.1016/j.mrfmmm.2004.07.022
Shin JK, Know SJ, Lee JK, Min HK (2006) Genetic diversity of maize kernel starch-synthesis genes with SNPs. Genome 49:1287–1296
Shure M, Wessler S, Fedoroff N (1983) Molecular identification and isolation of the waxy locus in maize. Cell 35:225–233
Singh BD, Singh AK (2015) Marker-assisted plant breeding: principles and practices. Springer, New Delhi, India. https://doi.org/10.1007/978-81-322-2316-0
Stamp P, Eicke S, JampatongS, Le-Huy H, Jompuk C, EscherF, Streb S (2016) Southeast Asian waxy maize (Zea mays L.), a resource for amylopectin starch quality types? Plant Genet Res 1–8. https://doi.org/10.1017/S1479262116000101
Talukder ZA, Muthusamy V, Chhabra R et al. (2022) Combining higher accumulation of amylopectin, lysine and tryptophan in maize hybrids through genomics-assisted stacking of waxy1 and opaque2 genes. Sci Rep 12:706. https://doi.org/10.1038/s41598-021-04698-3
Tong J, Han Z, Han A, Liu X, Zhang S, Fu B, Zhu Y (2016) Sdt97, a point mutation in 5’ untranslated region, confers semi-dwarfism in rice. G3-Genes Genom Genet 6(6):1491–1502
Vrinten PL, Nakamura T (2000) Wheat granule-bound starch synthase I and II are encoded by separate genes that are expressed in different tissues. Plant Physiol 122(1):255–264. https://doi.org/10.1104/pp.122.1.255
Wessler S, Baran G, Varagona M, Dellaporta S (1986) Excision of Ds produces waxy proteins with a range of enzymatic activities. EMBO J 5:2427
Wu XY, Wu S, Long W, Chen D, Zhou G, Du J, Cai Q, Huang X (2019) New waxy allele wx-Reina found in Chinese waxy maize. Genet Resour Crop Evol 66:885–895
Xiaoyang W, Dan C, Yuqing L, Weihua L, XinmingY XL, Juan D, Lihui L (2017) Molecular characteristics of two new waxy mutations in China waxy maize. Mol Breed 37:27
Yang L, Wang M, We Wang, Yang W (2013) Marker-assisted selection for pyramiding the waxy and opaque-16 genes in maize using crosses and backcross schemes. Mol Breed 31:767–775. https://doi.org/10.1007/s11032-012-9830-8
Zhang WL, Yang WP, Wang MC, Wang W, Zeng GP, Chen ZW (2013) Increasing lysine content of waxy maize through introgression of opaque-2 and opaque-16 genes using molecular assisted and biochemical development. PLoS One 8:e56227. https://doi.org/10.1371/journal.pone.0056227
Zunjare RU, Hossain F, Muthusamy V, Baveja A, Chauhan HS, Bhat JS, Thirunavukkarasu N, Saha S, Gupta HS (2018) Development of biofortified maize hybrids through marker-assisted stacking of β-Carotene hydroxylase, Lycopene- ε-Cyclase and Opaque2 genes. Front Plant Sci 9:178
Acknowledgements
The authors sincerely acknowledge BHEARD and ICAR-Indian Agricultural Research Institute, New Delhi, for the financial support. The first author wishes to thank USAID, USA, for providing the BHEARD fellowship to undertake his Ph.D. program at ICAR-IARI. We thank IARI for providing the required lab and field facilities. We thank IIMR, Ludhiana, and AICRP (Maize) for providing the off-season nursery at Hyderabad. The authors sincerely thank Dr. B.M. Prasanna, CIMMYT, for providing the waxy source germplasm from where the waxy donor was developed. The help of CCSHAU, Uchani for sharing the parental inbreds is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
Genotyping of populations: ZAT, designing of markers and standardization of PCR protocol: RC, estimation of amylopectin: ZAT and RC, development of mapping populations: VM, statistical analysis: RUZ, manuscript writing: ZAT and FH, designing of the experiment: FH.
Corresponding author
Ethics declarations
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare no competing interests.
Additional information
Communicated by: Izabela Pawłowicz
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Talukder, Z.A., Chhabra, R., Muthusamy, V. et al. Development of novel gene-based markers for waxy1 gene and their validation for exploitation in molecular breeding for enhancement of amylopectin in maize. J Appl Genetics 64, 409–418 (2023). https://doi.org/10.1007/s13353-023-00762-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-023-00762-y