
Abstract
Sulfonylureas are often used alone or in combination with other drugs for treating type 2 diabetes. Ipragliflozin is a sodium/glucose cotransporter 2 inhibitor that enhances urinary glucose excretion and improves glycemic control. We examined the efficacy and safety of ipragliflozin as an add-on to a sulfonylurea in Japanese type 2 diabetes patients with inadequate glycemic control in a phase III study. Patients were randomized, double-blind, to 50 mg ipragliflozin or placebo for 24 weeks, followed by a 28-week open-label extension in which all patients received either 50 or 100 mg ipragliflozin. The primary endpoint was the change in HbA1c from baseline to week 24 (last observation carried forward). Overall, 166 patients were prescribed ipragliflozin and 77 were prescribed placebo. The adjusted mean change in HbA1c from baseline to week 24 between the ipragliflozin and placebo groups was −1.14 % (P < 0.001). The reductions in fasting plasma glucose and body weight were significantly greater in the ipragliflozin group (placebo-adjusted mean change: −38.0 mg/dl and −1.32 kg, respectively; both, P < 0.001). These changes were maintained until the end of the open-label extension. There were more treatment-emergent adverse events in the ipragliflozin group than in the placebo group (75.9 vs. 61.8 %; P = 0.032). The most common adverse events with a higher incidence in the ipragliflozin group than in the placebo group were mild pollakiuria and thirst. Ipragliflozin as an add-on to a sulfonylurea significantly improved glycemic control and reduced body weight, with good tolerability, in Japanese type 2 diabetes patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sodium/glucose cotransporters (SGLTs) are responsible for the cotransporting of sodium and glucose in the intestine and kidney. The kidney-specific member of this family, SGLT2, is mainly expressed on the brush border membrane of the S1 and S2 segments of the proximal tubule [1] where it reabsorbs about 90 % of glucose from the renal filtrate [2] or as much as 180 g of glucose per day [3]. Because inhibition of SGLT2 increases urinary glucose excretion and thereby lowers blood glucose levels, several SGLT2 inhibitors have been developed for the treatment of type 2 diabetes [4, 5].
Previous pharmacokinetic and pharmacodynamic studies confirmed that ipragliflozin significantly enhances urinary glucose excretion without affecting plasma glucose levels in healthy individuals [6, 7]. In patients with type 2 diabetes, ipragliflozin reduced plasma glucose levels by enhancing urinary glucose excretion [8].
Consequently, several clinical trials have been performed, showing that ipragliflozin improved glycemic control and was well tolerated when used as monotherapy [9, 10] or in combination with metformin [11].
Metformin is widely used as the initial treatment for type 2 diabetes. However, sulfonylureas are also widely used as a first-line treatment in East Asian countries [12, 13]. If glycemic control remains inadequate, another oral antidiabetic drug or insulin can be added to ongoing therapy. Consequently, it is important to determine the efficacy and safety of newer drugs as an add-on to a sulfonylurea in Japanese patients with inadequate glycemic control.
An earlier study clearly demonstrated that the pharmacokinetics of ipragliflozin and sulfonylureas were not affected by their coadministration [14]; however, no studies have examined the medium- to long-term effects on glycemic control or safety of using ipragliflozin as an add-on to a sulfonylurea.
Therefore, the objective of this double-blind, placebo-controlled, phase III study was to evaluate the efficacy and safety of treatment with ipragliflozin as an add-on to a sulfonylurea for 24 weeks in Japanese patients with type 2 diabetes. We also included an open-label, 28-week extension period to examine the longer term efficacy and safety of this combination. This article focuses on the placebo-controlled part of the study because of its scientific importance, but also briefly describes the results obtained during the open-label extension period.
Methods
Patients
The main inclusion criteria were age ≥20 years, diagnosis of type 2 diabetes ≥12 weeks before the study, HbA1c of 7.4–9.9 % [National Glycohemoglobin Standardization Program (NGSP) units] at the start of the run-in period, fasting plasma glucose (FPG) ≥126 mg/dl, body mass index of 20.0–45.0 kg/m2, and treatment with sulfonylurea alone at a stable dose (≥1.25 mg/day glibenclamide, ≥40 mg/day gliclazide, or ≥1 mg/day glimepiride) for ≥4 weeks before the screening period.
Patients satisfying the following major criteria were excluded: proliferative diabetic retinopathy; dysuria; symptomatic urinary tract or genital infection; a serious cardiovascular event within 12 weeks; New York Heart Association class III or IV congestive heart failure; unstable psychiatric disorder; history of malignant tumors (except those who had not received treatment for ≥5 years and who were not considered to have a recurrence); severe gastrointestinal disease; serum creatinine (Cr) exceeding the upper limit of normal; urinary albumin/urinary Cr ratio >300 mg/g Cr; aspartate aminotransferase or alanine aminotransferase > twice the upper limit of normal; uncontrolled hypertension (systolic blood pressure >170 mmHg and/or diastolic blood pressure >95 mmHg); treatment with insulin <12 weeks before the screening period; or chronic use of adrenocorticosteroids, immunosuppressants, or a loop diuretic (short-term or temporary use of these drugs was allowed), among other criteria. All of the patients provided written informed consent at the time of enrollment.
Study design and treatments
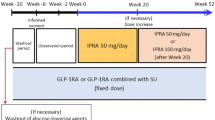
As illustrated in Fig. 1a, the study consisted of a 4-week screening period, a 2-week single-blind run-in period in which all patients received placebo, a double-blind 24-week treatment period (treatment period I), an open-label 28-week extension period (treatment period II), and a 4-week follow-up period. Patients who used another drug in combination with a sulfonylurea before the study underwent a washout period of ≥4 weeks after providing informed consent and before they entered the screening period.

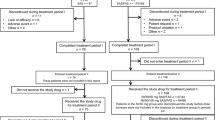
a Study design. b Patient disposition. *Includes one patient whose use of the double-blind study drug was unknown. †Includes two patients whose ipragliflozin dose was decreased from 100 to 50 mg during treatment period II. ‡Includes one patient whose ipragliflozin dose was decreased from 100 to 50 mg during treatment period II
At the end of the run-in period, patients were randomized (in a 2:1 ratio) to receive either 50 mg ipragliflozin or placebo once daily before breakfast. The randomization schedule was prepared by a central registration center. The patients and clinicians were kept blind to the treatment received in treatment period I until the data for treatment period I had been entered into the study database and locked. The placebo drug was identical in appearance and packaging to the active drug.
The sulfonylurea was continued at a constant dose from enrollment to the end of the treatment period or discontinuation. However, the dose could be changed in the follow-up period.
Patients were eligible to enter treatment period II if their HbA1c at week 20 was <8.4 % and was lower than the baseline value. Written informed consent was newly acquired at week 24 to participate in treatment period II. All patients were treated with ipragliflozin in an open-label manner.
At week 24, the ipragliflozin dose could be increased to 100 mg if the patient’s HbA1c at week 20 was ≥7.4 %. After a dose increase to 100 mg, the ipragliflozin dose could be reduced to 50 mg if there were safety concerns, such as hypoglycemia, but no further dose change was permitted.
The use of other antidiabetic drugs and continuous administration of drugs capable of affecting glucose metabolism were prohibited between the start of the screening period and the end of the treatment period or discontinuation. Continuous use of systemic corticosteroids, immunosuppressants, or loop diuretics was also prohibited.
Treatment compliance was assessed by the investigators who reviewed their patients’ diaries to determine the numbers of prescribed, returned, or lost study drugs.
The study was approved by institutional review boards at each participating site and was conducted in accordance with Good Clinical Practice, International Conference on Harmonization Technical Requirements for Registration of Pharmaceuticals for Human Use, as well as local laws and regulations.
Study objectives and assessments
The primary efficacy endpoint was change in HbA1c from baseline to week 24 (last observation carried forward; LOCF). Secondary efficacy endpoints included the proportion of HbA1c responders (<7.0 %) and the changes in FPG, fasting serum insulin (FSI), leptin, adiponectin, body weight, and waist circumference from baseline to week 24. Efficacy endpoints were also assessed in terms of the changes from baseline to week 52 (LOCF).
HbA1c was measured using an enzymatic assay, insulin using a microparticle enzyme immunoassay, leptin using a double-antibody radioimmunoassay, and adiponectin using a latex agglutination turbidimetry, all of which were performed by Mitsubishi Chemical Medience Corp. (Tokyo, Japan). HbA1c values were initially reported in Japan Diabetes Society (JDS) units, which were then converted into NGSP units using the following equation [15]: HbA1c (NGSP) (%) = 1.02 × HbA1c (JDS) (%) + 0.25 %.
Safety outcomes included treatment-emergent adverse events (TEAEs), laboratory parameters (measured using routine methods at Mitsubishi Chemical Medience Corp.), vital signs, 12-lead electrocardiograms, and estimated glomerular filtration rate (eGFR) [16]. TEAEs were recorded according to system organ class and preferred term (MedDRA version 12.1), along with the severity and relationship to the study drug. Most episodes of hypoglycemia were labeled as such by the investigators based on the patients’ subjective symptoms without blood glucose data at the onset of symptoms.
Statistical analysis
We planned to enroll 225 patients and randomize 150 to ipragliflozin and 75 to placebo to comply with the Japanese Guidelines for Clinical Evaluation of Oral Hypoglycemic Agents [17]. From a statistical perspective, the sample size was deemed large enough to show statistical superiority of ipragliflozin to placebo according to the results of a phase II study [10].
The primary and secondary efficacy endpoints were evaluated in the full analysis set, which was defined as all patients who received at least one dose of the study drug and in whom efficacy variables were measured at least once after starting administration. Safety analyses were done using the safety analysis set, which included all patients who received at least one dose of the study drug.
Data are presented as mean ± SD or n (%), unless otherwise specified. Baseline characteristics were compared between the ipragliflozin and placebo groups using the two-sample t test or Fisher’s exact test, as appropriate.
The primary endpoint (change in HbA1c from baseline to week 24) was evaluated using analysis of covariance (ANCOVA) with baseline HbA1c as a covariate and treatment group as a fixed effect. Efficacy analysis was based on the last observation carried forward method to impute missing data at the endpoint. For the sensitivity analysis, each demographic and baseline characteristic (including body weight at screening, which showed an imbalance between the treatment groups) was added to the primary analysis model as a covariate or a fixed effect. The analytical method used in the primary analysis was also applied to the per protocol set. In addition, subgroup analyses were conducted in patients stratified by sex, age, baseline HbA1c, baseline eGFR, BMI, sulfonylurea type, and whether or not a washout period was required.
Secondary efficacy endpoints at week 24 were compared using ANCOVA with the baseline value as a covariate and the treatment group as a fixed effect. For 52 weeks of treatment, changes in efficacy endpoints from baseline to week 52 were evaluated descriptively according to the ipragliflozin dose received. Changes in homeostasis model assessment of β cell function (HOMA-β) [18] were also assessed in post hoc analyses.
Safety variables were analyzed descriptively and are presented as the number of patients (%) within each group. eGFR was calculated from serum creatinine, age, and sex, using the modification of diet in renal disease equation modified for Japanese subjects [16]. Post hoc analyses using two-sample t-test were performed for laboratory parameters.
Subgroup analyses of efficacy (HbA1c, FPG, and body weight) and TEAEs were done after dividing patients into two subgroups according to their sulfonylurea dose as either a high dose (at least half of the highest approved dose; glibenclamide ≥5 mg/day, gliclazide ≥80 mg/day, or glimepiride ≥3 mg/day) or a low dose (less than half of the highest approved dose; glibenclamide <5 mg/day, gliclazide <80 mg/day, or glimepiride <3 mg/day).
For all analyses, P < 0.05 (two-tailed) was considered statistically significant.
Results
Patient disposition and characteristics
The study was conducted across 35 sites in Japan between September 2010 and April 2012. As shown in Fig. 1b, 386 patients provided informed consent, of whom 245 were randomized and 243 were prescribed ipragliflozin (n = 166) or placebo (n = 77). The baseline characteristics of both groups are shown in Table 1 and were generally comparable, except for body weight and body mass index at screening, which were significantly greater in the ipragliflozin group than in the placebo group.
The durations of exposure (mean ± SD) during treatment period I were 163.4 ± 22.23 and 149.4 ± 38.84 days in the ipragliflozin and placebo groups, respectively. The treatment compliance levels (mean ± SD) were 98.70 ± 2.428 and 99.23 ± 1.352 % in the ipragliflozin and placebo groups, respectively.
About 80 % of patients in both groups used glimepiride; the remainder used either glibenclamide or gliclazide.
Of 166 patients randomized to ipragliflozin in treatment period I, 128 entered treatment period II (Fig. 1b). Of these, 57 received 50 mg ipragliflozin in both periods and 71 received 50 mg ipragliflozin in treatment period I and 100 mg ipragliflozin in treatment period II.
Efficacy
Glycemic control
The mean changes in HbA1c from baseline to week 24 were −0.83 and 0.32 % in the ipragliflozin and placebo groups, respectively (Table 2). The adjusted mean difference was −1.14 % [95 % confidence interval (CI) −1.348, −0.936; P < 0.001]. Similar results were obtained in sensitivity analyses in which the baseline characteristic variables were individually included as a fixed factor in each analysis (data not shown). Analysis of the per protocol set yielded similar results. The mean change in HbA1c was also significantly greater in the ipragliflozin group than in the placebo group in subgroup analyses of patients stratified by prior washout, age (<65, ≥65 years), sex, baseline HbA1c (<8.0, ≥8.0 %), baseline eGFR (<90, ≥90 ml/min/1.73 m2), BMI at screening (<25, ≥25 kg/m2), or sulfonylurea type (glimepiride, gliclazide, and glibenclamide) (all P < 0.001; data not shown) except in patients using gliclazide (P = 0.052; data not shown). Among patients with HbA1c ≥8.0 % at baseline, the mean changes in HbA1c from baseline to week 24 were −0.98 and 0.23 % in the ipragliflozin (baseline: 8.68 %) and placebo (baseline: 8.76 %) groups, respectively. The changes in HbA1c in patients with HbA1c <8.0 % at baseline were −0.48 and 0.49 % in the ipragliflozin (baseline: 7.63 %) and placebo (baseline: 7.59 %) groups, respectively. The adjusted mean difference in HbA1c change was larger in patients with HbA1c ≥8.0 % (−1.26 %) than in patients with HbA1c <8.0 % (−0.95 %). The change in HbA1c was also significantly greater in the ipragliflozin group than in the placebo group after dividing patients into those who used a low (Supplementary Table 1) or a high (Supplementary Table 2) sulfonylurea dose.
Overall, 19.4 % (32/165) and 5.3 % (4/75) of patients in the ipragliflozin and placebo groups, respectively, achieved HbA1c <7.0 % at week 24. As shown in Fig. 2a, decreases in mean HbA1c were apparent in the ipragliflozin group as early as week 4 and HbA1c continued to decrease up to week 24. By contrast, the mean HbA1c increased in the placebo group until week 8 and thereafter started decreasing until week 24.

a Time course of HbA1c over 24 weeks according to treatment group. b Time course of HbA1c in the ipragliflozin group over 52 weeks according to the treatment received in treatment period II (50 or 100 mg ipragliflozin). Values are mean ± SD. HbA1c hemoglobin A1c, NGSP National Glycohemoglobin Standardization Program
Over 52 weeks of treatment, HbA1c declined progressively over the first 24 weeks of treatment and was maintained thereafter in patients who received ipragliflozin in both treatment periods (Fig. 2b). In patients who continued 50 mg ipragliflozin (50/50 mg group), HbA1c decreased from 8.16 % at baseline to 6.96 % at week 24 (mean change from baseline: −1.21 %) and then increased slightly to 7.15 % at week 52 (mean change from baseline: −1.02 %). In patients who switched to 100 mg ipragliflozin (50/100 mg group), HbA1c decreased from 8.59 % at baseline to 7.75 % at week 24 (mean change from baseline: −0.84 %) and to 7.59 % at week 52 (mean change from baseline: −1.00 %). The proportion of patients who achieved HbA1c <7.0 % was 50.9 % (29/57) at week 24 and decreased to 29.8 % (17/57) at week 52 in the 50/50 mg group but increased slightly from 0 % (0/71) to 9.9 % (7/71) in the 50/100 mg group.
FPG decreased by 41.4 mg/dl in the ipragliflozin group and by 1.0 mg/dl in the placebo group between baseline and week 24, corresponding to a placebo-adjusted mean change of −38.0 mg/dl (95 % CI −45.27, −30.75) in the ipragliflozin group (Table 2). Among patients treated with ipragliflozin, FPG declined progressively during the first 2 weeks of treatment and continued to decrease until week 12, then remained stable at about 140 mg/dl until week 24 (Fig. 3a). The decrease in FPG was also significantly greater in the ipragliflozin group than in the placebo group after dividing patients into subgroups according to their sulfonylurea dose (Supplementary Tables 1 and 2).

a Time course of FPG over 24 weeks according to treatment group. b Time course of FPG in the ipragliflozin group over 52 weeks according to the treatment received in treatment period II (50 or 100 mg ipragliflozin). Values are mean ± SD. FPG fasting plasma glucose
The reduction in FPG was also maintained over 52 weeks (Fig. 3b). In the 50/50 mg group, FPG decreased from 173.7 mg/dl at baseline to 129.4 mg/dl at week 24 (mean change from baseline: −44.3 mg/dl) and increased slightly to 139.9 mg/dl at week 52 (mean change from baseline: −33.8 mg/dl). In the 50/100 mg group, FPG decreased from 185.0 mg/dl at baseline to 140.7 mg/dl at week 24 (mean change from baseline: −44.4 mg/dl) and was 140.6 mg/dl at week 52 (mean change from baseline: −44.4 mg/dl).
Other secondary outcomes
As shown in Fig. 4a, the decrease in body weight was greater in the ipragliflozin group than in the placebo group. The placebo-adjusted mean change at week 24 was −1.32 kg (95 % CI −1.884, −0.754) (Table 2). The decrease in body weight was also significantly greater in the ipragliflozin group than in the placebo group after dividing patients into subgroups according to their sulfonylurea dose (Supplementary Tables 1 and 2). Among patients who used ipragliflozin in both treatment periods, body weight decreased progressively until about week 24, after which it stabilized at about −2 to −2.5 kg until week 52 (Fig. 4b). The mean changes from baseline to week 24 and week 52 were −2.44 and −2.18 kg, respectively, in the 50/50 mg group (baseline: 69.92 kg) and −2.16 and −2.38 kg, respectively, in the 50/100 mg group (baseline: 69.37 kg).

a Changes in body weight over 24 weeks according to treatment group. b Changes in body weight in the ipragliflozin group over 52 weeks according to the treatment received in treatment period II (50 or 100 mg ipragliflozin). Values are mean ± SD
The changes in FSI, leptin, adiponectin, and waist circumference from baseline to week 24 are presented in Table 2. Of these, only the change in adiponectin was significantly different between the ipragliflozin and placebo groups, with a placebo-adjusted change of 0.44 μg/ml (95 % CI 0.107, 0.781). The increase in adiponectin was maintained over 52 weeks (data not shown).
As shown in Table 2, HOMA-β increased significantly in the ipragliflozin group relative to the placebo group.
Safety
The incidence of TEAEs was significantly higher in the ipragliflozin group than in the placebo group during the 24-week double-blind period (treatment period I), but there was no significant difference in the incidence of drug-related TEAEs between the two groups (Table 3). The most common events with a higher incidence in the ipragliflozin group than in the placebo group were pollakiuria (14/166 vs. 1/76 patients) and thirst (12/166 vs. 1/76 patients). All of these events were classified as mild in severity. The incidence of TEAEs was not obviously influenced by the sulfonylurea dose (Supplementary Table 3).
Serious TEAEs (other than death) occurred in five patients in the ipragliflozin group (spinal column stenosis, proliferative retinopathy, intervertebral disc protrusion, and Parkinson’s disease in one patient each; and osteomyelitis, subcutaneous abscess, and hyperkeratosis in one patient) and in three patients in the placebo group (gastric polyps, thalamic infarction, and worsening of diabetes in one patient each) in treatment period I.
Overall, 9 patients in the ipragliflozin group and 11 in the placebo group discontinued the study because of TEAEs in treatment period I. In the placebo group, the most common TEAE leading to discontinuation was worsening of diabetes, which occurred in eight patients. In the ipragliflozin group, there were no TEAEs leading to discontinuation in two or more patients.
In terms of prespecified TEAEs, the incidence of TEAEs related to hypoglycemia was similar between the ipragliflozin (1.2 %, 2/166) and placebo groups (1.3 %, 1/76). The incidence of TEAEs related to urinary tract infection and genital infection was lower in the ipragliflozin group (1.2 and 0.6 %, respectively) than in the placebo group (3.9 and 3.9 %, respectively). However, the incidence of polyuria/pollakiuria was higher in the ipragliflozin group (9.6 %) than in the placebo group (2.6 %), but all of the events were classified as mild in severity. Two patients in the ipragliflozin group discontinued treatment because of a TEAE related to hypoglycemia or polyuria/pollakiuria in treatment period I. The incidence of hypoglycemia was not obviously influenced by the sulfonylurea dose.
Over the 52-week study, TEAEs occurred in 86.7 % (144/166) of patients initially randomized to ipragliflozin, including those who received 100 mg ipragliflozin (Table 3). The incidence of serious TEAEs and TEAEs leading to permanent discontinuation was 7.2 and 9.6 %, respectively. Most TEAEs were classified as mild in severity. One TEAE was classified as severe (idiopathic thrombocytopenic purpura), but it was not considered to be related to the study drug.
TEAEs related to hypoglycemia occurred in 7 (4.2 %) patients, urinary tract infection occurred in 5 (3.0 %), genital infection occurred in 3 (1.8 %), and polyuria and/or pollakiuria occurred in 17 (10.2 %). In addition, for 1 patient in the 50/100 mg group, the dose was reduced to 50 mg because of a mild TEAE related to hypoglycemia. The incidence of polyuria/pollakiuria did not increase in the 28-week open-label extension period (treatment period II).
Systolic/diastolic blood pressure significantly decreased by 5.5/2.9 mmHg in the ipragliflozin group and by 1.3/0.5 mmHg in the placebo group from baseline to week 24. Among patients treated with ipragliflozin in treatment period I, the reduction in systolic/diastolic blood pressure from baseline to week 52 was 3.8/2.2 mmHg (Table 4). The changes in systolic blood pressure (Supplementary Fig. 1) or diastolic blood pressure (data not shown) were not correlated with the change in body weight from baseline to the end of treatment period I.
There were no clinically relevant changes in pulse rate, eGFR, or other laboratory test values, although hematocrit, blood urea nitrogen, magnesium (serum and urine), and phosphate (serum and urine) increased slightly in the ipragliflozin group compared with the placebo group (Table 4). Triglyceride levels decreased and high-density lipoprotein cholesterol levels increased in the ipragliflozin group compared with the placebo group. Changes in other total cholesterol, free fatty acids, and low-density lipoprotein were comparable in both groups. Electrocardiogram abnormalities were noted in three patients (electrocardiogram T wave biphasic, angina pectoris and tachycardia) in the ipragliflozin group. These events were classified as mild in severity except for moderate angina pectoris in one patient, which led to study discontinuation. These events were not considered related to the study drug.
Discussion
Sulfonylureas are relatively inexpensive drugs with well-established clinical efficacy and safety profiles. Therefore, sulfonylureas are widely used to treat type 2 diabetes either as monotherapy or in combination with other drugs. However, sulfonylureas often show limited long-term efficacy as monotherapy [19, 20], requiring the addition of a second drug.
Several classes of drugs can be used in combination with sulfonylureas, including glucagon-like peptide-1 analogs and dipeptidyl peptidase-4 inhibitors, which promote insulin secretion; thiazolidinediones and metformin, which enhance insulin action; and other drugs, such as insulin and α-glucosidase inhibitors, which directly reduce circulating glucose levels [21].
SGLT2 inhibitors represent a novel class of drugs that is proposed as an insulin-independent approach for treating type 2 diabetes and may avoid some of the limitations associated with other drugs, including weight gain. Their independence of insulin means they are well suited for use in combination with sulfonylureas and may reduce the overall demand on insulin for glucose disposal from the blood, possibly enabling a reduction in the sulfonylurea dose during treatment with SGLT2 inhibitors. Treatment with an SGLT2 inhibitor could help to prevent the progression of β cell dysfunction, weight gain, and hypoglycemia that are frequently associated with sulfonylurea. In addition, the increase in urinary glucose excretion during treatment with an SGLT2 inhibitor achieves caloric loss and mild osmotic diuresis, which are associated with clinically significant body weight reductions. Despite the changes in total cholesterol, free fatty acids, and low-density lipoprotein being comparable in both groups, the improvements in triglyceride and HDL-C suggest that ipragliflozin may improve the overall lipid profile during longer term treatment. Although we did not assess body composition in this study, an earlier study investigated the changes in body composition in patients treated with dapagliflozin [22], another SGLT2 inhibitor. The results of that study suggest that the reduction in body weight is largely attributable to a decrease in fat mass. The potential effects of ipragliflozin on lean mass should be examined in future studies.
From this context, the present study was designed to demonstrate the efficacy and safety of using ipragliflozin in combination with ongoing sulfonylurea. Briefly, we found that ipragliflozin significantly improved glycemic control in terms of HbA1c and FPG at 24 weeks, and these improvements were maintained for up to 52 weeks in the open-label extension. HbA1c levels were higher in patients who received 50/100 mg ipragliflozin than in those who received 50/50 mg ipragliflozin, even though the changes in FPG and body weight after week 28 were similar in both groups. These inconsistencies may be related to differences in post-prandial glucose levels between the two groups. However, unfortunately, we only measured fasting glucose levels in the present study. Therefore, we are unable to discuss the possible effects of post-prandial glucose on the changes in HbA1c in this study. In addition, ipragliflozin was associated with improvements in adiponectin levels and body weight compared with placebo, and these improvements were maintained through to 52 weeks. We also found an increase in HOMA-β in the present study. However, this result should be interpreted with great caution and re-evaluated using other methods because HOMA-β is a function of FPG and fasting insulin levels, and SGLT2 inhibitors might elicit apparently favorable improvements in HOMA-β by reducing FPG, without actually improving β cell function. Reductions in blood pressure were also observed in ipragliflozin-treated patients over 52 weeks.
The results of this study were similar to those of an earlier monotherapy study in which treatment with 50 mg ipragliflozin resulted in significant placebo-adjusted reduction in HbA1c of 1.29 % over 12 weeks of treatment in Japanese patients with type 2 diabetes [10]. In this study, ipragliflozin significantly reduced FPG and body weight, and it tended to reduce systolic blood pressures [10].
In terms of safety, ipragliflozin was generally well tolerated in the present study as an add-on to a sulfonylurea, similar to the study of ipragliflozin alone [10]. Although the incidence of TEAEs was significantly higher in the ipragliflozin group than in the placebo group, there was no significant difference in the incidence of drug-related TEAEs between the two groups. The incidence of hypoglycemia, urinary tract infections, and genital infections was low, and it was similar between the ipragliflozin and placebo groups. Pollakiuria and polyuria were more common in the ipragliflozin group than in the placebo group, and these events were probably caused by the mild osmotic diuresis effect of SGLT2 inhibitors.
Similarly, Strojek et al. [23] examined the efficacy and safety of another SGLT2 inhibitor, dapagliflozin (2.5, 5, or 10 mg/day), in combination with glimepiride in a 24-week study. In that study, dapagliflozin was associated with significant reductions in HbA1c (2.5–10 mg/day vs. placebo: −0.58 to −0.82 vs. −0.13 %), FPG (−0.93 to −1.58 vs. −0.11 mmol/l), and body weight (−1.18 to −2.26 vs. −0.72 kg) compared with placebo. They also noted a higher incidence of hypoglycemic events in the dapagliflozin groups than in the placebo group (6.9–7.9 vs. 4.8 %), but an increase in these events is commonly observed when antidiabetic drugs are added to a sulfonylurea.
The results of our study and the study by Strojek et al. [23] indicate that SGLT2 inhibitors are effective and well tolerated when used in combination with a sulfonylurea and that SGLT2 inhibitors are promising for use in combination with other antidiabetic drugs because of their insulin-independent mode of action and low risk of hypoglycemia. In fact, we found that improvements in glycemic control and body weight in the ipragliflozin group were generally similar between patients using a low or a high sulfonylurea dose. Moreover, the sulfonylurea dose did not markedly influence the incidence of TEAEs, including hypoglycemia.
Some limitations of our study need to be discussed. Because the sulfonylurea dose was fixed during the study, we could not confirm whether combination with ipragliflozin could allow reductions of the sulfonylurea dose, which might minimize the risk of sulfonylurea-associated adverse events, especially hypoglycemia and weight gain. In addition, SGLT2 inhibitors improved glucose intolerance and increased glucose-stimulated insulin secretion in vivo, so they could preserve pancreatic β cell function [24]. This effect might prevent the secondary failure of a sulfonylurea. However, this possibility could not be demonstrated because the double-blind treatment period was short and there was no control group in treatment period II.
In conclusion, the results of the present phase III study show that using ipragliflozin as an add-on to ongoing sulfonylurea can produce significant improvements in glycemic control in terms of HbA1c and FPG as well as marked reductions in body weight. This combination was also well tolerated in our cohort of Japanese patients.
References
Sabolić I, Vrhovac I, Eror DB, Gerasimova M, Rose M, Breljak D, Ljubojević M, Brzica H, Sebastiani A, Thal SC, Sauvant C, Kipp H, Vallon V, Koepsell H. Expression of Na+-d-glucose cotransporter SGLT2 in rodents is kidney-specific and exhibits sex and species differences. Am J Physiol Cell Physiol. 2012;302:C1174–88.
Bakris GL, Fonseca VA, Sharma K, Wright EM. Renal sodium-glucose transport: role in diabetes mellitus and potential clinical implications. Kidney Int. 2009;75:1272–7.
Wright EM. Renal Na+-glucose cotransporters. Am J Physiol Renal Physiol. 2001;280:F10–8.
Nair S, Wilding JP. Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus. J Clin Endocrinol Metab. 2010;95:34–42.
Neumiller JJ, White JR Jr, Campbell RK. Sodium-glucose co-transport inhibitors: progress and therapeutic potential in type 2 diabetes mellitus. Drugs. 2010;70:377–85.
Veltkamp SA, Kadokura T, Krauwinkel WJ, Smulders RA. Effect of ipragliflozin (ASP1941), a novel selective sodium-dependent glucose co-transporter 2 inhibitor, on urinary glucose excretion in healthy subjects. Clin Drug Investig. 2011;31:839–51.
Kadokura T, Saito M, Utsuno A, Kazuta K, Yoshida S, Kawasaki S, Nagase I, Kageyama S. Ipragliflozin (ASP1941), a selective sodium-dependent glucose cotransporter 2 inhibitor, safely stimulates urinary glucose excretion without inducing hypoglycemia in healthy Japanese subjects. Diabetol Int. 2011;2:172–82.
Schwartz SL, Akinlade B, Klasen S, Kowalski D, Zhang W, Wilpshaar W. Safety, pharmacokinetic, and pharmacodynamic profiles of ipragliflozin (ASP1941), a novel and selective inhibitor of sodium-dependent glucose co-transporter 2, in patients with type 2 diabetes mellitus. Diabetes Technol Ther. 2011;13:1219–27.
Fonseca VA, Ferrannini E, Wilding JP, Wilpshaar W, Dhanjal P, Ball G, Klasen S. Active- and placebo-controlled dose-finding study to assess the efficacy, safety, and tolerability of multiple doses of ipragliflozin in patients with type 2 diabetes mellitus. J Diabetes Complicat. 2013;27:268–73.
Kashiwagi A, Kazuta K, Yoshida S, Nagase I. Randomized, placebo-controlled, double-blind glycemic control trial of novel sodium-dependent glucose cotransporter 2 inhibitor ipragliflozin in Japanese patients with type 2 diabetes mellitus. J Diabetes Investig. 2013. doi:10.1111/jdi.12156.
Wilding JP, Ferrannini E, Fonseca VA, Wilpshaar W, Dhanjal P, Houzer A. Efficacy and safety of ipragliflozin in patients with type 2 diabetes inadequately controlled on metformin: a dose-finding study. Diabetes Obes Metab. 2013;15:403–9.
Arai K, Matoba K, Hirao K, Matsuba I, Takai M, Takeda H, Kanamori A, Yamauchi M, Mori H, Terauchi Y. Present status of sulfonylurea treatment for type 2 diabetes in Japan: second report of a cross-sectional survey of 15,652 patients. Endocr J. 2010;57:499–507.
Pan C, Yang W, Jia W, Weng J, Tian H. Management of Chinese patients with type 2 diabetes, 1998–2006: the Diabcare-China surveys. Curr Med Res Opin. 2009;25:39–45.
Smulders RA, Zhang W, Veltkamp SA, van Dijk J, Krauwinkel WJ, Keirns J, Kadokura T. No pharmacokinetic interaction between ipragliflozin and sitagliptin, pioglitazone, or glimepiride in healthy subjects. Diabetes Obes Metab. 2012;14:937–43.
Kashiwagi A, Kasuga M, Araki E, Oka Y, Hanafusa T, Ito H, Tominaga M, Oikawa S, Noda M, Kawamura T, Sanke T, Namba M, Hashiramoto M, Sasahara T, Nishio Y, Kuwa K, Ueki K, Takei I, Umemoto M, Murakami M, Yamakado M, Yatomi Y, Ohashi H. International clinical harmonization of glycated hemoglobin in Japan: from Japan Diabetes Society to National Glycohemoglobin Standardization Program values. Diabetol Int. 2012;3:8–10.
Matsuo S, Imai E, Horio M, Yasuda Y, Tomita K, Nitta K, Yamagata K, Tomino Y, Yokoyama H, Hishida A. Revised equations for estimated GFR from serum creatinine in Japan. Am J Kidney Dis. 2009;53:982–92.
Director, Evaluation and Licensing Division, Pharmaceutical and Food Safety Bureau, Ministry of Health, Labour and Welfare. Guideline for clinical evaluation of oral hypoglycemic agents. Tokyo: Ministry of Health, Labour and Welfare; 2010.
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–9.
Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, Kravitz BG, Lachin JM, O’Neill MC, Zinman B, Viberti G. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355:2427–43.
Tan MH, Baksi A, Krahulec B, Kubalski P, Stankiewicz A, Urquhart R, Edwards G, Johns D, GLAL Study Group. Comparison of pioglitazone and gliclazide in sustaining glycemic control over 2 years in patients with type 2 diabetes. Diabetes Care. 2005;28:544–50.
Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M, Peters AL, Tsapas A, Wender R, Matthews DR. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012;35:1364–79.
Bolinder J, Ljunggren Ö, Kullberg J, Johansson L, Wilding J, Langkilde AM, Sugg J, Parikh S. Effects of dapagliflozin on body weight, total fat mass, and regional adipose tissue distribution in patients with type 2 diabetes mellitus with inadequate glycemic control on metformin. J Clin Endocrinol Metab. 2012;97:1020–31.
Strojek K, Yoon KH, Hruba V, Elze M, Langkilde AM, Parikh S. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with glimepiride: a randomized, 24-week, double-blind, placebo-controlled trial. Diabetes Obes Metab. 2011;13:928–38.
Jurczak MJ, Lee HY, Birkenfeld AL, Jornayvaz FR, Frederick DW, Pongratz RL, Zhao X, Moeckel GW, Samuel VT, Whaley JM, Shulman GI, Kibbey RG. SGLT2 deletion improves glucose homeostasis and preserves pancreatic beta-cell function. Diabetes. 2011;60:890–8.
Acknowledgments
This study was sponsored by Astellas. Medical writing and editorial support were funded by Astellas and provided by Dr. Nicholas D. Smith and ELMCOM™.
Conflict of interest
NA, TS, KK, AU, HO, SY, and EU are employees of Astellas Pharma, Inc., Japan. AK has acted as a consultant for Astellas Pharma, Inc., and has received consulting fees/honoraria from Astellas Pharma, Inc.
Author information
Authors and Affiliations
Corresponding author
Additional information
Clinicaltrials.gov identifier: NCT01242215.
EMIT: Study to show the efficacy of ipragliflozin in add-on therapy with sulfonylurea in type 2 diabetes mellitus patients.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Primary Investigators
Primary Investigators
Kazuo Yamagata (Sakajiri Naika Iin), Daishiro Yamada (Jiyugaoka Yamada Clinic of Internal Medicine), Fuminobu Okuguchi (Okuguchi Clinic of Internal Medicine), Takashi Naitou (Saka General Clinic), Hiroaki Seino (Seino Internal Medicine Clinic), Hiroshi Kouno (Jusendo General Hospital), Hirokazu Shoda (Seiwakai Shoda Hospital), Shuichi Fukuda (Wakakusa Clinic), Yoshio Ohashi (Tokyo Ekimae-building Clinic), Madoka Taguchi (Toshiba General Hospital), Mizuki Kaneshiro (Kaneshiro Diabetes Clinic), Koutaro Fujimoto (Fujimoto Clinic), Ikuro Matsuba (Matsuba Clinic), Takaaki Iwai (Sagamino Central Hospital), Akira Yamauchi (Medical Corp. Rikeikai Suruga Clinic), Shizuo Kajiyama (Kajiyama Clinic), Takehiko Kobayashi (Kobayashi Shinryosho), Kiyomitsu Ikeoka (Medical Corp. Ikeoka Clinic), Emi Kose (Sato Hospital), Bunrei Goto (Medical Corp. Gotou Medical Clinic), Kunihiro Ekawa (Saiseikai Wakayama Hospital), Tetsuji Okuno (Nippon Kokan Fukuyama Hospital), Nobuyuki Abe (Abe Diabetes Clinic), Makoto Kunisaki (Kunisaki Makoto Clinic, Medical Corp. Shinaikai), Syuichi Otabe (Otabe Internal Medicine Clinic), Hideaki Jinnouchi (Jinnouchi Hospital), Masahiko Takai (Medical Corp. Takai Internal Medicine Clinic), Tsuguyoshi Asano (Asano Kanamachi Clinic), Kazuo Satake (Fukui General Clinic), Kazunori Yokoyama (Nikko Memorial Hospital), Munenori Okamoto (Sapporo Century Hospital), Tamayuki Koizumi (Medical Corp. Pieta Association Ishikari Hospital), Mitsutoshi Kato (Kato Clinic of Internal Medicine), Osamu Tomonaga (Tomonaga Clinic), and Yasuhiro Hashiguchi (Tenpozan Internal Clinic).
About this article

Cite this article
Kashiwagi, A., Akiyama, N., Shiga, T. et al. Efficacy and safety of ipragliflozin as an add-on to a sulfonylurea in Japanese patients with inadequately controlled type 2 diabetes: results of the randomized, placebo-controlled, double-blind, phase III EMIT study. Diabetol Int 6, 125–138 (2015). https://doi.org/10.1007/s13340-014-0184-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13340-014-0184-9