
Abstract
Background: Hyperglycaemia is associated with serious complications, significant morbidity and death. Despite the availability of a wide range of therapeutic options, many patients with diabetes mellitus fail to achieve or maintain recommended glycaemic goals. Ipragliflozin (ASP1941) is a novel, selective inhibitor of the sodium-dependent glucose co-transporter 2, which is highly expressed in the proximal tubules of the kidneys. It suppresses renal glucose reabsorption and increases urinary glucose excretion (UGE), potentially providing an insulin-independent treatment option for type 2 diabetes.
Methods: This multiple ascending-dose study assessed the safety, tolerability, pharmacokinetics and pharmacodynamics of ipragliflozin in healthy subjects after single doses and multiple once-daily doses for 10 days (dose levels: 5–600 mg).
Results: Ipragliflozin was well tolerated following single and multiple once-daily oral dosing. Ipragliflozin was rapidly absorbed with a median time to reach the maximum plasma concentration of 1.3 hours after the last dose. The area under the plasma concentration-time curve increased proportionally with increasing dose. The mean elimination half-life was 12 hours following the last dose. Ipragliflozin dose dependently increased UGE up to a maximum of approximately 59 g (327 mmol) of glucose excreted over 24 hours following multiple doses, without affecting plasma glucose levels in healthy subjects.
Conclusion: Administration of ipragliflozin was well tolerated and resulted in a rapid, dose-dependent increase in glucosuria. Pharmacodynamic and pharmacokinetic data suggest that ipragliflozin is suitable for prolonged once-daily oral treatment.
Trial Registration: ClinicalTrials.gov Identifier: NCT01288898.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Type 2 diabetes mellitus is a chronic disease characterized by hyperglycaemia due to insulin resistance and β-cell dysfunction.[1] Hyperglycaemia in patients with type 2 diabetes has been associated with microvascular (nephropathy, retinopathy) and macrovascular (cardiovascular) complications and mortality.[2,3] Important aspects in the treatment of type 2 diabetes are therefore optimizing glucose regulation and reducing the risk of cardiovascular complications.
Most patients with type 2 diabetes require a combination of antihyperglycaemic drugs over the course of their disease to maintain glycaemic control.[4] A wide range of therapies are currently available, such as biguanides (metformin), sulfonylurea derivatives (glimepiride), thiazolidinediones (pioglitazone), dipeptidylpeptidase-4 (DPP-4) inhibitors (sitagliptin) and glucagon-like peptide-1 receptor agonists (exenatide). All of these therapies act on an insulin-dependent mechanism. Despite these treatment options, many patients do not achieve or maintain glycaemic targets[4–6] and/or experience limiting side effects, including weight gain, hypoglycaemia, fluid retention and gastrointestinal adverse events (AEs).[6,7] New strategies to control hyperglycaemia are essential, and drugs that act independently of insulin may be beneficial in the treatment of type 2 diabetes.
The kidneys play a major role in the regulation of plasma glucose levels via gluconeogenesis and glucose reabsorption/excretion.[8] In a healthy individual, approximately 180 g (1000 mmol) of plasma glucose is filtered through the kidneys each day, and almost all (>99%) of this glucose is reabsorbed via sodium-dependent glucose co-transporters (SGLTs).[9,10] SGLT2 is a high-capacity, low-affinity glucose transporter,[9,11] and a recent study in wild-type and SGLT2 knock-out mice has shown that SGLT2 mediates glucose reabsorption in the early proximal tubules of the kidneys.[12] SGLT2 is responsible for about 90% of glucose reabsorption from the kidneys, with the remaining 10% of glucose renal reabsorption being mediated by SGLT1, a high-affinity, low-capacity glucose/galactose transporter located in the S3 segment of the proximal tubules.[9,11] Although present in the kidneys, SGLT1 is primarily expressed in the small intestine and has a greater affinity for galactose than for glucose.[9,11] While SGLT1 gene mutations cause glucose-galactose malabsorption,[13] individuals with SGLT2 gene mutations seem to live normal lives, apart from exhibiting persistent glucosuria.[14]
Studies have shown a positive correlation between the extent of SGLT2 expression and plasma glucose levels in diabetes.[15–17] Inhibition of SGLT2 increases the excretion of glucose into the urine and could therefore lower plasma glucose levels in patients with type 2 diabetes. Moreover, the loss of calories due to increased glucose excretion may lead to body weight reduction.[18] As SGLT2 inhibition works independently of insulin, the risk of hypoglycaemia is reduced.[6] Therefore, selective and potent inhibition of SGLT2 in the kidneys provides a novel and promising insulin-independent strategy for the treatment of type 2 diabetes. Several SGLT2 inhibitors are currently in development.[6,10,19]
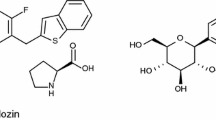
Ipragliflozin (ASP1941) is a novel, selective SGLT2 inhibitor (255-fold selectivity for human SGLT2 vs SGLT1)[20] being developed for the treatment of type 2 diabetes. The mechanism of action of ipragliflozin is presented in figure 1 and the structural formula is shown in figure 2. The efficacy of ipragliflozin, either as a monotherapy or in combination with metformin or pioglitazone, for controlling blood glucose levels by increasing urinary glucose excretion (UGE) has been demonstrated in several animal models of diabetes.[20,21] These preclinical studies have also indicated that ipragliflozin is associated with a low risk of hypoglycaemia,[20] and results in reduced urinary albumin excretion and increased pancreatic insulin content in diabetic mice.[21] Preclinical studies indicated that ipragliflozin is metabolized to multiple pharmacologically inactive metabolites[22,23] predominantly via glucuronidation by the uridine diphosphate-glucuronosyltransferase (UGT) enzyme UGT2B7.[24] In addition, preclinical studies showed that ipragliflozin does not significantly inhibit UGT enzymes[25] or cytochrome P450 (CYP) enzymes;[26] therefore, drug-drug interactions with concurrently administered drugs that are CYP substrates are not likely to occur.

Mechanism of action of ipragliflozin. SGLT = sodium-dependent glucose co-transporter.

Structural formula of ipragliflozin L-proline.
This multiple ascending-dose study was performed to evaluate the safety, tolerability, pharmacokinetics and pharmacodynamics of ipragliflozin in healthy subjects.
Methods
Subjects
Healthy male and female subjects aged 18–55 years with a body weight of 60–100 kg and a body mass index (BMI) of 20–30 kg/m2 were eligible for inclusion in this study (ClinicalTrials.gov Identifier: NCT01288898). Criteria for exclusion were a history of diabetes, fasting plasma glucose level >6.4 mmol/L (115 mg/dL), glycosylated haemoglobin A1c (HbA1c) level >6.2%, presence of renal glucosuria and/or proteinuria, history of any clinically significant disease or disorder, concurrent disease, and clinically significant deviations from the normal range in physical examination findings, vital signs, ECG parameters or clinical laboratory determinations. Women who were lactating, pregnant or of child-bearing potential not using a double barrier method of birth control were excluded from the study. The study was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice, and the International Conference on Harmonisation guidelines, and was approved by an institutional review board. All subjects provided written informed consent.
Design
This was a randomized, double-blind, placebo-controlled, sequential, multiple ascending-dose study. Ipragliflozin or matching placebo was administered orally as a single dose on day 1 and as multiple once-daily doses from day 5 to day 14 (10 days). Ipragliflozin or placebo was administered as a tablet formulation (1, 10 or 100 mg tablets). The investigated dose levels were 5, 30, 100, 300 and 600 mg. Only males were included for the 5, 30, 300 and 600 mg doses. At the 100 mg dose level, both males and females were included in order to explore sex effects. Each cohort consisted of eight subjects of whom six subjects received ipragliflozin and two subjects received placebo (n = 48). If a dose level (beginning with the lowest dose) was found to be safe and well tolerated, the dose was escalated to the next higher dose. On days 1 and 14, study medication was administered after a 9-hour fasting period. On all other days, study medication was administered approximately 30 minutes prior to the intake of a standardized breakfast.
Blood and urine samples for bioanalysis of ipragliflozin and glucose in plasma and urine were collected on days 1 and 14 at pre-dose, and during a time period of 96 hours after administration of the study medication.
Safety Assessments
Safety and tolerability assessments included physical examination (screening, day –1 and days 25–31), BP and pulse rate (screening, day –1, days 1–18, and days 25–31), 12–lead ECG (screening, day –1, days 1–5, days 8 and 11, days 14–18, and days 25–31), clinical laboratory tests consisting of blood samples for haematology and biochemistry and urine for urinalysis (screening, day –1, days 1–5, days 8 and 11, and days 14–17) and the nature, frequency and severity of AEs (assessed throughout the study as reported). As the target of SGLT2 inhibitors is the renal proximal tubule cell, changes in N-acetyl-β-D-glucosaminidase (NAG), β2-microglobulin and α1-microglobulin were assessed as potential markers of renal tubular damage.[27,28]
Bioanalytical Methods
Validated liquid chromatography-tandem mass spectrometry methods were used for measurement of ipragliflozin concentrations in plasma and urine.[29,30]
The overall-run variability and within-run variability for ipragliflozin were <5.8% and <4.6% for plasma and <15.0% and <10.8% for urine, respectively. Deviations of the nominal concentrations were less than −6.6% and +3.9% for plasma and urine, respectively. For ipragliflozin, the assay ranges representing the lower and upper limits of quantification in plasma and urine were 1–2000 and 2–5000 ng/mL, respectively. Concentrations of glucose in plasma and urine were routinely measured on a Synchron CX® Systems analyser (Beckman Coulter, Fullerton, CA, USA).
Pharmacokinetic and Pharmacodynamic Assessments
Pharmacokinetic assessments of ipragliflozin included the maximum plasma concentration (Cmax), the area under the plasma concentration-time curve (AUC), the time to reach Cmax (tmax), the elimination half-life (t1/2), the apparent total clearance from plasma after oral administration (CL/F), the cumulative amount of unchanged drug excreted into the urine (Ae), the renal clearance (CLR) and, after multiple administrations, the accumulation ratio (Rac). Pharmacodynamic measurements included plasma glucose levels, UGE and body weight. The pharmacodynamics of ipragliflozin were assessed by measurement of plasma glucose AUC from time zero to 24 hours (AUC24), the plasma concentration of glucose at the time of dosing (trough plasma concentration [Ctrough]), UGE and the CLR for glucose. In addition, total urine volume was recorded and the sodium concentrations in the urine were assessed.
Statistical Methods
Descriptive statistics included mean, standard deviation (SD), minimum and maximum (range), median and coefficient of variation for continuous variables; and frequencies and percentages for categorical data. Pharmacokinetic parameters were calculated by non-compartmental analysis using WinNonLin® version 5.0 (Pharsight Corp., Mountain View, CA, USA). Dose proportionality of Cmax and AUC for ipragliflozin in plasma, and a comparison between single- and multiple-dose pharmacokinetics of ipragliflozin in plasma and urine, were evaluated by ANOVA (power model) using SAS® version 9.1.3 (SAS Institute Inc., Cary, NC, USA). The relationship between the dose of ipragliflozin and the exposure to glucose in plasma and urine was analysed for both single and steady-state dosing using analysis of covariance (ANCOVA) of the log-transformed values. In addition, whether sex affected the pharmacokinetic and pharmacodynamic parameters of ipragliflozin was explored.
Results
Subject Demographics
A total of 48 subjects were enrolled and completed the study. The mean age ranged from 22.3 to 38.2 years among the subjects in the different dose cohorts of ipragliflozin, and was 38.1 years among the placebo subjects. In the ipragliflozin 100 mg cohort, the mean age of female subjects (24.8 years; range 22–35 years) was considerably lower than the mean age of male subjects (38.2 years; range 24–46 years). Mean BMI ranged from 21.9 to 26.8 kg/m2 for the subjects in the different ipragliflozin dose cohorts, and was 24.4 kg/m2 for the placebo group. Mean BMI was comparable between male and female subjects in the ipragliflozin 100 mg cohort. Of all subjects, 42 (87.5%) were Caucasian, three (6.3%) were Asian, two (4.2%) were Black, and ethnicity was not specified for one (2.1%).
Safety and Tolerability
Ipragliflozin was found to be safe and well tolerated in healthy subjects after single and multiple administrations up to the highest dose level (600 mg) evaluated in this study. An overview of treatment-emergent AEs (TEAEs) is shown in table I. TEAEs were reported in 23 out of 36 (63.9%) subjects who received ipragliflozin and 8 out of 12 (66.7%) subjects who received placebo. The incidence of these TEAEs did not appear to be dose-related, and sex did not appear to affect the incidence of TEAEs, based only on the small number of male and female subjects in the ipragliflozin 100 mg cohort. All TEAEs were mild in severity, except one moderate TEAE (vasovagal syncope) in one subject who received placebo. Treatment-related TEAEs occurred in 19 subjects (5/12 [41.7%] for placebo; 14/36 [38.9%] for ipragliflozin). The most frequently reported TEAEs included gastrointestinal disorders (5/12 [41.7%] for placebo; 7/36 [19.4%] for ipragliflozin), nervous system disorders (5/12 [41.7%] for placebo; 9/36 [25.0%] for ipragliflozin), and rash (1/12 [8.3%] for placebo; 5/36 [13.9%] for ipragliflozin). Headache was the most frequently reported nervous system disorder (3/12 [25.0%] for placebo; 7/36 [19.4%] for ipragliflozin). All rash events were considered not to be related to the study medication after evaluation by an independent dermatologist, except one possibly drug-related rash that occurred in a male subject treated with ipragliflozin 600 mg. One possibly drug-related renal TEAE (pollakiuria) was experienced by a subject who received ipragliflozin 300 mg and two renal TEAEs (pollakiuria and dysuria) were experienced by two subjects who received placebo. All three renal TEAEs were mild in severity. No hypoglycaemic events were reported, no serious AEs or deaths occurred, and none of the subjects discontinued due to AEs.

Summary of treatment-emergent adverse events (TEAEs) in healthy subjects treated with ipragliflozin or placeboa
With respect to kidney function markers, there were no clinically relevant changes from normal levels in blood urea, blood and urine creatinine levels, and in urinary NAG, β2-microglobulin or α1-microglobulin levels. Furthermore, no clinically relevant dose- or sex-related changes in mean systolic or diastolic BP, mean pulse or ECG parameters were observed.
Pharmacokinetics
The pharmacokinetic parameters of ipragliflozin in plasma and urine following single and multiple dosing are shown in table II. Mean ipragliflozin plasma concentration versus time curves following single (day 1) and 10-day multiple once-daily dosing (day 14) are shown in figure 3. Ipragliflozin was rapidly absorbed and Cmax was obtained within 2.3 hours after administration. Overall, the median tmax (range) among all ipragliflozin dose cohorts was 1.3 (1.0–1.5) hours after the first single dose, and 1.3 (1.0–2.3) hours after the last dose following multiple dosing. No statistically significant changes were found in mean Cmax and mean AUC for ipragliflozin after the first and last dose for each dose level (table II, figure 4). The increase in Cmax of ipragliflozin was slightly less than dose proportional after single (exponent of power model [β] = 0.91; 95% confidence interval [CI] 0.87, 0.95) and multiple dosing (β = 0.93; 95% CI 0.88, 0.97, p = 0.002) and the AUC increased dose proportionally following single (β = 1.03; 95% CI 0.98, 1.08) and multiple (β = 1.03; 95% CI 0.99, 1.07) dosing. Ipragliflozin did not significantly accumulate following 10-day multiple dosing compared with a single dose (Rac ∼1.16; equivalent to a 16% higher concentration after the last dose compared with the first dose, table II). The mean ± SD t1/2 of all ipragliflozin dose cohorts was 11.7 ± 1.3 hours after a single dose and 12.2 ± 1.3 hours after the last dose following multiple dosing. The mean ± SD CL/F was 14.1 ± 1.1 L/h following a single dose and 14.3 ± 1.0 L/h after the last dose of multiple dosing. Only minimal amounts of ipragliflozin were excreted into the urine as the parent compound (approximately 1% of the dose) at all dose levels. There appeared to be no major differences in the pharmacokinetics of ipragliflozin between male and female subjects, but it should be noted that sex effects were assessed only in the 100 mg cohort and group sizes were small (table II, figure 4).

Ipragliflozin plasma pharmacokinetic parameters in healthy subjects following single and multiple dosinga

Mean plasma concentration versus time curves for ipragliflozin after (a) single and (b) multiple dosing.

Mean ± SD ipragliflozin (a) maximum plasma concentration (Cmax) and (b) area under the plasma concentration-time curve (AUC) as a function of dose after single and multiple dosing.
Pharmacodynamics
Glucose pharmacodynamic parameters in plasma and urine after single and multiple dosing of ipragliflozin are presented in table III. No significant changes from baseline in plasma glucose AUC24 and plasma glucose levels at the time of ipragliflozin Ctrough were observed in any of the ipragliflozin treatment groups after single and multiple doses. Cumulative UGE increased significantly with increasing doses of ipragliflozin after both single and multiple dosing (figures 5a and b). A maximum level of UGE of approximately 70 g (390 mmol) over 24 hours was obtained with the 300 mg single dose (figures 5a and 6a). The UGE over 24 hours was slightly lower in female subjects than in male subjects (figure 5), but the glucose CLR over 24 hours was similar in females and males (table III).

Pharmacodynamic parameters of ipragliflozin as measured by changes in glucose parameters in plasma and urine following single and multiple dosinga

Mean ± SD cumulative urinary glucose excretion (UGE) as a function of ipragliflozin dose after (a) single and (b) multiple dosing.

(a) Mean ± SD urinary glucose excretion (UGE) over 24 hours vs the dose of ipragliflozin and (b) mean ± SD UGE over 24 hours vs the area under the plasma concentration-time curve (AUC) of ipragliflozin (semi-logarithmic scale) after both single and multiple dosing.
There was no indication of an effect of ipragliflozin, at any dose-level, on urine volume, both after single and multiple once-daily dosing (data not shown). These data need to be interpreted with caution, however, since the study was not controlled for fluid intake. Sodium concentrations in urine were assessed throughout the study and no significant differences were observed between the ipragliflozin and placebo groups.
Treatment with single and multiple doses of ipragliflozin in the dose range of 5–600 mg did not affect plasma glucose levels as compared with placebo in these healthy subjects. After the last dose, mean fasted plasma glucose levels were 5.00 mmol/L in the placebo group and 5.06, 4.97, 4.76, 4.96 and 4.89 mmol/L in the ipragliflozin 5, 30, 100, 300 and 600 mg cohorts, respectively.
Pharmacokinetic-Pharmacodynamic Relationships
The relationship between the dose of ipragliflozin and UGE after the first and last administration is shown in figure 6a. The relationship between the ipragliflozin AUC and UGE (semi-logarithmic scale) is presented in figure 6b. Ipragliflozin caused a clear dose-dependent increase in UGE (figure 6a). Similarly, UGE clearly increased with an increase in exposure (AUC) to ipragliflozin after both the first and the last administration (figure 6b).
Discussion
This multiple ascending-dose study demonstrated that ipragliflozin was safe and well tolerated in normoglycaemic healthy subjects following single and 10-day daily dosing up to a dose level of 600 mg. Administration of ipragliflozin resulted in a rapid, dose-dependent increase in glucosuria, inhibiting renal glucose resorption and resulting in UGE of up to 59 g/day (327 mmol) following multiple doses, without clinically relevant changes in plasma glucose levels.
Repeated administration of ipragliflozin did not result in an increase in the number of subjects with TEAEs or overall number of TEAEs, suggesting that ipragliflozin poses no serious safety concerns. No hypoglycaemic events occurred and, given that ipragliflozin acts independently of insulin, the risk of hypoglycaemia should be low. No clinically significant changes were detected in parameters of renal function and no subjects experienced urinary or genital tract infections. Urinary tract and genital infections, particularly vulvovaginal infections, secondary to increased glucosuria are considered to be a potential concern during treatment with SGLT2 inhibitors.[10,18,31] These infections are known to occur more frequently in women with type 2 diabetes than in the general population,[32,33] but evidence also suggests that glucosuria is not a risk factor for urinary tract infections.[33] Ongoing clinical trials in patients with type 2 diabetes will evaluate the incidence of these events during treatment with ipragliflozin.
Ipragliflozin has a C-glucoside linkage, which has been shown to confer resistance to ß-glucosidase-mediated degradation of dapagliflozin in the gastrointestinal tract.[34] The pharmacokinetic profile of ipragliflozin, i.e. rapid absorption, with a mean t1/2 of approximately 12 hours, and the favourable pharmacokinetic-pharmacodynamic relationship suggest that ipragliflozin is suitable for prolonged once-daily oral treatment. There appeared to be no major differences in pharmacokinetic parameters of ipragliflozin between male and female subjects. However, it should be noted that sex effects were evaluated only at the 100 mg dose and the number of subjects included in this analysis was small (six males and six females).
Administration of ipragliflozin resulted in a dose-dependent increase in UGE in healthy subjects following single and multiple dosing, which is consistent with data from preclinical studies in normal and diabetic mice.[20] Ipragliflozin doses of 300 mg and higher resulted in a UGE of approximately 70 g/day (390 mmol/day) after a single dose and 59 g/day (327 mmol/day) after multiple dosing, suggesting that glucose reabsorption by SGLT2 was inhibited by up to 40%, assuming that approximately 180 g of glucose is filtered by the kidneys per day. It is not known why the increase in UGE was somewhat higher after the first dose than after multiple doses of ipragliflozin. It is possible that after multiple doses a steady state is reached where both ipragliflozin and glucose bind to SGLT2, and the increase in UGE will be somewhat lower compared with the first dose. A similar study of dapagliflozin in healthy subjects reported a maximum UGE of 62 g/day after a single dose of 50 mg, and 55 g/day after multiple dosing (100 mg/day for 14 days).[35]
In patients with type 2 diabetes, the effects of SGLT2 inhibition on UGE appear to be more pronounced. Once-daily treatment with ipragliflozin 300 mg for 28 days resulted in a UGE mean change from baseline of approximately 90 g of glucose over 24 hours,[36] and once-daily treatment with dapagliflozin 100 mg for 14 days resulted in a UGE of approximately 70 g over 24 hours.[37]
In our view, the absence of complete inhibition of glucose resorption by SGLT2 inhibitors may be explained by assuming that not all SGLT2 receptors in the proximal tubules of the kidneys are blocked by ipragliflozin. It has also been hypothesized that this may be due to the action of additional mechanisms or transport molecules, such as SGLT1, or potential competition for binding to SGLT2 by increased tubular concentrations of glucose.[37]
Increases in UGE induced by ipragliflozin did not result in altered plasma glucose levels in this study, which is consistent with data from other studies of SGLT2 inhibitors in healthy subjects.[35,38] Healthy individuals maintain glucose homeostasis via a series of feedback loops and regulatory pathways involving a wide variety of factors including insulin, glucagon and sympathoadrenal mediators, many of which are impaired in patients with type 2 diabetes.[39] Gluconeogenesis accounts for the majority of glucose production in normal individuals with hypoglycaemia due to fasting.[40] Gluconeogenesis may also be responsible for maintaining normal plasma glucose levels in healthy subjects in response to a subtle reduction in plasma glucose due to increased UGE induced by ipragliflozin.
Conclusion
Ipragliflozin is a novel, selective SGLT2 inhibitor that is well tolerated and safe following a single dose and 10 days of multiple dosing in healthy subjects. Ipragliflozin is rapidly absorbed after oral administration and the AUC increases proportionally with increasing dose. Ipragliflozin increases UGE in a dose-dependent manner, without affecting plasma glucose levels. These characteristics make ipragliflozin a potentially suitable insulin-independent drug for the treatment of hyperglycaemia in type 2 diabetes patients.
References
Leahy JL. Pathogenesis of type 2 diabetes mellitus. Arch Med Res 2005; 36: 197–209
Moss SE, Klein R, Klein BE, et al. The association of glycemia and cause-specific mortality in a diabetic population. Arch Intern Med 1994; 154: 2473–9
Klein R. Hyperglycemia and microvascular and macro-vascular disease in diabetes. Diabetes Care 1995; 18: 258–68
Nathan DM, Buse JB, Davidson MB, et al., American Diabetes Association; European Association for Study of Diabetes. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy. A consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009; 32: 193–203
Saydah SH, Fradkin J, Cowie CC. Poor control of risk factors for vascular disease among adults with previously diagnosed diabetes. JAMA 2004; 291: 335–42
Nair S, Wilding JP. Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus. J Clin Endocrinol Metab 2010; 95: 34–42
Ismail-Beigi F, Craven T, Banerji MA, et al. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: an analysis of the ACCORD randomised trial. Lancet 2010; 376: 419–30
Marsenic O. Glucose control by the kidney: an emerging target in diabetes. Am J Kidney Dis 2009; 53: 875–83
Wright EM. Renal Na(+)-glucose cotransporters. Am J Physiol Renal Physiol 2001; 280: F10–8
Neumiller JJ, White Jr JR, Campbell RK. Sodium-glucose co-transport inhibitors: progress and therapeutic potential in type 2 diabetes mellitus. Drugs 2010; 70: 377–85
Jabbour SA, Goldstein BJ. Sodium glucose co-transporter 2 inhibitors: blocking renal tubular reabsorption of glucose to improve glycaemic control in patients with diabetes. Int J Clin Pract 2008; 62: 1279–84
Vallon V, Platt KA, Cunard R, et al. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol 2011; 22: 104–12
Turk E, Zabel B, Mundlos S, et al. Glucose/galactose malabsorption caused by a defect in the Na+/glucose cotransporter. Nature 1991; 350: 354–6
Santer R, Kinner M, Lassen CL, et al. Molecular analysis of the SGLT2 gene in patients with renal glucosuria. J Am Soc Nephrol 2003; 14: 2873–82
Vestri S, Okamoto MM, de Freitas HS, et al. Changes in sodium or glucose filtration rate modulate expression of glucose transporters in renal proximal tubular cells of rat. J Membr Biol 2001; 182: 105–12
Rahmoune H, Thompson PW, Ward JM, et al. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 2005; 54: 3427–34
Freitas HS, Anh e GF, Melo KF, et al. Na(+)-glucose transporter-2 messenger ribonucleic acid expression in kidney of diabetic rats correlates with glycemic levels: involvement of hepatocyte nuclear factor-1alpha expression and activity. Endocrinology 2008; 149: 717–4
List JF, Woo V, Morales E, et al. Sodium-glucose cotransport inhibition with dapagliflozin in type 2 diabetes. Diabetes Care 2009; 32: 650–7
Patel AK, Fonseca V. Turning glucosuria into a therapy: efficacy and safety with SGLT2 inhibitors. Curr Diab Rep 2010; 10: 101–7
Kurosaki E, Tahara A, Yokono M, et al. In vitro and in vivo pharmacological properties of ASP1941, a novel, potent and selective SGLT2 inhibitor [abstract no. 0570-P]. American Diabetes Association, 70th Scientific Sessions; 2010 June 25–29; Orlando (FL)
Takasu T, Tahara A, Yokono M, et al. ASP1941, a novel, potent and selective SGLT2 inhibitor, improves hemoglobin A1c and symptoms of diabetes in animal models [abstract no. 0562-P]. American Diabetes Association, 70th Scientific Sessions; 2010 June 25–29; Orlando (FL)
Data on file. Internal Report 1941-ME-0040. Identification of ASP1941 metabolites in humans, 12 March 2010. Astellas Pharma Inc., Kashima, Osaka
Data on file. Internal Report 1941-PH-0029. Inhibitory activities of human metabolites of ASP1941 on cellular uptake of methyl-α-D-glucopyranoside (AMG) in human Na+-glucose cotransporter (SGLT)-expressing cells, 12 April 2010. Astellas Pharma Inc., Tsukuba, Ibaraki
Data on file. Internal report 1941-ME-0045. Correlation of ASP1941 metabolic activity with UGT enzyme activities in human liver microsomes, 29 March 2010. Astellas Pharma Inc., Kashima, Osaka
Data on file. Internal Report 1941-ME-0055. Inhibitory effects of ASP1941 on the activity of UDP-glucuronosyltransferases using human liver microsomes, 09 December 2010. XenoTech, LLC, Lenexa (KS)
Data on file. Internal Report 1941-ME-0039. Time-dependent inhibitory effects of ASP1941 on the activity of CYP isozymes using human liver microsomes, 16 October 2009. Sekisui Medical Co., Ltd, Muramatsu, Ibaraki
Tassi C, Mancuso F, Feligioni L, et al. Expression modes of urinary N-acetyl-beta-D-glucosaminidase in patients with chronic renal insufficiency. Clin Chim Acta 2004; 346: 129–33
Lisowska-Myjak B. Serum and urinary biomarkers of acute kidney injury. Blood Purif 2010; 29: 357–65
Data on file. Internal Report 1941-ME-0009. Validation of a LC-MS/MS method for the determination of ASP1941 in human plasma, 04 December 2007. Astellas Pharma Europe B.V., Leiderdorp
Data on file. Internal Report 1941-ME-0010. Validation of a LC-MS/MS method for the determination of ASP1941 in human urine, 04 December 2007. Astellas Pharma Europe B.V., Leiderdorp
Bailey CJ, Gross JL, Pieters A, et al. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with metformin: a randomised, double-blind, placebo-controlled trial. Lancet 2010; 375: 2223–33
Sobel JD. Candidal vulvovaginitis. Clin Obstet Gynecol 1993; 36: 153–65
Geerlings SE, Stolk RP, Camps MJ, et al., Diabetes Women Asymptomatic Bacteriuria Utrecht Study Group. Risk factors for symptomatic urinary tract infection in women with diabetes. Diabetes Care 2000; 23: 1737–41
Meng W, Ellsworth BA, Nirschl AA, et al. Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. J Med Chem 2008; 51(5): 1145–9
Komoroski B, Vachharajani N, Boulton D, et al. Dapagliflozin, a novel SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharmacol Ther 2009; 85: 520–6
Schwartz S, Klasen S, Kowalski D, et al. ASP1941, a novel and selective inhibitor of sodium-glucose co-transporter 2 (SGLT2), reduces fasting plasma glucose in patients with type 2 diabetes mellitus [abstract no. 0566-P]. American Diabetes Association, 70th Scientific Sessions; 2010 June 25–29; Orlando (FL)
Komoroski B, Vachharajani N, Feng Y, et al. Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus. Clin Pharmacol Ther 2009; 85: 513–9
Kapur AR, O’Connor-Semmes RL, Hussey EK, et al. First human dose escalation study with remogliflozin etabonate (RE) in healthy subjects and in subjects with type 2 diabetes mellitus [abstract no. 509-P]. American Diabetes Association, 69th Scientific Sessions; 2009 June 5–9; New Orleans (LA)
Boyle PJ, Zrebiec J. Physiological and behavioral aspects of glycemic control and hypoglycemia in diabetes. South Med J 2007; 100: 175–82
Landau BR, Wahren J, Chandramouli V, et al. Contributions of gluconeogenesis to glucose production in the fasted state. J Clin Invest 1996; 98: 378–85
Acknowledgements
This study was funded by Astellas Pharma, Europe BV, Leiderdorp, the Netherlands. All authors are employees of Astellas Pharma. Ipragliflozin is under development by Astellas Pharma Inc. and Kotobuki Pharmaceutical Co., Ltd. The authors thank Sandra Mendes from Excerpta Medica, who provided writing/editorial assistance for this work, which was supported by Astellas.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Veltkamp, S.A., Kadokura, T., Krauwinkel, W.J.J. et al. Effect of Ipragliflozin (ASP1941), a Novel Selective Sodium-Dependent Glucose Co-Transporter 2 Inhibitor, on Urinary Glucose Excretion in Healthy Subjects. Clin. Drug Investig. 31, 839–851 (2011). https://doi.org/10.1007/BF03256922
Published:
Issue Date:
DOI: https://doi.org/10.1007/BF03256922