
Abstract
Fisetin and hesperetin, naturally occurring flavonoids, have been reported as novel antioxidants with chemopreventive/chemotherapeutic potential against various types of cancer. However, their mechanism of action in CML is still unknown. This particular study aims to evaluate the therapeutic potentials of fisetin and hesperetin and their effects on cell proliferation, apoptosis, and cell cycle progression in human K562 CML cells. The results indicated that fisetin and hesperetin inhibited cell proliferation and triggered programmed cell death in these cells. The latter was confırmed by mitochondrial membrane depolarization and an increase in caspase-3 activation. In addition to that, we have detected S and G2/M cell cycle arrests and G0/G1 arrest upon fisetin and hesperetin treatment, respectively. To identify the altered genes and genetic networks in response to fisetin and hesperetin, whole-genome microarray analysis was performed. The microarray gene profiling analysis revealed some important signaling pathways including JAK/STAT pathway, KIT receptor signaling, and growth hormone receptor signaling that were altered upon fisetin and hesperetin treatment. Moreover, microarray data suggested potential candidate genes for targeted CML therapy. Fisetin and hesperetin significantly modulated the expression of genes involved in cell proliferation and division, apoptosis, cell cycle regulation, and other significant cellular processes such as replication, transcription, and translation. In conclusion, our results suggest that fisetin and hesperetin as potential natural agents for CML therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic myeloid leukemia (CML) is a hematological malignancy derived from a balanced reciprocal translocation including the genes, Abelson murine leukemia virus (ABL) and breakpoint cluster region (BCR), which are located on the long arms of chromosome 9 and chromosome 22, t(9;22)(q34;q11), respectively [1]. This translocation creates the Philadelphia chromosome (Ph), which encodes the chimeric constitutively active BCR-ABL1 kinase protein [2]. BCR-ABL1 triggers the development of CML phenotype by activating various downstream signaling pathways responsible for uncontrolled cell proliferation and reduced apoptosis [3]. Despite imatinib and other tyrosine kinase inhibitors targeting BCR-ABL1 provide great success in CML treatment, resistance to these inhibitors, observed intolerance, and detrimental side effects still form therapeutic difficulties [4]. Therefore, alternative novel strategies are needed, and plant-derived natural substances are being investigated for their potentials in cancer prevention and treatment.
Recently, natural compounds obtained from fruits, vegetables, and herbs have been considered as potential chemopreventive and chemotherapeutic agents due to their non-toxic nature and usage for a long time without any adverse effects [5]. One of the compounds found in plants is flavonoids, naturally occurring polyphenolic compounds that possess various pharmacological features including antioxidative, anticarcinogenic, anti-inflammatory, antibacterial, immune-stimulating, and antiviral effects [6]. They have been reported to prevent the initiation, promotion, and progression of cancer by modulating various signaling pathways resulted in the control of cell growth and differentiation, apoptosis, angiogenesis, and metastasis [7].
Fisetin (3,3′,4′,7-tetrahydroxyflavone), a common flavonol molecule, found in various plants including strawberry, apple, persimmon, grape, onion, and cucumber has been reported to possess broad biological activities involving anticancer, antiangiogenic, anti-inflammatory, and anti-metastatic effects [8]. In the literature, there is a wide range of studies evaluating both in vitro and in vivo effects of fisetin on various cancer types including colon cancer [9], prostate cancer [10], pancreatic cancer [11], lung cancer [12], and multiple myeloma [13], but there is a gap related to investigation of the therapeutic potential of fisetin and its mode of actions in CML. Despite the underlying mechanisms of fisetin display variations in different cancer types, fisetin triggers its growth inhibitory effects by activating both the death receptor mediated and the intrinsic pathways of apoptosis [9, 10] in addition to alterations in cell cycle regulators [5] and inhibition of signaling pathways including PI3K/Akt and NF-κB [14]. In a recent study, fisetin has been shown to trigger apoptosis in human acute monocytic leukemia cells through induction of nitric oxide production, which results in double strand breaks [15]. Fisetin potentiated sorafenib-induced apoptosis of melanoma cells by inhibiting PI3K signaling pathway, which is characterized by decreases in PI3K expression and phosphorylation of MEK1/2, ERK1/2, AKT, and mTOR [16].
Hesperetin (3′,5,7-trihydroxy-4-methoxyflavonone) is aglycone form of hesperidin, which is commonly found in citrus species such as orange and lemon [17]. Hesperidin derived from diet is converted to hesperetin via deglycosylation by intestinal bacteria before absorption. Therefore, hesperidin could be a pro-drug, which is metabolized to hesperetin [18]. These citrus flavonones have been characterized by their antioxidant, anti-inflammatory, and anticancer actions [17]. Its anticancer activities have been shown to be mediated by inhibiting proliferation/inducing apoptosis and inhibiting angiogenesis in various cancers such as breast cancer, cervical cancer, and colon cancer [19–21]. Molecular mechanisms responsible for hesperetin-induced apoptosis include cell cycle arrest by regulating cell cycle regulators [22], increases in several caspases like caspase-3 and caspase-9 [21] and downregulation of antiapoptotic proteins [23]. Hesperetin triggered apoptosis in MCF-7 breast cancer cells through activation of ASK1/JNK pathway and accumulation of ROS [19]. Hesperetin was shown to inhibit the growth of gastric cancer cells both in vitro and in vivo [24]. In this study, hesperetin treatment resulted in an increase in the levels of AIF, Apaf-1, Cyt C, caspase-3, caspase-9, and Bax and a decrease in Bcl-2 levels. However, the underlying mechanisms of hesperetin in CML cells remain unidentified yet.
The purpose of this study is to investigate the modes of action of fisetin- and hesperetin-induced antileukemic effects on CML cells by evaluating their roles in cell growth, induction of apoptosis, changes in caspase-3 enzyme activity, mitochondrial membrane disruption, and cell cycle distribution. We also identified for the first time the genes and genetic networks which are altered in CML upon fisetin and hesperetin treatment.
Materials and methods
Chemicals
Fisetin (purity ≥98 %) and hesperetin (purity ≥95 %) were purchased from Santa Cruz Biotechnology, Inc (Heidelberg, Germany). Stock solutions (10 mM) of these agents dissolved in DMSO were prepared and kept at −20 °C. The final DMSO concentration does not exceed 0.1 % in culture which is not cytotoxic. Penicillin-streptomycin, RPMI 1640, and fetal bovine serum were purchased from Invitrogen (Paisley, UK). MTT reagent was obtained from Sigma (St. Louis, MO).
Cell lines and culture conditions
Human K562 CML cells purchased from German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany) were grown in RPMI 1640 growth medium supplemented with 10 % fetal bovine serum and 1 % penicillin-streptomycin at 37 °C in 5 % CO2.
Evaluation of antiproliferative effects by MTT assay
Growth inhibitory effects of fisetin and hesperetin on K562 cells were evaluated by the MTT cell proliferation assay. Briefly, 1 × 104 cells were treated with step-wise increasing concentrations of fisetin and hesperetin for 48 and 72 h. Then, the cells were incubated with 20 μl MTT for 3 h. The resulting formazan crystals were homogenized by adding 100 μl DMSO. The absorbance values were measured at 570 nm wavelength using an ELISA reader (Thermo Electron Corporation Multiskan Spectrum, Finland). Finally, the obtained cell proliferation plots were used to calculate the IC50 value (drug concentration inhibits cell growth by 50 %) of these agents.
AnnexinV-FITC/PI double staining to evaluate apoptosis
The movement of phosphatidylserine (PS) from the inner leaflet to the outer leaflet was measured (Annexin V-FITC Apoptosis Detection Kit, BioVision Research Products, USA) to determine apoptotic effects of fisetin and hesperetin on CML cells. Initially, increasing concentrations of fisetin and hesperetin were introduced into 5 × 105 cells for 72 h. After the washing of the cells twice with cold PBS and homogenization with 200 μl of 1× binding buffer, they were incubated with 2 μl of FITC-Annexin V and 2 μl of propidium iodide (PI) for 15 min at room temperature in the dark. Afterwards, samples were analyzed by flow cytometry (BD Facscanto Flowcytometry, Belgium).
Detection of changes in mitochondrial membrane potential
Alterations in mitochondrial membrane potential (MMP) were detected by JC-1 Mitochondrial Membrane Potential Detection Kit (Cayman Chemicals, USA) containing a unique cationic dye, JC-1. Briefly, 5 × 105 cells were collected by centrifugation at 180 g for 10 min after treatment for 72 h. Then, 30 μl of JC-1 dye was added to the cells and incubated for 30 min. Then, absorbance values were measured by fluorescence ELISA reader (Thermo Varioskan Spectrum, Finland). Healthy cells produce red color with an absorption/emission maxima of 560/595 nm, whereas apoptotic cells form green color with an absorption/emission maxima of 485/535 nm. The ratio of green to red intensities was calculated for all samples.
Detection of changes in caspase-3 activity
Caspase-3 activity is detected using a caspase-3 colorimetric assay kit (BioVision Research Products, USA). Briefly, the cells (5 × 105 cells/2 mL) were centrifugated at 180×g for 10 min after inducing apoptosis by fisetin and hesperetin. Then, the cells were lysed by adding 50 μl of chilled cell lysis buffer on ice for 10 min. Fifty microliters of supernatants, collected by centrifugation at 10,000×g for 1 min, was mixed with the reaction mixture containing 50 μl of 2× reaction buffer with 10 mM DTT and 5 μl of DEVD-pNA substrate in 96-well plates. Following incubation for 2 h at 37 °C, the absorbance values were detected at 405 nm wavelengths by ELISA reader. Total protein concentrations determined by Bradford method were used to normalize the absorbance values.
Determination of cell cycle distribution by flow cytometry
In short, 5 × 105 cells/2 mL treated with increasing concentrations of fisetin and hesperetin for 72 h were centrifugated at 260×g for 10 min, and the pellets were suspended in 1 mL cold PBS. Afterwards, the cells were fixed with cold ethanol and incubated overnight at −20 °C Then, the cells were collected at 260×g for 10 min and resuspended with 1 mL PBS containing 0.1 % triton X-100. One hundred microliters of RNase A (200 μg/mL) was added to these cells, and incubation was carried out at 37 °C for 30 min. Afterwards, the samples were treated with 100 μl PI (1 mg/mL) and incubated at room temperature for 15 min, and then analyzed by flow cytometry.
Analysis of changes in genome-wide gene expression profiles
The quality of total RNA (Nucleospin Total RNA isolation kit (Machery-Nagel, USA) isolated from K562 cells upon treatment with fisetin (50 and 100 μM) and hesperetin (100 and 150 μM) was assessed in terms of A260/A280 ratio, which should be between 1.9 and 2.1 for further analysis. Expression profiling is accomplished using the Illumina Human HT-12v4 beadchip microarrays (containing ∼47,000 transcripts: ∼30,000 genes) (Illumina, Inc., San Diego, CA). Firstly, 500 ng of total RNA was converted to cDNA, which was then biotin labeled by in vitro transcription process to generate biotin-labeled cRNA using the Illumina Total Prep RNA Amplification Kit (Ambion, USA) according to manufacturer’s instructions. Then, 1.5 μg of labeled cRNA was used for hybridization according to the Illumina whole-genome gene expression direct hybridization assay protocol. Afterwards, arrays were scanned using the Illumina BeadArray Reader to measure fluorescence intensity at each probe. The obtained images were processed and converted into signal intensities using the Illumina GenomeStudio software (Illumina, Inc.)
Statistical analyses
One-way analysis of variance (ANOVA) for MTT analyses and two-way ANOVA for Annexin V, MMP, caspase-3 activity, and cell cycle analyses were performed using GraphPad Prism software. The results were rendered statistically significant if P value is ≤0.05. The obtained data files were analyzed using the Illumina Genome Studio software to determine gene expression signal intensities. The resulting data sets were used to construct hierarchical gene clusters with the statistically significant (P < 0.05) genes by evaluating the “proximity” between them. Differentially expressed genes were determined when logarithmic gene expression ratios in three independent hybridizations were more than 1.5 or less than 0.66 and when the P values were less than 0.05. For each comparison, we obtained the list of differentially expressed genes constrained by P value <0.05 and at least 2.0-fold change. Gene ontology and pathway analyses were carried out by using the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Ingenuity Pathway Analysis (IPA; Ingenuity Systems, Redwood City, CA).
Results
Fisetin and hesperetin inhibit growth of K562 cells
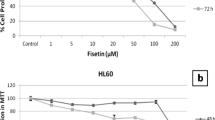
The growth inhibitory effect of fisetin and hesperetin on K562 cells was studied by performing MTT assay. Cells were treated with different concentrations of fisetin and hesperetin (1–200 μM) for different time points (48–72 h). Figure 1a shows the chemical structures of fisetin and hesperetin. Results showed that fisetin and hesperetin inhibited the cell growth effectively in a dose- and time-dependent manner. IC50 (inhibition of cell viability by 50 % at a particular concentration) values at 48 and 72 h were determined to be as 163 and 120 μM for fisetin (Fig. 1b) and 179 and 162 μM for hesperetin, respectively (Fig. 1c).

Chemical structures of fisetin and hesperetin (a). Cytotoxic effects of fisetin (b) and hesperetin (c) on K562 cells with statistical analysis. The IC50 values of fisetin and hesperetin were determined based on the cell proliferation plots. The data obtained from three independent experiments are presented as mean ± SEM. If the error bars are not seen, they are smaller than the thickness of the lines on the graphs. Statistical analysis was performed using one-way analysis of variance and P < 0.05 was considered to be significant
Fisetin and hesperetin induce apoptosis of K562 cells
In order to determine whether the growth inhibitory effect of fisetin and hesperetin is due to the induction of apoptosis, the cells were incubated with increasing concentrations of the agents for 72 h. The percentage of cells undergoing apoptosis upon treatment (50–200 μM) was detected by flow cytometry. The increase in the apoptotic cells was dose dependent in cells treated with fisetin or hesperetin. However, increasing concentrations of fisetin was more effective with 5.4-, 9.5-, and 11.6-fold increase than hesperetin with 1.27-, 1.8-, and 4.3-fold increases for 50, 100, and 200 μM respectively compared to untreated controls in K562 cells (Fig. 2a, b). Although both fisetin and hesperetin induce apoptosis in a dose-dependent manner, results of Annexin V/PI staining clearly demonstrated that fisetin triggers the highest induction of apoptosis.

Evaluation of apoptosis in K562 cells induced by fisetin and hesperetin (a) by FACS analysis via Annexin V-FITC/PI staining. Cells localized in Q4 quadrant display Annexin-positive/PI negative, early apoptotic cells while the cells gated in Q2 quadrant display Annexin-positive/PI positive, late apoptotic cells (b). The results derived from the means of three independent experiments are represented as mean ± SEM. If the error bars are not seen, they are smaller than the thickness of the lines on the graphs. Two-way analysis of variance was used to determine statistical significance and P < 0.05 was considered to be significant
Involvement of the mitochondria in fisetin- and hesperetin-induced apoptosis in K562 cells
Mitochondrial membrane permeabilization/depolarization is involved in apoptosis, which results in the release of cytochrome c and caspase activation [25]. The effects of fisetin and hesperetin on MMP were evaluated by using JC-1 dye-based method. The mitochondria of untreated control cells exhibited strong red JC-1 fluorescence, which is an indication of mitochondrial membrane polarization. However, fisetin-treated cells showed an increased green fluorescence, which is indicative of mitochondrial membrane depolarization. There were 1.03-, 1.25-, and 80-fold increases in the loss of MMP in K562 cells treated with 50, 100, and 200 μM fisetin, respectively, compared to untreated control cells (Fig. 3a). As shown in Fig. 3b, there were 1.7-, 2.4-, and 2.5-fold increases in loss of MMP in hesperetin-treated K562 cells in response to 50, 100, and 200 μM agents, respectively. Although hesperetin disrupted MMP in a dose-dependent manner, the highest concentration of fisetin treatment displayed the strongest effect on MMP in K562 cells.

Fold changes in MMP of K562 cells after treatment with defined concentrations of fisetin (a) and hesperetin (b). The results obtained from the means of three independent experiments are displayed as mean ± SEM. If not seen, they are smaller than the thickness of the lines on the graphs. P < 0.05 was considered to be statistically significant after the analysis using two-way analysis of variance
Fisetin and hesperetin increase caspase-3 activity in K562 cells
Next, we examined whether fisetin and hesperetin treatment activated caspase-3, which is an important marker of apoptosis. Following treatment of the cells with increasing concentrations of the agents for 72 h, the cleavage of the labeled substrate DEVD-pNA was used to measure the changes in caspase-3 enzyme activity. In fisetin-treated K562 cells, there were 1.3-, 1.6-, and 1.8-fold increases in caspase-3 enzyme activity in response to 50, 100, and 200 μM fisetin, respectively, as compared to untreated controls (Fig. 4). On the other hand, there were no significant changes in caspase-3 enzyme activity of K562 cells treated with 50 and 100 μM hesperetin but there was a significant (2.8-fold) increase in response to 200 μM hesperetin (Fig. 4).

Alterations in caspase-3 enzyme activity in response to fisetin and hesperetin in K562 cells. The data obtained from, the means of three independent experiments are presented as mean ± SEM. If not seen, they are smaller than the thickness of the lines on the graphs. P < 0.05 was considered to be statistically significant after the analysis using two-way analysis of variance
Fisetin arrested cell cycle at both S and G2/M phases while hesperetin at G0/G1 in K562 cells
In order to identify whether growth inhibitory effects of fisetin and hesperetin are due to the blockage in cell cycle progression, we evaluated their potential roles on cell cycle by flow cytometry and the percentages of cells in G0/G1, S, and G2 ⁄M phases were calculated. The cell cycle progression of fisetin-treated K562 cells was arrested at both S and G2/M phases, which was accompanied by a reduction in the G0/G1 phase in a dose-dependent manner (Fig. 5a). The percentage of the cells accumulating in S phase were 40.26 (Control), 48.04 (20 μM), and 77.045 % (50 μM) while the values at G2/M phase were 2.1 (Control), 4.68 (20 μM), and 8 (50 μM). Furthermore, the G0/G1 phase of the cell cycle distribution was 57,645 (Control), 47,285 (20 μM), and 15,955 % (50 μM). Hesperetin treatment with only a concentration of 200 μM (73.83 %) resulted in a significant increase in cell population at the G0/G1 phase of the cell cycle with respect to control (50.08 %) (Fig. 5b). Furthermore, there was a dose-dependent decrease in the S phase population of the cell cycle upon hesperetin treatment (Control: 46.78 %, 50 μM: 40.49 %, 100 μM: 40.39 %, 200 μM: 22.7 %).

Distribution of cell cycle phases in K562 cells after treatment with fisetin (a) and hesperetin (b). The results derived from the means of three independent experiments are shown as mean ± SEM. If not seen, they are smaller than the thickness of the lines on the graphs. Two-way analysis of variance was used to determine statistical significance and P < 0.05 was considered to be significant
Identification of genes differentially expressed in fisetin- and hesperetin-treated K562 cells
In order to determine the changes in the gene expression levels in K562 cells treated with fisetin (50 and 100 μM) and hesperetin (100 and 150 μM), hierarchical clustering of gene expression in untreated (Control), fisetin- and hesperetin-treated K562 cells was performed (Fig. 6). The isolated total RNA was amplified and converted to biotin-labeled cRNA, which was hybridized to microarray system containing approximately 30,000 genes.

Hierarchical gene clustering analysis was performed based on data obtained from gene expression analysis of K562 cells exposed to fisetin (upper panel) and hesperetin (lower panel)
A total of 553 and 1734 genes were significantly regulated (P < 0.05) in 50 and 100 μM fisetin-treated K562 cells, respectively. The number of upregulated genes were 286 whereas those of downregulated ones were 267 in 50 μM fisetin-treated cells. In 100 μM fisetin-treated cells, 806 genes were upregulated while 928 genes were downregulated. Both 50 and 100 μM fisetin treatment resulted in the upregulation of common genes such as nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (NFKBIA), Phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1/NOXA), cyclin-dependent kinase inhibitor 1A, p21 (CDKN1A), growth arrest and DNA damage inducible 45 beta (GADD45B), and several MT (metallothionein) genes (Table 1). On the other hand, it was clearly understood from Table 1 that different concentration of fisetin caused the upregulation of distinct genes in K562 cells. For instance, 50 μM fisetin upregulated nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, zeta (NFKBIZ) and Growth arrest and DNA damage inducible 45 alpha (GADD45A) while 100 μM fisetin treatment resulted in the upregulation of cyclin-dependent kinase inhibitor 2D, p19 (CDKN2D), TXNIP, and SMAD5. Furthermore, MYC (v-myc avian myelocytomatosis viral oncogene homolog), MYB (v-myb avian myeloblastosis viral oncogene homolog), C-KIT (v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog), several tubulin family members such as TUBA1A and TUBAL3 were examples of common downregulated genes after 50 and 100 μM fisetin treatment (Table 2). Treatment with 100 μM fisetin caused downregulation of several important genes such as Forkhead box K1 (FOXK1), Forkhead box A2 (FOXA2), B-cell lymphoma-61 extra large (BCL-XL), and members of ATP-binding cassette (ABC) family transporters (Table 2).
On the other hand, a total of 1659 and 1201 genes were significantly altered (P < 0.05) in 100 and 150 μM hesperetin-treated K562 cells, respectively. Treatment of K562 cells with 100 μM hesperetin resulted in the upregulation of 776 genes and the downregulation of 883 genes. The number of upregulated genes was 528, whereas that of downregulated was 673 in 150 μM hesperetin-treated K562 cells. As indicated in Table 3, dual specificity phosphatase 1 (DUSP1), CDKN1A, GADD45B, SPRR2D, and MT1F were the examples of common upregulated genes in 100 and 150 μM hesperetin treatment. On the other hand, 100 μM hesperetin lead to the upregulation of other genes such as CDKN1B/p27Kip1, CASP4, dual specificity phosphatase 5 (DUSP5), whereas, 150 μM hesperetin upregulated different genes such as MT1A, DUSP3, and NFKBIA (Table 3). STAT5A, TUBA1A, MYB, KIT, epithelial cell adhesion molecule (EPCAM), and proliferating cell nuclear antigen (PCNA) were the examples of common downregulated genes upon 100 and 150 μM hesperetin treatment (Table 4). Treatment with 100 μM hesperetin also resulted in the downregulation of eukaryotic translation elongation factor 1 gamma (EEF1G), polymerase (RNA) II (DNA directed) polypeptide B (POLR2B), and STAT3. Furthermore, 150 μM hesperetin treatment downregulated genes such as minichromosome maintenance complex component 10 (MCM10), ATP-binding cassette sub-family C member 4 (ABCC4), and polymerase (DNA directed), epsilon 2 (POLE2) (Table 4).
Identification of genetic networks affected by fisetin and hesperetin in K562 cells
IPA was performed to identify modulated networks based on the functional relationships between gene products by fisetin and hesperetin in K562 cells (Table 5). Apoptosis modulation and signaling, TP53 network, TNF-α signaling, KIT receptor signaling pathway, JAK/STAT signaling, adhesion networks, growth hormone receptor signaling, and angiogenesis were modulated networks in fisetin-treated K562 cells.
Hesperetin treatment in K562 cells resulted in alterations in translation initiation and elongation networks, replication networks, transcription networks, epidermal growth factor (EGF) signaling, JAK/STAT signaling, KIT receptor signaling pathway, growth hormone receptor signaling, and PI3K pathway (Table 6).
Discussion
Flavonoids are a significant member of polyphenolic compounds that are commonly found in the human diet. They are widely distributed in the plant species such as fruits and vegetables. Currently, there is an increased interest in polyphenolics, including flavonoids, since clinical and experimental studies show that they posses many therapeutic features including anticarcinogenic activities [7]. Fisetin and hesperetin, plant-derived flavonoids, have been shown to exert antioxidant, anti-inflammatory, and anticancer effects and found to induce apoptosis in various tumor cells by causing alterations in various cellular processes [26]. However, there are no studies on K562 CML cells and the mechanisms regulating fisetin- and hesperetin-induced apoptotic effects in CML cells remain elusive.
This study was conducted to examine the cytotoxic, cytostatic, and apoptotic effects of fisetin and hesperetin on K562 CML cells. The cells were treated with increasing concentrations of these agents [1–200 μM], and their cytotoxic effects were determined by MTT cell proliferation assay. The results of this assay revealed that fisetin and hesperetin decreased proliferation of CML cells in a dose- and time-dependent manner. On the other hand, Annexin V/PI double staining showed increases in apoptotic cell population in K562 cells in response to increasing concentrations of agents as compared to untreated controls. Based on the data obtained by flow cytometry, it could be concluded that fisetin triggers the strongest apoptosis in a dose-dependent manner. To elucidate the molecular mechanism responsible for apoptosis in K562 cells, we first checked the effects of fisetin and hesperetin on MMP. The results displayed that hesperetin treatment caused the loss of MMP in a dose-dependent manner while fisetin disrupted the MMP at the highest concentration. Mitochondria are known to be involved in all apoptotic pathways. In addition to that the structural and functional alterations in mitochondria are responsible for caspase-dependent apoptosis. Caspase-3 functions in the last step of caspase-mediated apoptosis, which results in the formation of apoptotic cell features such as DNA fragmentation [27]. Therefore, we next evaluated the changes in caspase-3 enzyme activity in the cells after fisetin and hesperetin treatment. Caspase-3 enzyme activity increased in the fisetin-treated K562 cells in a dose-dependent manner. However, hesperetin treatment caused significant changes in caspase-3 activity at the highest concentration (2.8-fold at 200 μM).
Similar to our results, fisetin was found to induce depolarization in mitochondrial membrane and caspase activation in HCT-116 colon cancer [28] and DU145, PC-3, and LNCaP prostate cancer cells [29]. U266 multiple myeloma cells and U967 leukemic cells underwent apoptosis in response to fisetin through caspase-3 activation [30, 31]. In a recent study, fisetin induced apoptosis in the human nonsmall cell lung cancer cell line, NCI-H460, via mitochondrial membrane depolarization and caspase-3 activation [32]. Similarly, hesperetin was displayed to attenuate mitochondrial membrane potential with increased expression of caspase-3, thus, causing apoptosis in SiHa cervical cancer cells [33]. In another study, hesperetin also induced apoptosis on HT-29 colon cancer cells by causing an increase in cleaved caspase-3 expression [34].
To understand the mechanisms underlying fisetin- and hesperetin-induced growth inhibitory effects, we also investigated the cytostatic property of fisetin and hesperetin on CML cells by flow cytometry. The cell cycle progression of fisetin-treated K562 cells was found to be arrested in both S and G2/M phases of the cell cycle while hesperetin blocked the cell cycle progression in G0/G1 phase with a significant accumulation of the cells at the highest concentration. In the literature, there are various studies showing the effects of these three flavonoids on the cell cycle in different cancer types. Similar to the data obtained in this study, fisetin induced G2/M arrest in human epidermoid carcinoma A431 cells [35], HT-29 colon cancer cells [36], and PC3 prostate cancer cells [37]. The G2/M checkpoint is important to prevent cells with damaged DNA from entering mitosis. Therefore, this checkpoint gives enough time to those cells in order to repair DNA and stop their proliferation. Thus, the G2/M phase arrest is one of the most critical checkpoints of many anticancer agents in cancer cells [36, 37]. We also determined that fisetin treatment caused S phase arrest in K562 CML cells for the first time. Hesperetin treatment resulted in significant cell cycle arrest in the G1 phase by downregulating Cdk-2 and Cdk-4 together with cyclin D in MCF-7 breast cancer cells similar to our results [22]. It is known that G1 arrest results in DNA repair or apoptosis [22]. Therefore, this arrest could inhibit cell proliferation and contribute the apoptotic process as a possible mechanism of hesperetin action in K652 CML cells.
We also performed genome-wide microarray analysis in K562 cells to identify the genes and networks that are involved in fisetin- and hesperetin-related antiproliferative, apoptotic, and cytostatic effects. The list of differentially expressed genes constrained by P value <0.05 and at least 2.0-fold change was indicated in the result part. As clearly seen in Table 1, NFKBIA, MT family genes, PMAIP1/NOXA, CDKN1A, and GADD45B were examples of the genes upregulated in both 50 and 100 μM fisetin-treated K562 cells with significant effects on cell growth and death. For instance, NFKBIA (also known as IκBα), a member of the NF-kappa-B inhibitor family, inhibits NF-κB pathway, which is involved in cell survival [38]. Therefore, upregulation of this gene by fisetin could suppress cell growth and induce apoptosis. The BH3-only protein PMAIP1 (NOXA), a p53 transcriptional target, has been reported to be involved in chemotherapeutic agent-induced apoptosis [39]. Its upregulation may result in apoptosis of K562 cells after fisetin treatment. This treatment upregulated CDKN1A (p21/WAF1/Cip1), one of the regulators involved in cell cycle control, and could induce growth arrest [40]. Fisetin induced upregulation of GADD45B participating in cell cycle arrest, DNA repair, cell survival, and apoptosis, which could be another important event in suppressing leukemic cell growth [41]. Metallothionin (MT) family genes such as MT1A, MT1G, and MT2A were significantly upregulated and are known to play roles in apoptosis [42]. Furthermore, each fisetin concentration resulted in upregulation of specific genes most of which act as tumor suppressor (Table 1). Moreover, fisetin treatment downregulated some common genes involved in significant cellular processes as shown in Table 2. For instance, C-KIT (proto-oncogenic cell surface receptor tyrosine kinase) and oncogenic transcription factors MYB and MYC are known to function in cell growth, proliferation, survival, migration, and apoptosis [43–45]. Their overexpression or constitutive activation is a common alteration in various cancer types including leukemias. Therefore, approaches or agents such as fisetin that targets them have a potential to inhibit leukemic growth. The expression of several tubulin family members like TUBA1A and TUBA1C also decreased after fisetin treatment, which could disrupt in cell division and thus induce apoptosis [46].
The genome-wide changes in the expression levels of genes after hesperetin treatment in K562 CML cells were summarized in Tables 3 and 4. As seen in Table 3, different hesperetin concentrations resulted in upregulation of common genes as well as concentration-specific alterations. For instance, a recent study has shown that the overexpression of dual specificity phosphatases (DUSP) family members was responsible for inducing apoptosis by inhibiting p38 MAPK and NF-κB pathways in a recent study [47]. Therefore, these genes could be potent targets of hesperetin-induced apoptosis. SPRR2D, small proline-rich proteins, have no or lower expression in cancerous cells, thus increasing SPRR2D expression could impair leukemic growth [48]. MTF1, a member of metallothionin family, was found to increase dramatically. The product of this gene plays a role in apoptosis [42] and its overexpression could increase apoptotic cell death in K562 cells. Several cell cycle regulators including p21, p19, and GADD45 family members were also upregulated, which could block cell cycle progression and impair cell growth.
Hesperetin treatment in K562 cells resulted in downregulation of several important genes as well (Table 4). In addition to the genes that were downregulated by fisetin such as tubulin family members (TUBA1A, TUBAL3), MYC, MYB, and KIT, we have found that hesperetin treatment leads to the downregulation of unique set of genes. These unique genes included kinesins such as KIF1A and KIF11, in addition to factors involved in replication such as MCM family members, POLD1 and PCNA. The E2F transcription factors like E2F2 and E2F3 and factors involved in translation such as EIF2S2, EEF1G, and EIF4G1 were also included in the downregulated genes. Based on the data in Table 4, hesperetin induced the inhibition of K562 cells by targeting critical cellular processes such as cell division, replication, transcription, and translation that have vital importance for cell survival.
To identify the genetic networks, IPA was performed as well as gene clustering. The determined networks were found to be involved in the cell survival, proliferation, cell death, cell cycle, in addition to cell signaling pathways such as JAK/STAT and KIT receptor signaling. They were also involved in adhesion, replication, transcription, and translation as summarized in Tables 5 and 6. These results conform with the gene clustering analysis.
Conclusions
In conclusion, this is the first report demonstrating that fisetin and hesperetin could act as a potent chemotherapeutic drug against CML by exerting their pleiotropic effects on the cells. The present study indicated that fisetin and hesperetin may have therapeutic potential in CML cells due to the inhibition of cell proliferation, induction of apoptosis, and cell-cycle arrest. Moreover, the genetic systems identified in this study provide a deep understanding about some important signaling networks that are altered by fisetin and hesperetin treatment while giving a list of potential genes that could be a significant target for CML therapy. Xenograft CML model might be developed through subcutaneous injection of CML cells into nude mice and tumor development could be evaluated after fisetin and hesperetin treatment. Therefore, in vivo administration of fisetin and hesperetin into K562 mice models could suggest that these natural flavonoids might have clinical usage in leukemia treatment. Furthermore, fisetin and hesperetin could be used to overcome resistance against tyrosine kinase inhibitors in resistant CML cells and the effects of their combination with these inhibitors could be investigated. These in vitro and in vivo studies could open the way to figure out whether fisetin and hesperetin might be used as novel chemotherapeutics in CML treatment.
References
Quinta’s-Cardama A, Cortes JE. Molecular biology of bcr-abl1–positive chronic myeloid leukemia. Blood. 2009;113:1619–30.
Deininger MVN, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96:3343–56.
Salesse S, Verfaillie CM. BCR/ABL: from molecular mechanisms of leukemia induction to treatment of chronic myeloid leukemia. Oncogene. 2002;21:8547–59.
Melo JV, Chuah C. Novel agents in CML therapy: tyrosine kinase inhibitors and beyond. Hematology Am Soc Hematol Educ Program. 2008;2008:427–35.
Khan N, Afaq F, Syed DN, Mukhtar H. Fisetin, a novel dietary flavonoid, causes apoptosis and cell cycle arrest in human prostate cancer LNCaP cells. Carcinogenesis. 2008;29:1049–56.
Sak K. Cytotoxicity of dietary flavonoids on different human cancer types. Pharmacogn Rev. 2014;8:122–46.
Ravishankar D, Rajora AK, Greco F. Osborn HMFlavonoids as prospective compounds for anti-cancer therapy. Int J Biochem Cell Biol. 2013;45:2821–31.
Khan N, Syed DN, Ahmad N, Mukhtar H. Fisetin: a dietary antioxidant for health promotion. Antioxid Redox Signal. 2013;19:151–62.
Lim do Y, Park JH. Induction of p53 contributes to apoptosis of HCT-116 human colon cancer cells induced by the dietary compound fisetin. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1060–8.
Szliszka E, Helewski KJ, Mizgala E, Krol W. The dietary flavonol fisetin enhances the apoptosis-inducing potential of TRAIL in prostate cancer cells. Int J Oncol. 2011;39:771–9.
Murtaza I, Adhami VM, Hafeez BB, Saleem M, Mukhtar H. Fisetin, a natural flavonoid, targets chemoresistant human pancreatic cancer AsPC-1 cells through DR3 mediated inhibition of NF-κB. Int J Cancer. 2009;2:2465–73.
Khan N, Afaq F, Khusro FH, Mustafa Adhami V, Suh Y, Mukhtar H. Dual inhibition of phosphatidylinositol 3-kinase/Akt and mammalian target of rapamycin signaling in human nonsmall cell lung cancer cells by a dietary flavonoid fisetin. Int J Cancer. 2012;130:1695–705.
Jang KY, Jeong SJ, Kim SH, Jung JH, Kim JH, Koh W, et al. Activation of reactive oxygen species/AMP activated protein kinase signaling mediates fisetin induced apoptosis in multiple myeloma U266 cells. Cancer Lett. 2012;319:197–202.
Adhami VM, Syed DN, Khan N, Mukhtar H. Dietary flavonoid fisetin: a novel dual inhibitor of PI3K/Akt and mTOR for prostate cancer management. Biochem Pharmacol. 2012;84:1277–81.
Ash D, Subramanian M, Surolia A, Shaha C. Nitric oxide is the key mediator of death induced by fisetin in human acute monocytic leukemia cells. Am J Cancer Res. 2015;5:481–97.
Pal HC, Baxter RD, Hunt KM, Agarwal J, Elmets CA, Athar M, Afaq F1. Fisetin, a phytochemical, potentiates sorafenib-induced apoptosis and abrogates tumor growth in athymic nude mice implanted with BRAF-mutated melanoma cells. Oncotarget. 2015; [Epub ahead of print].
Benavente-Garcia O, Castillo J. Update on uses and properties of citrus flavonoids: new findings in anticancer, cardiovascular, and anti-inflammatory activity. J Agric Food Chem. 2008;56:6185–205.
Nielsen IL, Chee WS, Poulsen L, Offord-Cavin E, Rasmussen SE, Frederiksen H, et al. Bioavailability is improved by enzymatic modification ofthe citrus flavonoid hesperidin in humans: a randomized, double-blind, crossover trial. J Nutr. 2006;136:404–8.
Palit S, Kar S, Sharma G, Das PK. Hesperetin induces apoptosis in breast carcinoma by triggering accumulation of ROS and activation of ASK1/JNK pathway. J Cell Physiol. 2015;230:1729–39.
Alshatwi AA, Ramesh E, Periasamy VS, Subash-Babu P. The apoptotic effect of hesperetin on human cervical cancer cells is mediated through cell cycle arrest, death receptor, and mitochondrial pathways. Fundam Clin Pharmacol. 2013;27:581–92.
Sivagami G, Vinothkumar R, Bernini R, Preethy CP, Riyasdeen A, Akbarsha MA, et al. Role of hesperetin [a natural flavonoid] and its analogue on apoptosis in HT-29 human colon adenocarcinoma cell line-A comparative study. Food Chem Toxicol. 2012;50:660–71.
Choi EJ. Hesperetin induced G1-phase cell cycle arrest in human breast cancer MCF-7 cells: involvement of CDK4 and p21. Nutr Cancer. 2007;59:115–9.
Chen YC, Shen SC, Lin HY. Rutinoside at C7 attenuates the apoptosis-inducing activity of flavonoids. Biochem Pharmacol. 2003;66:1139–50.
Zhang J, Wu D, Vikash, Song J, Wang J, Yi J, Dong W. Hesperetin induces the apoptosis of gastric cancer cells via activating mitochondrial pathway by increasing reactive oxygen species. Dig Dis Sci. 2015, [Epub ahead of print].
Can G, Ekiz A, Baran Y. Imatinib induces autophagy through BECLIN-1 and ATG5 genes in chronic myeloid leukemia cells. Hematology. 2011;16:95–9.
Chen CM, Hsieh YH, Hwang JM, Jan HJ, Hsieh SC, Lin SH, et al. Fisetin suppresses ADAM9 expression and inhibits invasion of glioma cancer cells through increasedphosphorylation of ERK1/2. Tumour Biol. 2015;36:3407–15.
Solmaz S, Adan Gokbulut A, Cincin B, Ozdogu H, Boga C, Cakmakoglu B, et al. Therapeutic potential of apigenin, a plant flavonoid, for imatinib sensitive and resistant chronic myeloid leukemia cells. Nutr Cancer. 2014;66:599–612.
Lim do Y, Park JH. Induction of p53 contributes to apoptosis of HCT-116 human colon cancer cells induced by the dietary compound fisetin. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1060–8.
Szliszka E, Helewski KJ, Mizgala E, Krol W. The dietary flavonol fisetin enhances the apoptosis-inducing potential of TRAIL in prostate cancer cells. Int J Oncol. 2011;39:771–9.
Jang KY, Jeong SJ, Kim SH, Jung JH, Kim JH, Koh W, et al. Activation of reactive oxygen species/AMP activated protein kinase signaling mediates fisetin induced apoptosis in multiple myeloma U266 cells. Cancer Lett. 2012;319:197–202.
Monasterio A, Urdaci MC, Pinchuk IV, López-Moratalla N, Martínez-Irujo JJ. Flavonoids induce apoptosis in human leukemia U937 cells through caspase- and caspase-calpain-dependent pathways. Nutr Cancer. 2004;50:90–100.
Kang KA, Piao MJ, Hyun JW. Fisetin induces apoptosis in human nonsmall lung cancer cells via a mitochondria-mediated pathway. In Vitro Cell Dev Biol Anim. 2015;51:300–9.
Alshatwi AA, Ramesh E, Periasamy VS, Subash-Babu P. The apoptotic effect of hesperetin on human cervical cancer cells is mediated through cell cycle arrest, death receptor, and mitochondrial pathways. Fundam Clin Pharmacol. 2013;27:581–92.
Sivagami G, Vinothkumar R, Bernini R, Preethy CP, Riyasdeen A, Akbarsha MA, et al. Role of hesperetin (a natural flavonoid) and its analogue on apoptosis in HT-29 human colon adenocarcinoma cell line-A comparative study. Food Chem Toxicol. 2012;50:660–71.
Pal HC, Sharma S, Elmets CA, Athar M, Afaq F. Fisetin inhibits growth, induces G2/M arrest and apoptosis of human epidermoid carcinoma A431 cells: role of mitochondrial membrane potential disruption and consequent caspases activation. Exp Dermatol. 2013;22:470–5.
Lu X, Jung J, Cho HJ, Lim DY, Lee HS, Chun HS, et al. Fisetin inhibits the activities of cyclin-dependent kinases leading to cell cycle arrest in HT-29 human colon cancer cells. J Nutr. 2015;135:2884–90.
Haddad AQ, Venkateswaran V, Viswanathan L, Teahan SJ, Fleshner NE, Klotz LH. Novel antiproliferative flavonoids induce cell cycle arrest in human prostate cancer cell lines. Prostate Cancer Prostatic Dis. 2006;9:68–76.
Li F, Sethi G. Targeting transcription factor NF-κB to overcome chemoresistance and radioresistance in cancer therapy. Biochim Biophys Acta. 1805;2010:167–80.
Zhao X, Liu X, Su L. Parthenolide induces apoptosis via TNFRSF10B and PMAIP1 pathways in human lung cancer cells. J Exp Clin Cancer Res. 2014;33:1–11.
Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–14.
Liebermann DA, Tront JS, Sha X, Mukherjee K, Hadley AM, Hoffman B. Gadd45 stress sensors in ,malignancy and leukemia. Crit Rev Oncog. 2011;16:129–40.
Lu DD, Chen YC, Zhang XR, Cao XR, Jiang HY, Yao L. The relationship between metallothionein-1F (MT1F) gene and hepatocellular carcinoma. Yale J Biol Med. 2003;76:55–62.
Liang J, Wu YL, Chen BJ, Zhang W, Tanaka Y, Sugiyama H. The C-kit receptor-mediated signal transduction and tumor-related diseases. Int J Biol Sci. 2013;9:435–43.
George OL, Ness SA. Situational awareness: regulation of the myb transcription factor in differentiation, the cell cycle and oncogenesis. Cancers (Basel). 2014;6:2049–71.
Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11.
Leandro-García LJ, Leskelä S, Landa I, Montero-Conde C, López-Jiménez E, Letón R, et al. Tumoral and tissue-specific expression of the major human beta-tubulin isotypes. Cytoskeleton (Hoboken). 2010;67:214–23.
Gil-Araujo B, Toledo Lobo MV, Gutiérrez-Salmerón M, Gutiérrez-Pitalúa J, Ropero S, Angulo JC, et al. Dual specificity phosphatase 1 expression inversely correlates with NF-κB activity and expression in prostate cancer and promotes apoptosis through a p38 MAPK dependent mechanism. Mol Oncol. 2014;8:27–38.
Anisowicz A, Sotiropoulou G, Sager R. Re-expression of SPR1 in breast cancer cells by phorbol 12-myristate 13-acetate (PMA) or UV irradiation is mediated by the AP-1 binding site in the SPR1 promoter. Mol Med. 1999;5:526–41.
Acknowledgements
We would like to thank Dr. Mona El Khatib for professional help with the English editing of the article.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Adan, A., Baran, Y. Fisetin and hesperetin induced apoptosis and cell cycle arrest in chronic myeloid leukemia cells accompanied by modulation of cellular signaling. Tumor Biol. 37, 5781–5795 (2016). https://doi.org/10.1007/s13277-015-4118-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-4118-3