
Abstract
Ipragliflozin is a novel oral sodium–glucose cotransporter 2 (SGLT2) inhibitor under development for the treatment of type 2 diabetes. We examined its efficacy and safety as an add-on to pioglitazone in Japanese patients with inadequately controlled diabetes. Japanese type 2 diabetes patients were randomized to 24 weeks of treatment with 50 mg ipragliflozin or placebo in a double-blind manner. At week 24, patients with hemoglobin (Hb)A1c <8.4 % were permitted to continue open-label ipragliflozin in a 28-week extension period. The primary endpoint was the change in HbA1c from baseline to week 24 (with last observation carried forward). Ninety-eight and 54 patients were randomized to ipragliflozin or placebo, respectively, and were prescribed the study drug. The mean HbA1c change from baseline to week 24 was −0.64 % and 0.22 % in the ipragliflozin and placebo groups, respectively, and the adjusted mean difference between the two groups was −0.88 % (P < 0.001). Changes in fasting plasma glucose (FPG) and body weight were significantly greater in the ipragliflozin group (both P < 0.001). Among patients who continued ipragliflozin in the extension period, reductions in HbA1c, FPG, and body weight were maintained until week 52. The incidence of treatment-emergent adverse events was not significantly different between the two groups. The most common event with a higher incidence in the ipragliflozin group than in the placebo group was pollakiuria (12/97 vs. 0/54 patients). Ipragliflozin improved glycemic control, promoted weight reduction, and had a good safety profile as an add-on to pioglitazone in Japanese type 2 diabetes patients.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Sodium–glucose cotransporters (SGLTs) are a family of transport proteins that cotransport sodium and glucose in the same directions. One of the main members of this family is SGLT2, which is specifically expressed in the kidney, predominantly in the S1 and S2 segments of the proximal tubule on the brush border membrane [1]. SGLT2 is responsible for approximately 90 % of renal glucose reabsorption [2]. Based on these functional properties of SGLT2, it was considered that SGLT2 inhibitors could be used to treat type 2 diabetes [3]. In addition, SGLT2 inhibition is insulin-independent, which may contribute to a low risk of hypoglycemic events.
Ipragliflozin is a highly selective SGLT2 inhibitor, and pharmacokinetic/pharmacodynamic studies have demonstrated that it enhances urinary glucose excretion without affecting plasma glucose levels in healthy subjects [4, 5]. Meanwhile, in subjects with type 2 diabetes, ipragliflozin in a dose dependent manner increased urinary glucose excretion and, consequently, decreased plasma glucose levels [6].
There have been reports of several clinical studies of ipragliflozin in which it was used as monotherapy [7, 8] or as an add-on to metformin [9]. These clinical trials confirmed that inhibition of SGLT2 led to significant reductions in hemoglobin (Hb)A1c and fasting plasma glucose (FPG) in patients with type 2 diabetes. These studies also indicated that ipragliflozin was well tolerated.
In clinical practice, metformin is widely used as the first-line treatment for type 2 diabetes based on the recommendations of the American Diabetes Association and European Association for the Study of Diabetes [10]. However, other classes of drugs may be used alone or in combination with other types of antidiabetic drugs. For example, pioglitazone is used because it improves insulin resistance and shows good tolerability during long-term use [11]. Nevertheless, some patients have shown inadequate responses to pioglitazone, necessitating the next step in treatment options.
A combination of ipragliflozin and pioglitazone might be useful because their actions are complementary. In particular, ipragliflozin enhances glucose excretion and pioglitazone enhances insulin sensitivity, and the former reduces body weight and induces osmotic diuresis whereas the latter is associated with weight gain and fluid retention. Combination therapy with ipragliflozin and pioglitazone appears to be a viable option because they do not exhibit pharmacokinetic interactions [12].
From this context, the objective of the present study was to determine the efficacy and safety of ipragliflozin when used as an add-on to pioglitazone for 24 weeks in a double-blind, placebo-controlled manner. We also included a 28-week open-label extension period, allowing for 52 weeks of treatment, to examine the longer-term efficacy and safety of this combination in patients with type 2 diabetes. This article focuses on the placebo-controlled part of the study because of its scientific importance, but also briefly describes the results of the open-label extension period.
Methods
Patients
The main inclusion criteria for this study were: age ≥20 years, diagnosis of type 2 diabetes ≥12 weeks before entering the screening period, treatment with pioglitazone monotherapy at an approved dose (15, 30, or 45 mg) for ≥4 weeks, HbA1c (National Glycohemoglobin Standardization Program value) of 7.4–9.9 % with a change of ≤1 % during the 4-week screening period, and body mass index of 20.0–45.0 kg/m2.
Patients with any of the following conditions were excluded: proliferative diabetic retinopathy; dysuria; symptomatic urinary tract/genital infection; a serious cardiovascular event within 12 weeks; history of heart failure; unstable psychiatric disorders; history of malignant tumors (unless the patient did not require treatment for ≥5 years before enrollment); severe gastrointestinal disease; serum creatinine (Cr) exceeding the upper limit of normal; urinary albumin/Cr ratio >300 mg/g Cr; aspartate aminotransferase or alanine aminotransferase more than 2 times the upper limit of normal; systolic blood pressure ≥170 mmHg or diastolic blood pressure ≥95 mmHg; treatment with insulin ≤12 weeks before entering the screening period; or chronic use of corticosteroids, immunosuppressants, or loop diuretics (short-term or temporary use of these drugs was allowed), among other criteria.
All of the patients provided written informed consent before enrollment.
Study design and treatments
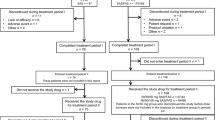
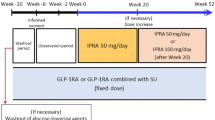
This study consisted of a 4-week screening period, a 2-week single-blind placebo run-in period, a 24-week double-blind treatment period (treatment period I), a 28-week open-label extension period (treatment period II), and a 4-week follow-up period (Fig. 1a). All patients had been on pioglitazone monotherapy at a stable dose for ≥4 weeks before screening. Its dose could not be changed until the end of treatment (i.e., week 52 or discontinuation). Patients who were using another hypoglycemic agent together with pioglitazone underwent an additional 4-week washout period before entering the screening period.

a Study design. b Patient disposition. *Includes one patient whose use of study drug was unknown. †Includes one patient whose ipragliflozin dose was reduced to 50 mg after being increased to 100 mg in treatment period II
At the end of the run-in period, patients were randomized in a double-blind manner to either 50 mg ipragliflozin or placebo (2:1 ratio), which was taken once daily before breakfast. Randomization was performed by a central registration center. The patients and clinicians were kept blind to the treatment received in treatment period I until the data for treatment period I had been entered into the study database and locked. The study drugs and their packaging were identical in appearance.
Patients could enter treatment period II if their HbA1c at week 20 was <8.4 % and was lower than the baseline value. Patients who entered treatment period II provided additional informed consent and received ipragliflozin in an open-label manner.
At week 24, the ipragliflozin dose could be increased to 100 mg if the patient’s HbA1c at week 20 was ≥7.4 %. The ipragliflozin dose could be reduced to 50 mg if there were safety concerns, such as hypoglycemia, but no further dose change was permitted.
Other antidiabetic drugs and continuous administration of drugs capable of affecting glucose metabolism were prohibited until the end of treatment (i.e., week 52 or discontinuation). However, corticosteroids, immunosuppressants, or loop diuretics could be used topically or temporarily.
Treatment compliance was assessed in terms of the numbers of study drugs prescribed, returned, or lost between each visit.
The protocol, case report forms, and patient consent forms were approved by institutional review boards at each participating site. The study was conducted in accordance with Good Clinical Practice, International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, as well as local laws and regulations.
This study was registered in the ClinicalTrials.gov registry (NCT01225081).
Efficacy and safety outcomes
The primary efficacy endpoint was change in HbA1c from baseline to week 24 with the last observation carried forward (LOCF). Secondary efficacy endpoints included the changes in FPG, fasting serum insulin (FSI), leptin, adiponectin, body weight, and waist circumference from baseline to week 24 (LOCF). Efficacy endpoints were also assessed in terms of the changes from baseline to week 52 (LOCF).
Major efficacy endpoints (HbA1c, FPG, and body weight) were measured at each visit, at weeks 2 (except HbA1c) and 4, and then every 4 weeks thereafter (weeks 4–52).
Safety variables included treatment-emergent adverse events (TEAEs), laboratory tests, vital signs, 12-lead electrocardiography, and estimated glomerular filtration rate (eGFR) [13]. Safety variables were generally assessed at each visit. Electrocardiography was performed at 4–12-week intervals during the study. TEAEs were classified according to system organ class and preferred term using MedDRA version 12.1.
HbA1c (enzymatic assay), insulin (microparticle enzyme immunoassay), leptin (double-antibody radioimmunoassay), and adiponectin (latex agglutination assay) assays as well as laboratory tests were performed by Mitsubishi Chemical Medience Corporation. HbA1c values were initially reported in Japan Diabetes Society (JDS) units, which were then converted into NGSP units using the following equation [14]: HbA1c (NGSP) (%) = 1.02 × HbA1c (JDS) (%) + 0.25 %.
Statistical analysis
We planned to enroll 150 patients and randomize 100 to ipragliflozin and 50 to placebo so that we could evaluate safety and comply with Japanese Guidelines for Clinical Evaluation of Oral Hypoglycemic Agents [15]. The sample size was deemed large enough to show statistical superiority of ipragliflozin over placebo according to the results of a Phase II study [8].
For this study, all efficacy analyses were conducted using the full analysis set (FAS), which consisted of all patients who received at least one dose of the study drug and with at least one efficacy variable measured in treatment period I after starting administration of the study drug. The safety analysis set (SAF) consisted of all patients who received at least one dose of the study drug.
Baseline characteristics were compared between the two treatment groups using the two-sample t test or Fisher’s exact test, as appropriate.
Changes in the primary/secondary efficacy endpoints from baseline (i.e., start of treatment period I) to week 24 were assessed using analysis of covariance (ANCOVA) with the baseline value as a covariate and treatment group as a fixed effect. For efficacy analyses, the LOCF method was used to impute missing data at week 24/52. Changes in efficacy endpoints from baseline to week 52 (LOCF) were analyzed descriptively.
Safety variables were analyzed descriptively and are presented as the number of patients (%) within each group. The eGFR was calculated from serum creatinine, age, and sex, using the Modification of Diet in Renal Disease equation modified for Japanese subjects. Post hoc analyses were conducted using the two-sample t test to compare laboratory parameters between the two groups.
For all analyses, P < 0.05 (two-tailed) was considered statistically significant.
Results
Patients
The first patient entered the study in September 2010 and the final evaluation was in April 2012. The disposition of patients is summarized in Fig. 1b. Of 152 patients randomized (ipragliflozin, n = 98; placebo, n = 54), 133 completed treatment period I. Eighty-four patients entered treatment period II, of which 69 were treated with ipragliflozin and 15 were treated with placebo in treatment period I.
The baseline characteristics of patients in the ipragliflozin and placebo groups were similar, with no significant imbalances between the two groups (Table 1), except for the duration of diabetes, which was numerically greater in the placebo group. Almost all of the patients were using either 15 or 30 mg pioglitazone at baseline (Table 1).
During treatment period I, the mean ± SD duration of exposure was 161.3 ± 28.68 and 151.6 ± 38.96 days in the ipragliflozin and placebo groups, respectively. Treatment compliance was good, with over 98 % of patients complying with study drug administration.
Efficacy in the 24-week double-blind period (treatment period I)
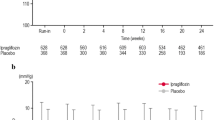
The mean HbA1c change from baseline to week 24 was −0.64 and 0.22 % in the ipragliflozin and placebo groups, respectively (Table 2). The adjusted mean difference was −0.88 % (95 % confidence interval [CI]: −1.108 to −0.648; P < 0.001), indicating an obvious effect of ipragliflozin on lowering HbA1c. As shown in Fig. 2a, decreases in HbA1c were apparent in the ipragliflozin group as early as week 4 and continued until week 24. By contrast, HbA1c increased progressively in the placebo group until week 16, and thereafter decreased until week 24.

a Time-course of hemoglobin A1c (HbA1c) over 24 weeks according to treatment group (mean ± SD). b Time-course of HbA1c in the ipragliflozin group over 52 weeks according to the treatment received in treatment period II (50 or 100 mg ipragliflozin). Values are mean ± SD. NGSP national glycohemoglobin standardization program
In the sensitivity analyses, we performed ANCOVA in which sex, age, washout before screening, baseline eGFR, BMI, and daily pioglitazone dose were added separately as covariates or fixed effects to the model used in the primary analysis. The placebo-adjusted mean change in HbA1c in the ipragliflozin group was statistically significant in all of these models (P < 0.001).
We also conducted analyses in subgroups of patients divided by baseline characteristics, sex, age (<65 vs. ≥65 years), HbA1c (<8.0 % vs. ≥8.0 %), washout before screening, baseline eGFR (<90 vs. ≥90 mL/min/1.73 m2), BMI (<25 vs. ≥25 kg/m2), and the daily pioglitazone dose (<30 vs. ≥30 mg). In these subgroup analyses, the placebo-adjusted changes in HbA1c from baseline to week 24 were statistically significant in all subgroups (P < 0.001). Of note, the placebo-adjusted mean difference in HbA1c was larger in patients with HbA1c ≥8.0 % than in patients with HbA1c <8.0 % (−0.95 % vs. −0.69 %). The mean changes in HbA1c from baseline to week 24 in the ipragliflozin and placebo groups were –0.73 % and 0.22 %, respectively, in patients with HbA1c ≥8.0 % (baseline: 8.61% and 8.66 %), and were –0.48 % and 0.23 %, respectively, in patients with HbA1c <8.0 % (baseline: 7.61 % and 7.74 %).
None of the patients in the ipragliflozin and placebo groups had HbA1c <7.0 % at baseline. At week 24, the proportions of patients with HbA1c <7.0 % were 12.4 % (12/97) in the ipragliflozin group and none in the placebo groups.
As shown in Table 2, the decreases in FPG (adjusted mean difference: −41.0 mg/dL; P < 0.001), body weight (−2.79 kg; P < 0.001), and waist circumference (−2.00 cm; P = 0.004) were significantly greater in the ipragliflozin group than in the placebo group. The mean change in FPG from baseline to week 24 was −36.4 mg/dL in the ipragliflozin group compared with 6.1 mg/dL in the placebo group. In the ipragliflozin group, the decrease in FPG was particularly evident at week 2 of treatment, and the reduction observed at this time was maintained until week 24 (Fig. 3a). The mean change in body weight from baseline was −2.29 kg in the ipragliflozin group versus 0.51 kg in the placebo group. Decreases in body weight were apparent at 2 weeks after treatment and continued until week 24 (Fig. 4a). The change in leptin from baseline to week 24 was significantly greater in the ipragliflozin group than in the placebo group (P < 0.001). However, the adjusted mean change in adiponectin was not significantly different between the two groups (P = 0.280). Post hoc analyses were performed to examine the change in homeostatic model assessment of β cell function (HOMA-β) [16], which increased in the ipragliflozin group compared with the placebo group.

a Time-course of fasting plasma glucose (FPG) over 24 weeks according to treatment group (mean ± SD). b Time-course of FPG in the ipragliflozin group over 52 weeks according to the treatment received in treatment period II (50 or 100 mg ipragliflozin). Values are mean ± SD

a Changes in body weight over 24 weeks according to treatment group (mean ± SD). b Changes in body weight in the ipragliflozin group over 52 weeks according to the treatment received in treatment period II (50 or 100 mg ipragliflozin). Values are mean ± SD
Efficacy over 52 weeks of treatment (treatment periods I and II)
The time-courses of HbA1c and FPG in patients who entered treatment period II are shown in Figs. 2b and 3b, respectively, while the changes in body weight from baseline through to week 52 are shown in Fig. 4b for patients treated with ipragliflozin in both treatment periods. The reduction in HbA1c from baseline to week 24 (−0.87 ± 0.494 %) was maintained at week 52 (−0.93 ± 0.436 %) in those who continued 50 mg ipragliflozin in treatment period II (50/50 mg group). HbA1c decreased further, albeit only slightly, in those who switched to 100 mg ipragliflozin (50/100 mg group) (−0.75 ± 0.526 % at week 24; −0.98 ± 0.743 % at week 52). Overall, 44.4 % (16/36) and 15.2 % (5/33) of patients in the 50/50 mg group and the 50/100 mg group achieved HbA1c < 7.0 % at week 52, having increased from 33.3 % (12/36) and 0.0 % (0/33), respectively, at week 24 (none had HbA1c < 7.0 % at baseline). The reductions in FPG and body weight observed at week 24 were maintained until week 52 (Figs. 3b, 4b). Tendencies for HbA1c, FPG and body weight to reduce in treatment period II were also observed in patients randomized to placebo in treatment period I and who started ipragliflozin (50 mg, n = 5; 100 mg, n = 10) in treatment period II (data not shown).
Safety
In the 24-week double-blind period (treatment period I), TEAEs occurred in 72.2 % and 68.5 % of patients in the ipragliflozin and placebo groups, respectively, which was not statistically significant (P = 0.710, Fisher’s exact test) (Table 3). The incidence of drug-related TEAEs was significantly greater in the ipragliflozin group (25.8 %) than in the placebo group (9.3 %) (P = 0.018, Fisher’s exact test). Serious TEAEs were reported in one patient in the ipragliflozin group (clavicle fracture) and in two patients in the placebo group (actinic keratosis and Bowen’s disease in one patient and cataract operation in one patient). TEAEs resulted in study discontinuation in more patients in the placebo group (11.1 %, 6/54) than in the ipragliflozin group (2.1 %, 2/97). Most (5/6) of the discontinuations in the placebo group and one discontinuation in the ipragliflozin group were due to worsening of diabetes. The other patient that discontinued in the ipragliflozin group had generalized pruritus and urinary tract infection. The most common TEAEs were nasopharyngitis, pollakiuria, and thirst. Most TEAEs were classified as mild in severity.
In terms of specific TEAEs, TEAEs related to hypoglycemia, urinary tract infection, genital infection, and polyuria/pollakiuria occurred in 1 (1.0 %), 3 (3.1 %), 2 (2.1 %), and 13 (13.4 %) patients, respectively, in the ipragliflozin group, and in 0, 1 (1.9 %), 0, and 0 patients, respectively, in the placebo group. The incidence of polyuria/pollakiuria was higher in the ipragliflozin group than in the placebo group but all of the events were mild in severity.
In the entire 52-week study, TEAEs occurred in 81.4 % (79/97) of patients initially randomized to ipragliflozin (Table 3). Serious TEAEs and TEAEs leading to permanent discontinuation both occurred in 3.1 % of patients. Most TEAEs were mild in severity. TEAEs related to hypoglycemia, urinary tract infection, genital infection, and polyuria/pollakiuria occurred in 1 (1.0 %), 4 (4.1 %), 4 (4.1 %), and 13 (13.4 %) patients, respectively.
After entering treatment period II (28-week open-label extension period), one patient experienced a serious urinary tract infection (pyelonephritis) that led to discontinuation. This event resolved after treatment with antibiotics. All of the urinary tract and genital infections occurred in females. The incidence of polyuria/pollakiuria did not increase in treatment period II.
In addition, the safety profile during ipragliflozin treatment in patients who were treated with placebo in treatment period I and with ipragliflozin in treatment period II was similar to that in patients who were treated with ipragliflozin in both treatment periods I & II (data not shown).
The changes in systolic and diastolic blood pressure from baseline to week 24 were −5.9 mmHg and −4.1 mmHg, respectively, in the ipragliflozin group and −2.5 mmHg and +1.7 mmHg in the placebo group. The changes in the ipragliflozin group were maintained until week 52 (Table 4).
The changes in several laboratory parameters are also presented in Table 4. As in an earlier study [8], there were changes in some laboratory parameters. In particular, hematocrit, blood urea nitrogen, magnesium (serum and urine), and phosphate (urine) were significantly greater in the ipragliflozin group than in the placebo group. However, ipragliflozin did not affect serum total cholesterol, free fatty acid, or creatinine levels, nor did it affect eGFR. Triglyceride, aspartate aminotransferase, and alanine aminotransferase significantly decreased in the ipragliflozin group compared with the placebo group. High-density lipoprotein–cholesterol increased in the ipragliflozin group. There were no clinically relevant changes in laboratory parameters at week 52.
Discussion
This study revealed that 24 weeks of treatment with 50 mg ipragliflozin significantly improved glycemic control (HbA1c and FPG), and reduced FSI, leptin, body weight, and waist circumference in Japanese patients who continued treatment with pioglitazone at the dose used before the study. These improvements were maintained for up to 52 weeks in patients randomized to ipragliflozin in treatment period I. We also observed an increase in HOMA-β in the present study. However, this result should be interpreted with caution and verified using other methods because HOMA-β is a function of FPG and fasting insulin levels. SGLT2 inhibitors might elicit apparent improvements in HOMA-β by reducing FPG without actually improving β cell function. Blood pressure also decreased slightly in the ipragliflozin group in treatment period I and was maintained over 52 weeks.
Dapagliflozin, another SGLT2 inhibitor, reduced HbA1c and FPG, and prevented weight gain when administered in combination with pioglitazone [17]. After 24 weeks of treatment, the mean decrease in HbA1c was –0.42 % in patients treated with placebo compared with −0.82 % and −0.97 % in patients treated with 5 mg or 10 mg dapagliflozin, corresponding to placebo-subtracted reductions of −0.40 % and −0.55 %, respectively. The mean decrease in FPG was −5.5 mg/dL in the placebo group compared with −24.9 and −29.6 mg/dL in the 5 mg and 10 mg dapagliflozin groups, respectively. Body weight increased by 1.64 kg in the placebo group, increased by 0.09 kg in the 5 mg dapagliflozin group, and decreased by 0.14 kg in the 10 mg dapagliflozin group. The differences in changes in efficacy parameters between placebo and ipragliflozin in our study were greater than the differences between placebo and dapagliflozin in that study [17]. However, differences in study design/patient populations possibly contributed to the differences in the results of these two studies.
Ipragliflozin and dapagliflozin were associated with decreases in body weight in other studies (e.g., in combination with metformin [9, 18] or sulfonylurea [19]). It is thought that SGLT2 inhibitors have mild diuretic effects and promote fat loss, which contributes to the reduction of body weight [20]. By contrast, pioglitazone is often associated with weight gain in clinical trials, making it less favorable than metformin and incretin secretagogues [21]. Fluid retention contributes to the increase in body weight associated with pioglitazone, and is of particular concern in older or female patients. Thus, SGLT2 inhibitors and pioglitazone have antagonistic effects on fluid balance. Accordingly, when used in combinations, edema as a side effect of pioglitazone might be reduced by treatment with SGLT2 inhibitors.
In the present study, only one patient in the ipragliflozin group experienced a hypoglycemia-related TEAE. Similarly, hypoglycemia occurred in only 3/141 patients treated with 5 mg dapagliflozin plus pioglitazone [17]. The low risk of hypoglycemia during treatment with SGLT2 inhibitors is probably due to their insulin-independent mechanism of action. Genital/urinary tract infections are a concern in patients treated with a SGLT2 inhibitor because SGLT2 inhibitors increase urinary glucose, which may facilitate infection. According to a 12-week study of dapagliflozin monotherapy in Japanese patients with type 2 diabetes, genital infections and urinary tract infections were reported in 0–1.8 % and 0–3.8 % of patients, respectively, in the dapagliflozin group versus 0 % and 1.9 %, respectively, of patients in the placebo group [22]. Genital and urinary tract infections occurred in 2.1 % and 3.1 % of patients, respectively, in the ipragliflozin group, and in 0 % and 1.9 %, of patients, respectively, in the placebo group in the 24-week randomized treatment phase in this study; thus, the incidence rates of these events were comparable to those in the dapagliflozin study. Furthermore, all of the TEAEs related to these infections in the ipragliflozin group were mild except for one moderate event (pyelonephritis) that recovered after discontinuation of the study drug and antibiotics. Polyuria and pollakiuria, which are also relatively common in trials of SGLT2 inhibitors, are probably related to drug-induced osmotic diuresis. All of the TEAEs related to polyuria/pollakiuria were mild and none led to discontinuation of the study drug.
In conclusion, the present study showed that adding ipragliflozin to ongoing pioglitazone was associated with significant improvements in glycemic control, reductions in body weight, and had a tolerable safety profile. Our results provide support for its use as an add-on to pioglitazone in Japanese patients with inadequate glycemic control, extending the results of the prior study in Japan showing its efficacy as monotherapy [8].
References
Sabolić I, Vrhovac I, Eror DB, Gerasimova M, Rose M, Breljak D, Ljubojević M, Brzica H, Sebastiani A, Thal SC, Sauvant C, Kipp H, Vallon V, Koepsell H. Expression of Na+-d-glucose cotransporter SGLT2 in rodents is kidney-specific and exhibits sex and species differences. Am J Physiol Cell Physiol. 2012;302:C1174–88.
Bakris GL, Fonseca VA, Sharma K, Wright EM. Renal sodium-glucose transport: role in diabetes mellitus and potential clinical implications. Kidney Int. 2009;75:1272–7.
Nair S, Wilding JP. Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus. J Clin Endocrinol Metab. 2010;95:34–42.
Kadokura T, Saito M, Utsuno A, Kazuta K, Yoshida S, Kawasaki S, Nagase I, Kageyama S. Ipragliflozin (ASP1941), a selective sodium-dependent glucose cotransporter 2 inhibitor, safely stimulates urinary glucose excretion without inducing hypoglycemia in healthy Japanese subjects. Diabetol Int. 2011;2:172–82.
Veltkamp SA, Kadokura T, Krauwinkel WJ, Smulders RA. Effect of ipragliflozin (ASP1941), a novel selective sodium-dependent glucose co-transporter 2 inhibitor, on urinary glucose excretion in healthy subjects. Clin Drug Investig. 2011;31:839–51.
Schwartz SL, Akinlade B, Klasen S, Kowalski D, Zhang W, Wilpshaar W. Safety, pharmacokinetic, and pharmacodynamic profiles of ipragliflozin (ASP1941), a novel and selective inhibitor of sodium-dependent glucose co-transporter 2, in patients with type 2 diabetes mellitus. Diabetes Technol Ther. 2011;13:1219–27.
Fonseca VA, Ferrannini E, Wilding JP, Wilpshaar W, Dhanjal P, Ball G, Klasen S. Active- and placebo-controlled dose-finding study to assess the efficacy, safety, and tolerability of multiple doses of ipragliflozin in patients with type 2 diabetes mellitus. J Diabetes Complicat. 2013;27:268–73.
Kashiwagi A, Kazuta K, Yoshida S, Nagase I. Randomized, placebo-controlled, double-blind glycemic control trial of novel sodium-dependent glucose cotransporter 2 inhibitor ipragliflozin in Japanese patients with type 2 diabetes mellitus. J Diabetes Investig. 2013;. doi:10.1111/jdi.12156.
Wilding JP, Ferrannini E, Fonseca VA, Wilpshaar W, Dhanjal P, Houzer A. Efficacy and safety of ipragliflozin in patients with type 2 diabetes inadequately controlled on metformin: a dose-finding study. Diabetes Obes Metab. 2013;15:403–9.
Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M, Peters AL, Tsapas A, Wender R, Matthews DR. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012;35:1364–79.
Dormandy J, Bhattacharya M, van Troostenburg de Bruyn AR. Safety and tolerability of pioglitazone in high-risk patients with type 2 diabetes: an overview of data from PROactive. Drug Saf. 2009;32:187–202.
Smulders RA, Zhang W, Veltkamp SA, van Dijk J, Krauwinkel WJ, Keirns J, Kadokura T. No pharmacokinetic interaction between ipragliflozin and sitagliptin, pioglitazone, or glimepiride in healthy subjects. Diabetes Obes Metab. 2012;14:937–43.
Matsuo S, Imai E, Horio M, Yasuda Y, Tomita K, Nitta K, Yamagata K, Tomino Y, Yokoyama H, Hishida A. Revised equations for estimated GFR from serum creatinine in Japan. Am J Kidney Dis. 2009;53:982–92.
Kashiwagi A, Kasuga M, Araki E, Oka Y, Hanafusa T, Ito H, Tominaga M, Oikawa S, Noda M, Kawamura T, Sanke T, Namba M, Hashiramoto M, Sasahara T, Nishio Y, Kuwa K, Ueki K, Takei I, Umemoto M, Murakami M, Yamakado M, Yatomi Y, Ohashi H. International clinical harmonization of glycated hemoglobin in Japan: from Japan Diabetes Society to National Glycohemoglobin Standardization Program values. Diabetol Int. 2012;3:8–10.
Director, Evaluation and Licensing Division, Pharmaceutical and Food Safety Bureau, Ministry of Health, Labour and Welfare. Guideline for clinical evaluation of oral hypoglycemic agents. Tokyo: Ministry of Health, Labour and Welfare; 2010.
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–9.
Rosenstock J, Vico M, Wei L, Salsali A, List JF. Effects of dapagliflozin, an SGLT2 inhibitor, on HbA(1c), body weight, and hypoglycemia risk in patients with type 2 diabetes inadequately controlled on pioglitazone monotherapy. Diabetes Care. 2012;35:1473–8.
Bailey CJ, Gross JL, Hennicken D, Iqbal N, Mansfield TA, List JF. Dapagliflozin add-on to metformin in type 2 diabetes inadequately controlled with metformin: a randomized, double-blind, placebo-controlled 102-week trial. BMC Med. 2013;11:43.
Strojek K, Yoon KH, Hruba V, Elze M, Langkilde AM, Parikh S. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with glimepiride: a randomized, 24-week, double-blind, placebo-controlled trial. Diabetes Obes Metab. 2011;13:928–38.
Bolinder J, Ljunggren Ö, Kullberg J, Johansson L, Wilding J, Langkilde AM, Sugg J, Parikh S. Effects of dapagliflozin on body weight, total fat mass, and regional adipose tissue distribution in patients with type 2 diabetes mellitus with inadequate glycemic control on metformin. J Clin Endocrinol Metab. 2012;97:1020–31.
Campbell IW. Comparing the actions of older and newer therapies on body weight: to what extent should these effects guide the selection of antidiabetic therapy? Int J Clin Pract. 2010;64:791–801.
Kaku K, Inoue S, Matsuoka O, Kiyosue A, Azuma H, Hayashi N, Tokudome T, Langkilde AM, Parikh S. Efficacy and safety of dapagliflozin as a monotherapy for type 2 diabetes mellitus in Japanese patients with inadequate glycaemic control: a phase II multicentre, randomized, double-blind, placebo-controlled trial. Diabetes Obes Metab. 2013;15:432–40.
Acknowledgments
Ipragliflozin is under development by Astellas Pharma Inc. and Kotobuki Pharmaceutical Co., Ltd. This study was sponsored by Astellas Pharma Inc. Medical writing and editorial support was funded by Astellas Pharma Inc., and was provided by Dr. Nicholas D. Smith and ELMCOM™.
Conflict of interest
TS, NA, KK, AU, SY, and EU are employees of Astellas Pharma Inc. AK has acted as a consultant for Astellas Pharma Inc. and has received consulting fees/honoraria from Astellas Pharma Inc.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
Primary investigators: Kazuo Yamagata (Sakajiri Naika Iin), Hideki Kuribayashi (Aohitokusa Kuribayashi Clinic), Daishiro Yamada (Jiyugaoka Yamada Clinic of Internal Medicine), Fuminobu Okuguchi (Okuguchi Clinic of Internal Medicine), Hiroshi Kouno (Jusendo General Hospital), Shuichi Fukuda (Wakakusa Clinic), Hirokazu Shoda (Seiwakai Shoda Hospital), Hideto Ishii (Asano Internal Medicine Clinic, Medical Corporation Yukeikai), Tomio Tsukazaki (Aozora Total Clinic), Shinya Minagawa (Minagawa Clinic), Madoka Taguchi (Toshiba General Hospital), Katsuhiko Yamada (Kousei Medical Clinic), Masahiro Sugawara (Sugawara Clinic), Koki Shin (Shin Clinic), Yoshio Ohashi (Tokyo Ekimae-building Clinic), Takaaki Iwai (Sagamino Central Hospital), Ikuro Matsuba (Matsuba Clinic), Ichitaro Takada (Takada Internal Medicine Clinic), Mizuki Kaneshiro (Kaneshiro Diabetes Clinic), Yoichi Koizumi (Komoro Kosei General Hospital), Takuro Ichikawa (Ichikawa Clinic), Kotaro Kawai (Shimada Municipal Hospital), Naoko Hirahara (Shimada Municipal Hospital), Kiyomitsu Ikeoka (Medical Corporation Ikeoka Clinic), Sadahiro Sempuku (Senpuku Clinic), Tetsuji Okuno (Nippon Kokan Fukuyama Hospital), Yasuo Toh (Healthcare Corporations Association Kunwa-Kai Aiwa Clinic), Daigaku Uchida (Medical Corporation Hotaruno Hakuyukai), Tsuguyoshi Asano (Asano Kanamachi Clinic), Makoto Sugiura (Specified Medical Corporation Tokoharu and Touei Hospital), Tamayuki Koizumi (Medical Corporation Pieta Association Ishikari Hospital), Munenori Okamoto (Sapporo Century Hospital), Masahiko Takai (Medical Corporation Takai Internal Medicine Clinic), Yoshiyuki Arai (Arai Clinic), Masakazu Kato (Kato Internal Medicine Clinic), Kazunori Yokoyama (Nikko Memorial Hospital), Kazuo Satake (Fukui General Clinic), Yasuhiro Hashiguchi (Tenpozan Internal Clinic).
About this article

Cite this article
Kashiwagi, A., Shiga, T., Akiyama, N. et al. Efficacy and safety of ipragliflozin as an add-on to pioglitazone in Japanese patients with inadequately controlled type 2 diabetes: a randomized, double-blind, placebo-controlled study (the SPOTLIGHT study). Diabetol Int 6, 104–116 (2015). https://doi.org/10.1007/s13340-014-0182-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13340-014-0182-y