
Abstract
Endothelial progenitor cells (EPCs) have recently been shown to promote the angiogenic switch in solid neoplasms, thereby promoting tumour growth and metastatisation. The genetic suppression of EPC mobilization from bone marrow prevents tumour development and colonization of remote organs. Therefore, it has been assumed that anti-angiogenic treatments, which target vascular endothelial growth factor (VEGF) signalling in both normal endothelial cells and EPCs, could interfere with EPC activation in cancer patients. Our recent data, however, show that VEGF fails to stimulate tumour endothelial colony-forming cells (ECFCs), i.e. the only EPC subtype truly belonging to the endothelial lineage. The present article will survey current evidence about EPC involvement in the angiogenic switch: we will focus on the controversy about EPC definition and on the debate around their actual incorporation into tumour neovessels. We will then discuss how ECFC insensitivity to VEGF stimulation in cancer patients could underpin their well-known resistance to anti-VEGF therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bone marrow (BM)-derived haematopoietic and endothelial progenitor cells (BMDCs) do not only serve as mere bystanders during tumour vascularisation but play an active role during the angiogenic switch both at the primary lesion site and at the more distant metastatic deposits [1–11]. Moreover, certain chemotherapy drugs and vascular-disrupting agents (VDA) cause an acute endothelial progenitor cell (EPC) spike in the peripheral blood, which underpins the vascular rebound and tumour relapse often reported after such treatments [12, 13]. Recent work conducted both by us [14, 15] and by other research groups [16, 17] has further unveiled that tumour microenvironment may finely reprogram BM-derived EPCs to support neoplastic growth and metastatisation. Therefore, EPC recruitment has been proposed among the key mechanisms responsible for the growing failure of anticancer therapies, including the most recent anti-VEGF drugs [18, 19]. Importantly, we have recently demonstrated that tumour EPCs are insensitive to VEGF [14], thereby questioning VEGFR-2 suitability in anti-angiogenic therapy [2, 20]. The present article will first survey the evidence in favour of EPC contribution to tumour vasculature; we will then speculate how the use of normal, rather than patient-derived, EPCs could have hampered the search for the most suitable target for anti-angiogenic treatments.
Identification of circulating EPC in human adult subjects: a never ending story?
Following the seminal paper by Asahara and coworkers, published in Science in 1997 [21], an increasing burden of investigations on human circulating EPCs has been carried out. However, already at a first lecture, it becomes clear that the method employed to isolate EPCs from the peripheral blood in scientific publications is not unique and, in turn, the identification of EPCs is far from being homogenous in the different manuscripts. This diversity, besides being responsible for an ambiguity in the identification and enumeration of EPCs, makes also very difficult, if not impossible in some cases, to compare the results obtained in different studies. To review all the issues that have been raised in the field of identification of human circulating EPCs, it should be first clarified how can be defined an EPC. An “operational” definition of EPC, based on the properties that are usually attributed to a progenitor cell, could be “a cell able to self renew, proliferate and differentiate to generate a large progeny of mature endothelial cells in vitro and/or to participate to neovessel formation in vivo”. Two main approaches have been proposed for isolation and enumeration of circulating EPCs: (i) immunophenotyping and (ii) cell culture.
-
a)
Immunophenotyping: Soon after their first description, there have been many attempts to define an immunophenotypic pattern which could identify an EPC. This resulted in the publication of hundreds of papers that enumerated, characterized and correlated to various clinical parameters or outcomes, the frequency of circulating EPCs [22]. However, due to the overlapping expression of some surface proteins between haematopoietic and endothelial lineages (among the most frequently used: CD34, CD133, VEGFR-2), a unique pattern of surface molecule has never been described [23]. Moreover, EPC distinction from mature endothelial cells is also a matter of discussion, due to the lack of specific marker(s) that can discriminate between the two cell types. Thus, up to now, a satisfying, unambiguous signature of cell surface molecules that can be used for EPC identification has not been accepted unanimously by the scientific community [24]. Nevertheless, flow cytometric identification of EPCs remains an attractive approach since it is fast and requires small amount of blood; however, only the search for novel surface markers will push further the use of flow cytometry for EPC identification.
-
b)
Cell culture: Cell culture represents the second methodological approach that has been developed for isolating and expanding ex vivo circulating EPCs. In this field, three different types of cultures have been used in the last 15 years. The first attempt to culture circulating EPCs was described by Asahara and coworkers [21] and modified by Hill and coworkers in 2003 [25]. By this method, mononuclear cells from 5–10 ml of peripheral blood were first cultured for 2 days on culture dishes to get rid of adherent cells. The non-adherent fraction was then plated in a specific commercial medium onto fibronectin-treated Petri dishes, and putative endothelial colonies of round cells surrounded by flat elongating cells were obtained after 5–7 days. These colonies were named CFU-Hill (aka CFU-End or CFU-EC). However, at a deeper investigation, it turned out that the cells present in the colony were of haematopoietic origin [26–28]; moreover, the ability of CFU-Hill to expand and proliferate extensively in vitro (one of the pre-requisites for a cell to be qualified as “progenitor”) was never demonstrated. Thus, CFU-Hill does not represent true EPCs, according to the definition given above. A second way to isolate EPCs in culture is based on the concept that the EPC population resides in the adherent fraction of circulating mononuclear cells. These are plated in culture dishes in endothelial growth medium, and after 2 days, the non-adherent fraction is discarded and cultures are fed with fresh medium. The adherent cells that remain attached display, after 3–4 further days of culture, a morphology resembling the spindle-shape cells described in the CFU-Hill and express markers that are typical of the endothelial lineage, such as, for instance, von Willebrand factor (vWf), VE-cadherin, CD31 and Tie2 [29–31]. Moreover, these cells were shown to promote vascularisation in animal models of hindlimb ischemia [29], and therefore, they were considered bona fide EPCs and termed circulating angiogenic cells (CACs). Interestingly, these cells never formed real colonies in vitro nor they could be expanded, two features that distinguish progenitor cells. As a matter of fact, an in-depth analysis of their phenotype confirmed that CACs were haematopoietic by clonal lineage tracking, exactly as it occurred with CFU-Hill [27].
Lin et al. in 2000 [32] and Ingram et al. in 2004 [33] described the isolation from the adherent fraction of circulating mononuclear cells both of adult subjects and of umbilical cord blood of a new class of cells that could be ascribed to the category of EPCs and that were termed endothelial colony-forming cells (ECFCs) [33]. From the mononuclear cells adherent to collagen-I-coated culture dishes, discrete cobblestone-like colonies of flat cells resembling mature endothelial cells originated after 10–15 days of culture grew to confluence and could be expanded up to 20 passages. Because of their late appearance in culture compared to CACs or CFU-Hills, these colonies have also been called “late-outgrow EPCs” [33, 32, 22]. Cells forming ECFC-derived colonies were proven to be phenotypically indistinguishable from cultured endothelial cells and possess de novo vessel-forming ability [27]; in addition, they satisfied the definition of a “true” progenitor cell, being able to self-renew (at least for a number of passages), to proliferate and differentiate into mature endothelial cells and to generate in vivo new vessels. Their faithful belonging to endothelial lineage was proved by the lack of expression of haematopoietic markers (CD45 and CD14) and by the expression of typical endothelial markers (VE-cadherin, CD146, vWf, CD31). Finally, clonal analysis of ECFCs from patients with an acquired mutation of the JAK2 gene in the haematopoietic lineage showed the absence of the mutation in ECFC-derived cells. On the contrary, the mutation was detected in CACs and CFU-Hills derived from the same patients [27, 22].
In conclusion, although a role can be demonstrated for CFU-Hill and CACs in experimental vasculogenesis, these must be considered haematopoietic in origin and therefore not representing a surrogate of EPCs. On the other side, ECFCs display all the characteristics of endothelial progenitor cells and represent up to now the best surrogate of this elusive population of progenitor cells.
Evidence in favour of EPC contribution to the establishment of tumour vasculature
The angiogenic switch is turned on when the delicate balance between pro- and anti-angiogenic factors is tipped in favour of neovessel formation by tumour microenvironment. This process is regulated by the hypoxia-inducible factor-1α (HIF-1α), a transcription factor that is activated under the hypoxic conditions of a growing tumour and drives the expression of VEGF, EGF, bFGF, and SDF-1α, interleukin-8 (IL-8), CCL2 and CCL5 [34, 6]. Once released into circulation, these soluble factors promote local angiogenesis by stimulating nearby endothelial cells to sprout towards the neoplasm; at the same time, they mobilize BMDCs and target them to the nascent vasculature [2, 1, 35, 5, 11, 10, 36]. A remarkable pro-angiogenic activity has been acknowledged to several populations of BM-derived haematopoietic cells, such as CXCR4+ VEGFR1+ hemangiocytes, Tie2-expressing monocytes, CD45+/CD11b+ myeloid cells, F4/80+ CD11b+ tumour-associated macrophages (TAMs), GR1+ CD11b+ “myeloid-derived suppressor cells” (MDSCs), and infiltrating neutrophils and mast cells [1, 37]. These cells sustain tumour growth perivascularly by paracrine liberation of growth factors and cytokines but do not incorporate within vessel lumen. Conversely, EPCs may provide the building blocks for neovessel formation as well as secrete instructive signals for neighbouring endothelial cells [1–3, 10, 11]. The earlier demonstration that EPCs are involved in tumour angiogenesis was provided by Lyden and coworkers, who found that BM transplantation rescued growth and metastatisation of two distinct syngenic tumour models (B6RV2 lymphoma and Lewis lung carcinoma or LLC) xenografted in the angiogenic defective Id1+/+ Id3−/− mutant mice [38]. Tumour vascularisation under these conditions was associated to the recruitment of VEGFR-1+ myeloid cells and VEGFR-2+ EPCs from reconstituted BM. The same authors documented that the engraftment of β-galactosidase-positive (lacZ) BM from Rosa26 mice, which express lacZ in all tissues, recapitulated angiogenesis in Id1+/− Id3−/− mice implanted with B6RV2 tumours [38]. Later work indeed demonstrated that Id1 is a reliable EPC marker and drives their egression from BM [39], while it is not expressed by myeloid cells [40]. This feature underlies the defective angiogenic process observed in Id1+/− mutants [39]. Subsequently, fluorescence in situ hybridization (FISH) of sex chromosomes in individuals who developed cancer after BM transplantation with donors of the opposite sex detected BM-derived endothelial cells throughout tumour vasculature, and their percentage was ranged from 1 to 12 % depending on the malignancy [41]. More recently, Nolan and coworkers used BMDCs isolated from GFP+ mice and injected into lethally irradiated syngenic wild-type recipient to investigate EPC contribution to tumour angiogenesis [4]. Reconstituted animals were xenografted with three distinct tumour types, i.e. LLC, B6RV2 and melanoma, and then examined at various stages of tumour development by using endothelial (VE-cadherin, CD31, endoglin and VCAM), haematopoietic (CD11b, CD45RB, CD41) and progenitor (CD133) markers. GFP expression, in turn, ensured BM origin of vessel cells. These authors first found that BM-derived GFP+ cells were recruited at the periphery of LLC at the early stages of tumour growth (days 4–6) prior to the sprouting of endothelial cells from nearby capillaries. These cells were identified as EPCs based on their morphological and phenotypic characterization. When LLC tumours were inspected at later stages (6–8 days), they showed chimeric vessels comprising both non-BM-derived cells and BM-derived GFP+ EPCs. Importantly, high-resolution stereo-confocal microscopy confirmed that GFP+ cells did not occupy a perivascular location, while optical sectioning of multiple z-stacks (30-μm resolution) displayed that BM-derived endothelial cells possess a single nucleus and that CD31 (indicative of endothelial origin) and GFP signals derive from the same individual cell. This proved that tumour endothelial cells could actually originate from BM-mobilized EPCs. Intriguingly, flow cytometric analysis revealed that the percentage of BM-derived EPCs (GFP+ VE-cadherin+ CD31low CD11b−) decreases from 25–35 % in the early phase of tumour development (4–6 days) to 6–8 % at later stages (6–8 days), while the fraction of local non-BM-derived endothelial cells (GFP− VE-cadherin+ CD31low CD11b−) increased to 65–75 % at days 10–14 [4]. The same results were found in a transgenic breast cancer mouse model (MMTV-PyMT) [4]. Thus, EPCs play a crucial role during the initial steps of tumour vascularisation. The MMTV-PyMT transgenic mice were further exploited to assess EPC contribution to the dynamics of vessel assembly that turns dormant micrometastases into lethal macrometastases [7]. By using the same procedure described in their seminal paper [4], Gao and coworkers focussed on the angiogenic switch in lung metastases that spontaneously develop in this breast cancer model. They found that micrometastases formed by week 12 and were poorly vascularised, as shown by the lack of CD31+ vessels. Nevertheless, macrometastases that appeared at week 16 were positive to CD31 staining and displayed luminally incorporated BM-derived GFP+ endothelial cells in about 11 % of neovessels [4]. This means that EPCs home to micrometastatic foci and contribute to neovessel formation, thereby sustaining the macrometastatic transition. The low percentage of EPC engraftment suggested that, apart from a structural role, they drive the angiogenic switch in a paracrine manner. The same findings were obtained by analysing lung metastases in LLC xenograft mice; again, by using this model, the authors further found that many BM-derived GFP+ cells are recruited to micrometastases, but confocal microscopy analysis revealed that only endothelial cells (GFP+ CD31+) integrated into neovessels. More specifically, BM-derived EPCs were recruited at the outer rim of the metastatic lesion, while haematopoietic stem cells (HSCs) adopted a perivascular location [4]. Consistent with these observations, the acute and conditional short hairpin RNA (shRNA)-mediated genetic ablation of Id1 in BM-derived EPCs did not decrease the number of micrometastatic lesions but prevented the macrometastatic transformation in LLC xenografts [7]. Finally, the use of Id1 promoter to drive GFP expression enabled Mellick and coworkers to selectively track EPC homing from BM to LLC tumours in the xenograft murine model employed in their previous work [7, 4]. Again, high-resolution microscopy revealed that BM-derived EPCs (Id1+/GFP+ VE-cadherin+ CD31low) were recruited at the periphery of early nonvascularised tumours (days 6–8). The quantification of luminally incorporated Id1+/GFP+ EPCs by fluorescence-activated cell sorting (FACS) upon systemic administration of isolectin IB4 revealed that 9 % of tumour neovessels within later tumours (days 8–12) incorporated BM-derived endothelial cells [40]. In this same study, the authors used the Id1 proximal promoter (pr/p) to drive the expression of the suicide gene herpes simplex virus-thymidine kinase in BM-derived EPCs; this manoeuvre led to a notable reduction in EPC frequency and impaired tumour (LLC and B6RV2) growth and vascularisation [40]. Along with many other parallel studies (listed in Table 1), these reports reinforced the concept that EPCs sustain the angiogenic switch in primary tumours and micro-to-macrometastatic transition at secondary lesions. An alternative approach consisted in assessing the engraftment and contribution of exogenous EPCs to tumour development in human xenograft models, including those for renal cellular carcinoma (RCC), hepatocellular carcinoma and LLC [42–46]. Unlike the previous investigations, however, these studies failed to validate the endothelial phenotype of the injected cells, by relying on rather unspecific markers (i.e. CD133, CD34, VEGFR-2) that also feature HSCs (see above). More recently, human ECFCs, which are regarded as truly endothelial precursors, were probed for their ability to specifically home to sites of tumour angiogenesis in mice bearing an array of distinct cancer types. For instance, upon intravenous injection into lethally irradiated mice, DiI-labelled ECFCs target LLC lung, but not the kidney or liver, metastases; they are mainly found at the periphery of lung metastases, rather than in the centre, and integrate within neovessels, albeit most of them adopt a perivascular location [47]. Subsequently, Bieback and coworkers evaluated the extent of ECFC recruitment to rat C6 glioma xenograft, which is the most suitable model for the study of glioblastoma multiforme. By using the dorsal skinfold chamber model associated to intravital multifluorescence videomicroscopy, they found that DiI-stained ECFCs strongly interacted (adhesion and extravasation) with tumour vasculature, while human umbilical vein endothelial cells (HUVECs) and CD34+ MNCs were much less active [48]. These preliminary findings lend strong support to the tenet that ECFCs represent the most suitable subtype to unveil the molecular mechanisms driving EPC-based tumour neovascularisation [2, 3, 49, 50, 20].
How to solve the controversy about EPC contribution to tumour vascularisation
Despite the undoubted evidence in favour of EPC involvement in tumour growth and metastatisation, several authors questioned their participation to the angiogenic switch [50, 37]. This is why several studies failed to evidence a measurable amount of luminally incorporated EPCs within tumour vessels. For instance, De Palma et al. transduced BMDCs with lentiviral vectors expressing the GFP gene under the control of the specific endothelial Tie2 promoter, which was followed by BM implantation into several subcutaneous tumour models [51]. High-resolution microscopic inspection detected only rare GFP+ CD31+ endothelial cells in tumour cells, which were instead abundant of GFP+ CD45+ CD11b+ CD31− monocytes and pericyte progenitors, with a preferential perivascular location [51]. Subsequently, Göthert et al. [52] generated an endothelial-specific inducible transgenic model to assess the BM origin of tumour endothelium. They took advantage from the fact that the 5′ enhancer element of the transcription factor stem cell leukemia-1 (SCL-1) drives the expression of this gene within the endothelial lineage. Therefore, by using a tamoxifen-inducible reporter system (laZ) driven by its 5′ endothelial enhancer, they found that LLC and B6RV2 vasculature lacks BM-derived endothelial cells but is supported by local angiogenesis [52]. Other studies also could not demonstrate EPC incorporation in the endothelial layer of several primary and metastatic tumours [53–56]. An additional proof of evidence that has been carried against EPC contribution to the angiogenic switch is their rarity in some tumour models, which display only 1–2 % of EPC-derived neovessels (Table 1). The passionate debate arose about this issue might be easily reconciled when taking a few key considerations in account. As recalled by Gao et al. [1] and in [50], EPCs are recruited to tumour periphery prior to vasculature formation [7, 4], acquire an endothelial phenotype and are luminally engrafted into a subset of neovessels in early tumours. At later stages, these chimeric structures are diluted/replaced by local host-derived sprouting vessels, which would explain the low involvement described in already developed tumours by other investigators [55, 56, 51, 52, 57]. Accordingly, De Palma et al. [51] assessed EPC incorporation at week 4, while Göthert et al. [52] at 14 days post-implantation, which are fully compatible with the data described in [4] and [7]. EPC recruitment preceding the engagement of host vasculature suggests that these cells play a key role in the stimulation of non-BM-derived endothelial cells sprouting from nearby capillaries. Nevertheless, by using two distinct transgenic mouse models of de novo tumorigenesis, i.e. Rip-Tag5 that develops pancreatic islet cancer and Alb-Tag that develops liver cancer, Spring et al. [58] could not identify BM-derived GFP+ endothelial cells in pre-neoplastic lesions, while up to 30 % of tumour-associated vessels were green at more advanced stages. This report highlights another feature that should be borne in mind when discussing the role served by EPC during tumour development, i.e. the stage-specific engagement of EPC might depend on tumour type [1]. More in general, the overall EPC contribution to the angiogenic switch could be tumour-type dependent. This hypothesis is corroborated by the work carried out on mice heterozygous for the tumour suppressor Pten (Pten+/−) that exhibit a wide array of malignancies, such as uterine carcinomas (UC), pheochromocytomas, lymph hyperplasia and prostate interepithelial neoplasias. Ruzinova et al. [59] discovered that Pten+/− spontaneous lymph hyperplasia lacked BM-derived EPCs, identified as lacZ+ VEGFR-2+ cells in animals transplanted with BM from Rosa26 mice, while these cells were easily detectable on 15–20 % of UC neovessels. Finally, it is worth of recalling that the field has long been flawed by the wrong interpretation of the term EPC [50, 60, 3]. As already mentioned throughout this article, several studies claimed to evaluate EPC frequency in cancer patients or to evaluate their contribution to tumour growth and development in xenograft models [42–46]; yet, these reports identified EPCs based on the selection of a panel of surface antigens that is inadequate to detect truly endothelial progenitors [1, 24, 60, 50, 3, 2]. As aforementioned, these cells were more likely to belong to the haematopoietic lineage that may certainly sustain the angiogenic switch in a paracrine manner, but does not provide structural support to tumour vasculature. On the other hand, cancer development and metastatisation are impaired in Id mutant mice [38, 40, 7, 61]. Likewise, Plummer et al. achieved a decrease in circulating EPCs, tumour (LLC and breast cancer) size and vessel density through the genetic suppression of the miRNA-processing enzyme, Dicer, specifically in BM [16]. Therefore, there is ample experimental evidence to conclude that truly endothelial precursors play a crucial role in cancer development and metastatisation (Fig. 1).

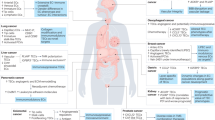
Endothelial progenitor cells support tumour growth. Early avascular tumours release pro-angiogenic cytokines (VEFG and SDF-1α) to stimulate EPC mobilization from bone marrow. Once in circulation, EPCs are targeted to the growing tumour, where they promote the formation of a vascular network by engrafting within the endothelial lining. EPCs, in turn, secrete further growth factors and cytokines, which stimulate adjacent capillaries to undergo angiogenesis so that, at later stages, they are diluted by local endothelial cells
VEGF-driven EPC incorporation into tumoral endothelium in xenograft murine models
VEGF has long been known to stimulate EPC proliferation, survival, tubulogenesis and homing both in vitro and in vivo [62–67]. In particular, VEGF released in circulation upon an ischemic insult redirects circulating EPCs to either the infarcted myocardium or the obstructed limb arteries, depending on the injury site [34, 66, 68]. Therefore, it was obvious to assume that VEGF exerts the same functions during the early phases of the angiogenic switch, during which growing tumours secrete high levels of this, as well as many other growth factors. The question then arises as to whether such hypothesis is supported by any experimental evidence in human subjects. Unfortunately, most of the work conducted to untangle this crucial aspect of cancer biology has been carried out in murine models. For instance, neutralizing antibodies (DC101) against VEGFR-2, the receptor isotype whereby VEGF activates both mature endothelial cells and their more immature progenitors [69, 14, 70], reduced LLC and B6RV2 tumour growth and angiogenesis in the neoplastic model described in [38]. However, this study could not address whether the recruitment of truly endothelial precursors was affected in DC101-treated animals (see also Discussion in [40]). Interestingly, the VEGFR-2 inhibitor ZD6474 blocked EPC mobilization in nontumour-bearing mice challenged with VEGF, while it was ineffective in LLC-implanted animals [71]. More recently, Mellick et al. harnessed the Id1 reporter constructs described above to demonstrate that EPC-specific VEGFR-2 knockdown produces a loss of EPC function, dampens LLC and B6V2 tumour development, and reduces vessel density [40]. Additionally, VEGF was found to trigger the EPC spike induced by VDA in mice bearing PC3 human prostate cancer [72] and MeWo tumour melanoma [12]. More specifically, CA-4-P induced two EPC peaks in the peripheral blood of tumours xenografted with PC3, one occurring a few hours after the administration and the second one only 3–4 days later [72]. The second increase in EPC levels was selectively abrogated by sunitinib, a multi-targeted receptor tyrosine kinase (RTK) inhibitor [72]. Conversely, OXi-4503, another VDA widely employed in cancer therapy, induced only one EPC spike in mice implanted with PC3 tumours: again, EPC mobilization was abrogated upon DC101 injection [12]. This study further unveiled that OXi-4503 did not mobilize EPCs when the tumour was grown in Id1+/− Id3−/− mutant mice [12], which display EPC mobilization defects. Altogether, these studies give substantial credit to the tenet that VEGF activates tumour EPCs by binding to VEGFR-2, just as it does in healthy cells. Nevertheless, the xenograft models employed in these studies consist in rapidly growing tumours that do not recapitulate the multistep and heterogeneous process of carcinogenesis that occurs in human patients over years [6]. The simplest and most therapeutically relevant approach is, therefore, to study the effect of VEGF on EPCs isolated from cancer patients.
VEGF fails to induce pro-angiogenic Ca2+ oscillations in tumoral ECFCs: implications for resistance to anti-angiogenic treatments
An increase in intracellular Ca2+ concentration ([Ca2+]i) is a ubiquitous signalling mode whereby virtually any cell type controls a multitude of functions, including cell cycle progression, cytokinesis, migration, cytoskeleton remodelling, gene expression, ATP synthesis, protein folding, autophagy and programmed cell death [73, 74]. It has long been known that Ca2+ signals regulate angiogenesis by controlling all the key steps of vessel remodelling, including endothelial proliferation, permeability, motility and interaction with the extracellular matrix [49, 75–78]. A sustained elevation in [Ca2+]i encodes cell death by leading to mitochondrial Ca2+ overload and induction of the apoptotic cascade. Conversely, repetitive intracellular Ca2+ spikes (or oscillations) are more suitable for cell survival by favouring mitochondrial bioenergetics and triggering the transcriptional programme responsible for cell proliferation [79, 80]. Consistently, intracellular Ca2+ oscillations mediate the pro-angiogenic action of many, if not all, growth factors on mature endothelial cells [81, 75, 82, 34, 83, 84]. Likewise, we have recently demonstrated that VEGF stimulates human ECFCs to undergo proliferation and tubulogenesis through an oscillatory increase in [Ca2+]i [62, 63, 70]. Briefly, VEGFR-2 recruits phospholipase C-γ (PLCγ) to cleave phosphatidylinositol 4,5-bisphosphate (PIP2), a minor phospholipid component of plasma membrane, into the two intracellular second messengers, inositol-1,4,5-trisphosphate (InsP3) and diacylglycerol (DAG). InsP3, in turn, mobilizes intracellular Ca2+ by gating InsP3 receptors in the endoplasmic reticulum (ER), the most abundant endothelial Ca2+ reservoir, thereby causing one to four consecutive Ca2+ spikes [62, 85]. The consequent fall in intraluminal Ca2+ load is detected by stromal interaction protein 1 (Stim1), which functions as ER Ca2+ sensor and translocates into ER-plasma membrane juxtaposed sites, termed puncta, in response to store depletion; herein, Stim1 physically engages the Ca2+-permeable channels, Orai1 and transient receptor potential canonical 1 (TRPC1), to bring about Ca2+ inflow [14, 85, 86]. This peculiar mode of store-operated Ca2+ entry (SOCE) is the most widespread mode of Ca2+ influx in mature endothelial cells as well and sustains the pro-angiogenic response to VEGF [86, 87]. SOCE refills ER Ca2+ pool and maintains the rhythmic InsP3-dependent Ca2+ spikes throughout VEGF stimulation in healthy ECFCs [62, 88]. VEGF-induced Ca2+ oscillations, in turn, promote ECFC proliferation and in vitro tubulogenesis by stimulating the nuclear translocation of the Ca2+-sensitive transcription factor, NF-κB (Fig. 2) [62]. No study has hitherto assessed whether VEGF does stimulate ECFCs, or any other EPC subtype, isolated from peripheral blood of cancer patients. We sought to address this therapeutically relevant issue by focussing on ECFCs deriving from patients early diagnosed with metastatic RCC (mRCC); as a consequence, these subjects were yet to be exposed to any pharmacological treatment. RCC is a chemoresistant and hypervascularised tumour where anti-angiogenic drugs—often administered in sequence because of its peculiar sensitivity to this therapeutic strategy—represent the standard of care for treatment without the need of combination with chemo- or radio-therapy [89, 2]. VEGF signalling in these patients is inhibited by using either humanized monoclonal anti-VEGF antibodies (Bevacizumab [Avastin]) or multi-targeted receptor tyrosine kinase inhibitors (Sunitinib [Sutent], Pazopanib [Votrient], Sorafenib [Nexavar] and Vandetanib [Zactima]). Unfortunately, anti-VEGF drugs do not significantly enhance overall survival (OS) in these patients, who become resistant after a few months free of disease and ultimately dye because of the metastatic relapse [89, 90, 2, 6]. As VEGF is regarded as the most druggable target in mRCC, we expected to record aberrant Ca2+ oscillations in ECFCs isolated from these individuals (mRCC-ECFCs). Unexpectedly, we could not detect any increase in [Ca2+]i in these cells [14]. Additionally, VEGF-induced Ca2+-dependent gene expression is absent in these cells (Fig. 2), despite the fact the VEGFR-2 is normally expressed and auto-phosphorylated upon VEGF binding [20]. Accordingly, we found that ER Ca2+ levels are significantly decreased in mRCC-ECFCs as compared to their healthy counterparts; in addition, InsP3Rs are dramatically down-regulated and InsP3-evoked Ca2+ release is compromised, although it is still coupled to SOCE [14]. It is, therefore, conceivable that the remodelling of the Ca2+ machinery in mRCC-ECFCs prevents the stimulating effect of VEGF. This feature could have a tremendous impact on the therapeutic efficacy of anti-angiogenic treatments. Endothelial cells isolated from the primary tumour are quite sensitive to VEGF, which is required for them to proliferate, survive to pro-apoptotic insults and form capillary-like structures in vitro [91]. Thus, anti-VEGF drugs will cause significant tumour shrinkage at the beginning of the therapy by blocking local angiogenesis; this, however, will rapidly lead to hypoxia-induced secretion of further growth factors and cytokines, e.g. VEGF, bFGF, EGF, SDF-1α and angiopoietins, and recruitment of BMDCs to the collapsing tumour. Herein, ECFCs, and perhaps all the other EPC subgroups, will not be affected by the therapeutic inhibition of VEGFR-2, which does not deliver pro-angiogenic signals to these cells. Consequently, ECFCs will be incorporated within tumour vasculature and fuel the formation of new blood vessels, thereby favouring tumour rebound. This adaptive mechanism would enable the tumour to circumvent the anti-angiogenic strategy by reducing its dependence on VEGF. This scenario is fully compatible with the modes of resistance to anti-angiogenic therapies suggested by Bergers and Hanahan [19], by Carmeliet and coworkers [92] and by Ellis and Hicklin [93]. These results further imply that signalling pathways other than VEGF activate mRCC-ECFCs; such hypothesis is corroborated by the well-known up-regulation of multiple pro-angiogenic factors that may readily substitute for each other in cancer patients. These include the already mentioned bFGF, EGF, angiopoietins, as well as placental growth factor (PGF), osteopontin, granulocyte colony-stimulating factor (G-CSF) and ephrins [92, 19]. Consistently, in a large fraction of kidney cancer patients who developed resistance to sunitinib, the metastatic progression was preceded by an increase in the circulating levels of bFGF, hepatocyte growth factor (HGF) and interleukin-6 (IL-6) [94]. The reduced signalling capability of VEGFR-2 in mRCC-ECFCs is entirely consistent with the model recently put forward by Lyden and coworkers [9, 95], who proposed that cancer cells secrete a multitude of soluble factors to orchestrate a sophisticated network of interactions with tumour microenvironment. This molecular cross-talk educates tumour-associated host cells, including local endothelial cells and BMDCs, to facilitate and support disease progression and metastatisation, while protecting the malignant milieu against intense pharmacologic interventions [9, 95]. For instance, the oncoprotein MET, which functions as tyrosine kinase receptor for HGF, is up-regulated in BMDCs by melanoma-derived exosomes [17], which are small vesicles (40–100 nm) derived from the luminal membrane of late endosomes/multivesicular bodies [9]. This novel mode of horizontal transfer of information between the tumour and its remote targets augments the pro-metastatic behaviour of BMDCs by favouring cancer growth and dissemination [17]. Interestingly, renal cancer stem cells release microvesicles (MVs) containing mRNAs and miRNAs that stimulate angiogenesis and lung metastases in xenografted SCID mice [96]. In agreement with the notion that tumour cells reprogram EPCs towards a more aggressive phenotype, gene expression analysis of FACS-isolated EPCs from tumour samples in Gao’s study [7] disclosed the up-regulation of an array of pro-angiogenic genes, including growth factors, tyrosine kinase receptors, cytokines and ECM modifiers. Moreover, 17-β-estradiol supplementation of ovariectomized mice was found to augment EPC-induced production of angiogenic factors [97]. Finally, genome-wide deep sequencing of siRNAs conducted on FACS-sorted EPCs from xenografted LLC tumours showed the up-regulation of miR-10b and miR-196 [16], which target homeobox D10 (HOXD10), an important regulator of endothelial cell proliferation and angiogenesis [98, 16]. Albeit future studies are mandatory to provide substantial evidence in favour of this hypothesis, we speculate that renal cancer cells hijack the normal signalling pathways recruited downstream of VEGFR-2 in ECFCs within the wider context of their systemic influence on the host organism. This mechanism is likely to contribute to the intrinsic or acquired resistance to anti-VEGF drugs observed in mRCC patients; targeting this interaction is predicted to improve the clinical outcome of these patients.

VEGF fails to activate tumour EPCs. VEGF stimulates pro-angiogenic Ca2+ oscillations in healthy human EPCs, thereby promoting the nuclear translocation of NF-κB and inducing EPC proliferation, tubulogenesis and, perhaps, differentiation. Conversely, VEGF is ineffective on tumour-derived EPCs
Conclusions
The initial enthusiasm raised by pre-clinical investigations on established murine models of different human cancers has not been followed by a successful translation into oncological practice. Anti-VEGF treatment does not produce any objective benefit in a minority of the patients, while in the others, an initial improvement, in the form of tumour shrinkage or stasis, is inevitably followed by disease rebound and progression until patient’s death [19, 93]. The failure of anti-VEGF drugs depends on our poor understanding of tumour vascular biology, as we have recently discussed in [20]. VEGF-dependent angiogenesis does not support neovessel formation in all human cancers, as shown by recent work on GBM and lung adenocarcinoma, which heavily rely on vasculogenic mimicry and vessel cooption, respectively, to gain access to the vascular system [99]. Moreover, it is now evident that tumours exert long-distance systemic effects with the aim to create an environment that supports survival, proliferation and metastatisation of neoplastic cells. Recent evidence provided both by us [15, 14] and by others [16, 9, 17, 7] indicate that BMDCs, including HPCs and ECFCs, are reprogrammed towards a more aggressive and VEGF-independent phenotype. In particular, our data clearly show that ECFCs derived from mRCC patients are insensitive to VEGF, which might explain why current anti-angiogenic treatments either encounter intrinsic refractoriness from the early beginning or rapidly lead to adaptive resistance. Future studies will have to assess whether anti-VEGF drugs are effective in xenograft murine models of kidney cancer injected with normal, but not mRCC-derived, ECFCs. Moreover, it will be important to ascertain whether VEGF fails to activate ECFCs in other types of angiogenic human cancers, such as breast carcinoma. More in general, these data warrant extreme caution when translating data obtained on healthy BMDCs into clinical application. Based on these evidences, it is mandatory to examine whether also tumour patient-derived haematopoietic cells, as well as the other EPC subtypes described in the literature (e.g. CFU-ECs and CACs), become insensitive to VEGF. Gaining access to BMDCs isolated from real patients, rather than relying on murine models that cannot recapitulate the complex biology of human tumours, will likely shed novel light on the mechanisms of resistance to anti-angiogenic treatments and disclose alternative, more realistic targets for a successful therapy.
References
Gao DC, Nolan D, McDonnell K, Vahdat L, Benezra R, Altorki N, et al. Bone marrow-derived endothelial progenitor cells contribute to the angiogenic switch in tumor growth and metastatic progression. Biochim Biophys Acta. 2009;1796(1):33–40. doi:10.1016/j.bbcan.2009.05.001.
Moccia F, Dragoni S, Poletto V, Rosti V, Tanzi F, Ganini C, et al. Orai1 and transient receptor potential channels as novel molecular targets to impair tumor neovascularisation in renal cell carcinoma and other malignancies. Anticancer Agents Med Chem. 2014;14(2):296–312.
Moccia F, Lodola F, Dragoni S, Bonetti E, Bottino C, Guerra G, et al. Ca2+ signalling in endothelial progenitor cells: a novel means to improve cell-based therapy and impair tumour vascularisation. Curr Vasc Pharmacol. 2014;12(1):87–105.
Nolan DJ, Ciarrocchi A, Mellick AS, Jaggi JS, Bambino K, Gupta S, et al. Bone marrow-derived endothelial progenitor cells are a major determinant of nascent tumor neovascularization. Genes Dev. 2007;21(12):1546–58. doi:10.1101/gad.436307.
Coghlin C, Murray GI. Current and emerging concepts in tumour metastasis. J Pathol. 2010;222(1):1–15. doi:10.1002/path.2727.
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307. doi:10.1038/nature10144.
Gao DC, Nolan DJ, Mellick AS, Bambino K, McDonnell K, Mittal V. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science. 2008;319(5860):195–8. doi:10.1126/science.1150224.
Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438(7069):820–7. doi:10.1038/nature04186.
Peinado H, Lavotshkin S, Lyden D. The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol. 2011;21(2):139–46. doi:10.1016/j.semcancer.2011.01.002.
Marçola M, Rodrigues CE. Endothelial progenitor cells in tumor angiogenesis: another brick in the wall. Stem Cells Int. 2015;2015:832649. doi:10.1155/2015/832649.
Moschetta M, Mishima Y, Sahin I, Manier S, Glavey S, Vacca A, et al. Role of endothelial progenitor cells in cancer progression. Biochim Biophys Acta. 2014;1846(1):26–39. doi:10.1016/j.bbcan.2014.03.005.
Shaked Y, Ciarrocchi A, Franco M, Lee CR, Man S, Cheung AM, et al. Therapy-induced acute recruitment of circulating endothelial progenitor cells to tumors. Science. 2006;313(5794):1785–7. doi:10.1126/science.1127592.
Shaked Y, Henke E, Roodhart JM, Mancuso P, Langenberg MH, Colleoni M, et al. Rapid chemotherapy-induced acute endothelial progenitor cell mobilization: implications for antiangiogenic drugs as chemosensitizing agents. Cancer Cell. 2008;14(3):263–73. doi:10.1016/j.ccr.2008.08.001.
Lodola F, Laforenza U, Bonetti E, Lim D, Dragoni S, Bottino C, et al. Store-operated Ca2+ entry is remodelled and controls in vitro angiogenesis in endothelial progenitor cells isolated from tumoral patients. PLoS One. 2012;7(9):e42541. doi:10.1371/journal.pone.0042541.
Dragoni S, Laforenza U, Bonetti E, Reforgiato M, Poletto V, Lodola F, et al. Enhanced expression of Stim, Orai, and TRPC transcripts and proteins in endothelial progenitor cells isolated from patients with primary myelofibrosis. PLoS One. 2014;9(3):e91099. doi:10.1371/journal.pone.0091099.
Plummer PN, Freeman R, Taft RJ, Vider J, Sax M, Umer BA, et al. MicroRNAs regulate tumor angiogenesis modulated by endothelial progenitor cells. Cancer Res. 2013;73(1):341–52. doi:10.1158/0008-5472.CAN-12-0271.
Peinado H, Alečković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18(6):883–91. doi:10.1038/nm.2753.
Bertolini F, Marighetti P, Martin-Padura I, Mancuso P, Hu-Lowe DD, Shaked Y, et al. Anti-VEGF and beyond: shaping a new generation of anti-angiogenic therapies for cancer. Drug Discov Today. 2011;16(23-24):1052–60. doi:10.1016/j.drudis.2011.08.007.
Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603. doi:10.1038/nrc2442.
Moccia F, Poletto V. May the remodelling of the Ca(2+) toolkit in endothelial progenitor cells derived from cancer patients suggest alternative targets for anti-angiogenic treatment? Biochim Biophys Acta. 2014. doi:10.1016/j.bbamcr.2014.10.024.
Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275(5302):964–7.
Prater DN, Case J, Ingram DA, Yoder MC. Working hypothesis to redefine endothelial progenitor cells. Leukemia. 2007;21(6):1141–9. doi:10.1038/sj.leu.2404676.
Case J, Mead LE, Bessler WK, Prater D, White HA, Saadatzadeh MR, et al. Human CD34+AC133+VEGFR-2+ cells are not endothelial progenitor cells but distinct, primitive hematopoietic progenitors. Exp Hematol. 2007;35(7):1109–18. doi:10.1016/j.exphem.2007.04.002.
Yoder MC. Human endothelial progenitor cells. Cold Spring Harb Perspect Med. 2012;2(7):a006692.
Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348(7):593–600. doi:10.1056/NEJMoa022287.
Rehman J, Li J, Orschell CM, March KL. Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation. 2003;107(8):1164–9.
Yoder MC, Mead LE, Prater D, Krier TR, Mroueh KN, Li F, et al. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007;109(5):1801–9. doi:10.1182/blood-2006-08-043471.
Hirschi KK, Ingram DA, Yoder MC. Assessing identity, phenotype, and fate of endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2008;28(9):1584–95. doi:10.1161/ATVBAHA.107.155960.
Kalka C, Masuda H, Takahashi T, Kalka-Moll WM, Silver M, Kearney M, et al. Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc Natl Acad Sci U S A. 2000;97(7):3422–7. doi:10.1073/pnas.070046397.
Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, et al. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001;89(1):E1–7.
Dimmeler S, Aicher A, Vasa M, Mildner-Rihm C, Adler K, Tiemann M, et al. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest. 2001;108(3):391–7. doi:10.1172/JCI13152.
Lin Y, Weisdorf DJ, Solovey A, Hebbel RP. Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest. 2000;105(1):71–7. doi:10.1172/JCI8071.
Ingram DA, Mead LE, Tanaka H, Meade V, Fenoglio A, Mortell K, et al. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood. 2004;104(9):2752–60.
Moccia F, Bonetti E, Dragoni S, Fontana J, Lodola F, Romani RB, et al. Hematopoietic progenitor and stem cells circulate by surfing on intracellular Ca2+ waves: a novel target for cell-based therapy and anti-cancer treatment? Curr Signal Transduction Ther. 2012;7(2):161–76.
de la Puente P, Muz B, Azab F, Azab AK. Cell trafficking of endothelial progenitor cells in tumor progression. Clin Cancer Res. 2013;19(13):3360–8. doi:10.1158/1078-0432.CCR-13-0462.
Tzeng HE, Chen PC, Lin KW, Lin CY, Tsai CH, Han SM, et al. Basic fibroblast growth factor induces VEGF expression in chondrosarcoma cells and subsequently promotes endothelial progenitor cell-primed angiogenesis. Clin Sci (Lond). 2015;129(2):147–58. doi:10.1042/CS20140390.
Patenaude A, Parker J, Karsan A. Involvement of endothelial progenitor cells in tumor vascularization. Microvasc Res. 2010;79(3):217–23. doi:10.1016/j.mvr.2010.01.007.
Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7(11):1194–201. doi:10.1038/nm1101-1194.
Nair R, Teo WS, Mittal V, Swarbrick A. ID proteins regulate diverse aspects of cancer progression and provide novel therapeutic opportunities. Mol Ther. 2014;22(8):1407–15. doi:10.1038/mt.2014.83.
Mellick AS, Plummer PN, Nolan DJ, Gao D, Bambino K, Hahn M, et al. Using the transcription factor inhibitor of DNA binding 1 to selectively target endothelial progenitor cells offers novel strategies to inhibit tumor angiogenesis and growth. Cancer Res. 2010;70(18):7273–82. doi:10.1158/0008-5472.CAN-10-1142.
Peters BA, Diaz LA, Polyak K, Meszler L, Romans K, Guinan EC, et al. Contribution of bone marrow-derived endothelial cells to human tumor vasculature. Nat Med. 2005;11(3):261–2. doi:10.1038/nm1200.
Asahara T, Takahashi T, Masuda H, Kalka C, Chen D, Iwaguro H, et al. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J. 1999;18(14):3964–72. doi:10.1093/emboj/18.14.3964.
Jung SY, Choi JH, Kwon SM, Masuda H, Asahara T, Lee YM. Decursin inhibits vasculogenesis in early tumor progression by suppression of endothelial progenitor cell differentiation and function. J Cell Biochem. 2012;113(5):1478–87. doi:10.1002/jcb.24085.
Zhu H, Shao Q, Sun X, Deng Z, Yuan X, Yu D, et al. The mobilization, recruitment and contribution of bone marrow-derived endothelial progenitor cells to the tumor neovascularization occur at an early stage and throughout the entire process of hepatocellular carcinoma growth. Oncol Rep. 2012;28(4):1217–24. doi:10.3892/or.2012.1944.
Ahn JB, Rha SY, Shin SJ, Jeung HC, Kim TS, Zhang X, et al. Circulating endothelial progenitor cells (EPC) for tumor vasculogenesis in gastric cancer patients. Cancer Lett. 2010;288(1):124–32. doi:10.1016/j.canlet.2009.06.031.
Yu P, Ge YZ, Zhao Y, Wu JP, Wu R, Zhou LH, et al. Identification and significance of mobilized endothelial progenitor cells in tumor neovascularization of renal cell carcinoma. Tumour Biol. 2014;35(9):9331–41. doi:10.1007/s13277-014-2205-5.
Wei J, Jarmy G, Genuneit J, Debatin KM, Beltinger C. Human blood late outgrowth endothelial cells for gene therapy of cancer: determinants of efficacy. Gene Ther. 2007;14(4):344–56. doi:10.1038/sj.gt.3302860.
Bieback K, Vinci M, Elvers-Hornung S, Bartol A, Gloe T, Czabanka M, et al. Recruitment of human cord blood-derived endothelial colony-forming cells to sites of tumor angiogenesis. Cytotherapy. 2013;15(6):726–39. doi:10.1016/j.jcyt.2013.01.215.
Moccia F, Tanzi F, Munaron L. Endothelial remodelling and intracellular calcium machinery. Curr Mol Med. 2014;14(4):457–80.
Yoder MC, Ingram DA. The definition of EPCs and other bone marrow cells contributing to neoangiogenesis and tumor growth: is there common ground for understanding the roles of numerous marrow-derived cells in the neoangiogenic process? Biochim Biophys Acta. 2009;1796(1):50–4. doi:10.1016/j.bbcan.2009.04.002.
De Palma M, Venneri MA, Roca C, Naldini L. Targeting exogenous genes to tumor angiogenesis by transplantation of genetically modified hematopoietic stem cells. Nat Med. 2003;9(6):789–95. doi:10.1038/nm871.
Göthert JR, Gustin SE, van Eekelen JA, Schmidt U, Hall MA, Jane SM, et al. Genetically tagging endothelial cells in vivo: bone marrow-derived cells do not contribute to tumor endothelium. Blood. 2004;104(6):1769–77. doi:10.1182/blood-2003-11-3952.
Wickersheim A, Kerber M, de Miguel LS, Plate KH, Machein MR. Endothelial progenitor cells do not contribute to tumor endothelium in primary and metastatic tumors. Int J Cancer. 2009;125(8):1771–7. doi:10.1002/ijc.24605.
Machein MR, Renninger S, de Lima-Hahn E, Plate KH. Minor contribution of bone marrow-derived endothelial progenitors to the vascularization of murine gliomas. Brain Pathol. 2003;13(4):582–97.
Purhonen S, Palm J, Rossi D, Kaskenpää N, Rajantie I, Ylä-Herttuala S, et al. Bone marrow-derived circulating endothelial precursors do not contribute to vascular endothelium and are not needed for tumor growth. Proc Natl Acad Sci U S A. 2008;105(18):6620–5. doi:10.1073/pnas.0710516105.
Rajantie I, Ilmonen M, Alminaite A, Ozerdem U, Alitalo K, Salven P. Adult bone marrow-derived cells recruited during angiogenesis comprise precursors for periendothelial vascular mural cells. Blood. 2004;104(7):2084–6. doi:10.1182/blood-2004-01-0336.
Duda DG, Cohen KS, Kozin SV, Perentes JY, Fukumura D, Scadden DT, et al. Evidence for incorporation of bone marrow-derived endothelial cells into perfused blood vessels in tumors. Blood. 2006;107(7):2774–6. doi:10.1182/blood-2005-08-3210.
Spring H, Schüler T, Arnold B, Hämmerling GJ, Ganss R. Chemokines direct endothelial progenitors into tumor neovessels. Proc Natl Acad Sci U S A. 2005;102(50):18111–6. doi:10.1073/pnas.0507158102.
Ruzinova MB, Schoer RA, Gerald W, Egan JE, Pandolfi PP, Rafii S, et al. Effect of angiogenesis inhibition by Id loss and the contribution of bone-marrow-derived endothelial cells in spontaneous murine tumors. Cancer Cell. 2003;4(4):277–89.
Basile DP, Yoder MC. Circulating and tissue resident endothelial progenitor cells. J Cell Physiol. 2014;229(1):10–6. doi:10.1002/jcp.24423.
Li H, Gerald WL, Benezra R. Utilization of bone marrow-derived endothelial cell precursors in spontaneous prostate tumors varies with tumor grade. Cancer Res. 2004;64(17):6137–43. doi:10.1158/0008-5472.CAN-04-1287.
Dragoni S, Laforenza U, Bonetti E, Lodola F, Bottino C, Berra-Romani R, et al. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells. 2011;29(11):1898–907. doi:10.1002/stem.734.
Dragoni S, Laforenza U, Bonetti E, Lodola F, Bottino C, Guerra G, et al. Canonical transient receptor potential 3 channel triggers vascular endothelial growth factor-induced intracellular Ca2+ oscillations in endothelial progenitor cells isolated from umbilical cord blood. Stem Cells Dev. 2013;22(19):2561–80. doi:10.1089/scd.2013.0032.
Song Y, Li X, Wang D, Fu C, Zhu Z, Zou MH, et al. Transcription factor Krüppel-like factor 2 plays a vital role in endothelial colony forming cells differentiation. Cardiovasc Res. 2013;99(3):514–24. doi:10.1093/cvr/cvt113.
Yu D, Chen W, Ren J, Zhang T, Yang K, Wu G, et al. VEGF-PKD1-HDAC7 signaling promotes endothelial progenitor cell migration and tube formation. Microvasc Res. 2014;91:66–72. doi:10.1016/j.mvr.2013.10.006.
Ben-Shoshan J, George J. Endothelial progenitor cells as therapeutic vectors in cardiovascular disorders: from experimental models to human trials. Pharmacol Ther. 2007;115(1):25–36. doi:10.1016/j.pharmthera.2007.03.012.
Roncalli JG, Tongers J, Renault MA, Losordo DW. Endothelial progenitor cells in regenerative medicine and cancer: a decade of research. Trends Biotechnol. 2008;26(5):276–83. doi:10.1016/j.tibtech.2008.01.005.
Zampetaki A, Kirton JP, Xu Q. Vascular repair by endothelial progenitor cells. Cardiovasc Res. 2008;78(3):413–21. doi:10.1093/cvr/cvn081.
Koch S, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med. 2012;2(7):a006502. doi:10.1101/cshperspect.a006502.
Potenza DM, Guerra G, Avanzato D, Poletto V, Pareek S, Guido D, et al. Hydrogen sulphide triggers VEGF-induced intracellular Ca(2+) signals in human endothelial cells but not in their immature progenitors. Cell Calcium. 2014. doi:10.1016/j.ceca.2014.07.010.
Beaudry P, Force J, Naumov GN, Wang A, Baker CH, Ryan A, et al. Differential effects of vascular endothelial growth factor receptor-2 inhibitor ZD6474 on circulating endothelial progenitors and mature circulating endothelial cells: implications for use as a surrogate marker of antiangiogenic activity. Clin Cancer Res. 2005;11(9):3514–22. doi:10.1158/1078-0432.CCR-04-2271.
Taylor M, Billiot F, Marty V, Rouffiac V, Cohen P, Tournay E, et al. Reversing resistance to vascular-disrupting agents by blocking late mobilization of circulating endothelial progenitor cells. Cancer Discov. 2012;2(5):434–49. doi:10.1158/2159-8290.CD-11-0171.
Clapham DE. Calcium signaling. Cell. 2007;131(6):1047–58. doi:10.1016/j.cell.2007.11.028.
Parekh AB. Decoding cytosolic Ca2+ oscillations. Trends Biochem Sci. 2011;36(2):78–87. doi:10.1016/j.tibs.2010.07.013.
Moccia F, Berra-Romani R, Tanzi F. Update on vascular endothelial Ca2+ signalling: a tale of ion channels, pumps and transporters. World J Biol Chem. 2012;3(7):127–58. doi:10.4331/wjbc.v3.i7.127.
Moccia F, Berra-Romani R, Tanzi F. Ca2+ signalling in damaged endothelium and arterial remodelling: Do connexin hemichannels provide a suitable target to prevent in-stent restenosis? Curr Drug Ther. 2012;7(4):268–80. doi:10.2174/157488512804999109.
Munaron L, Pla AF. Endothelial calcium machinery and angiogenesis: understanding physiology to interfere with pathology. Curr Med Chem. 2009;16(35):4691–703.
Fiorio Pla A, Gkika D. Emerging role of TRP channels in cell migration: from tumor vascularization to metastasis. Front Physiol. 2013;4:311. doi:10.3389/fphys.2013.00311.
Ivanova H, Vervliet T, Missiaen L, Parys JB, De Smedt H, Bultynck G. Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim Biophys Acta. 2014. doi:10.1016/j.bbamcr.2014.03.007.
Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4(7):517–29. doi:10.1038/nrm1155.
Moccia F, Berra-Romani R, Tritto S, Signorelli S, Taglietti V, Tanzi F. Epidermal growth factor induces intracellular Ca2+ oscillations in microvascular endothelial cells. J Cell Physiol. 2003;194:139–50. doi:10.1002/jcp.10198.
Berra-Romani R, Raqeeb A, Torres-Jácome J, Guzman-Silva A, Guerra G, Tanzi F, et al. The mechanism of injury-induced intracellular calcium concentration oscillations in the endothelium of excised rat aorta. J Vasc Res. 2012;49(1):65–76. doi:10.1159/000329618.
Zhu LP, Luo YG, Chen TX, Chen FR, Wang T, Hu Q. Ca2+ oscillation frequency regulates agonist-stimulated gene expression in vascular endothelial cells. J Cell Sci. 2008;121(15):2511–8. doi:10.1242/jcs.031997.
Moccia F, Dragoni S, Cinelli M, Montagnani S, Amato B, Rosti V, et al. How to utilize Ca2+ signals to rejuvenate the repairative phenotype of senescent endothelial progenitor cells in elderly patients affected by cardiovascular diseases: a useful therapeutic support of surgical approach? BMC Surg. 2013;13 Suppl 2:S46. doi:10.1186/1471-2482-13-S2-S46.
Moccia F, Dragoni S, Lodola F, Bonetti E, Bottino C, Guerra G, et al. Store-dependent Ca(2+) entry in endothelial progenitor cells as a perspective tool to enhance cell-based therapy and adverse tumour vascularization. Curr Med Chem. 2012;19(34):5802–18.
Li J, Cubbon RM, Wilson LA, Amer MS, McKeown L, Hou B, et al. Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ Res. 2011;108(10):1190–8. doi:10.1161/circresaha.111.243352.
Abdullaev IF, Bisaillon JM, Potier M, Gonzalez JC, Motiani RK, Trebak M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ Res. 2008;103(11):1289–99. doi:10.1161/01.res.0000338496.95579.56.
Dragoni S, Guerra G, Fiorio Pla A, Bertoni G, Rappa A, Poletto V, et al. A functional transient receptor potential vanilloid 4 (TRPV4) channel is expressed in human endothelial progenitor cells. J Cell Physiol. 2015;230(1):95–104. doi:10.1002/jcp.24686.
Escudier B, Szczylik C, Porta C, Gore M. Treatment selection in metastatic renal cell carcinoma: expert consensus. Nat Rev Clin Oncol. 2012;9(6):327–37. doi:10.1038/nrclinonc.2012.59.
Porta C, Paglino C, Imarisio I, Canipari C, Chen K, Neary M et al. Safety and treatment patterns of multikinase inhibitors in patients with metastatic renal cell carcinoma at a tertiary oncology center in Italy. BMC Cancer. 2011;11. doi::10.1186/1471-2407-11-105.
Bussolati B, Deambrosis I, Russo S, Deregibus MC, Camussi G. Altered angiogenesis and survival in human tumor-derived endothelial cells. FASEB J. 2003;17(9):1159–61. doi:10.1096/fj.02-0557fje.
Loges S, Schmidt T, Carmeliet P. Mechanisms of resistance to anti-angiogenic therapy and development of third-generation anti-angiogenic drug candidates. Genes Cancer. 2010;1(1):12–25. doi:10.1177/1947601909356574.
Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8(8):579–91. doi:10.1038/nrc2403.
Porta C, Paglino C, Imarisio I, Ganini C, Sacchi L, Quaglini S, et al. Changes in circulating pro-angiogenic cytokines, other than VEGF, before progression to sunitinib therapy in advanced renal cell carcinoma patients. Oncology. 2013;84(2):115–22. doi:10.1159/000342099.
Barcellos-Hoff MH, Lyden D, Wang TC. The evolution of the cancer niche during multistage carcinogenesis. Nat Rev Cancer. 2013;13(7):511–8. doi:10.1038/nrc3536.
Grange C, Tapparo M, Collino F, Vitillo L, Damasco C, Deregibus MC, et al. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res. 2011;71(15):5346–56. doi:10.1158/0008-5472.CAN-11-0241.
Suriano R, Chaudhuri D, Johnson RS, Lambers E, Ashok BT, Kishore R, et al. 17Beta-estradiol mobilizes bone marrow-derived endothelial progenitor cells to tumors. Cancer Res. 2008;68(15):6038–42. doi:10.1158/0008-5472.CAN-08-1009.
Myers C, Charboneau A, Cheung I, Hanks D, Boudreau N. Sustained expression of homeobox D10 inhibits angiogenesis. Am J Pathol. 2002;161(6):2099–109. doi:10.1016/S0002-9440(10)64488-4.
Vasudev NS, Reynolds AR. Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions. Angiogenesis. 2014;17(3):471–94. doi:10.1007/s10456-014-9420-y.
Garcia-Barros M, Paris F, Cordon-Cardo C, Lyden D, Rafii S, Haimovitz-Friedman A, Fuks Z, Kolesnick R. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300:1155–9.
Acknowledgments
This work was supported by a grant from the Cariplo Foundation [2010-0807] and by a grant from the Associazione Italiana per la Ricerca sul Cancro (AIRC; Milan, Italy), Special Program Molecular Clinical Oncology 5x1000 to AIRC-Gruppo Italiano Malattie Mieloproliferative (AGIMM) project #1005, both awarded to VR.
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Additional information
Mariapia Cinelli passed away during the preparation of this manuscript.
Rights and permissions
About this article

Cite this article
Moccia, F., Zuccolo, E., Poletto, V. et al. Endothelial progenitor cells support tumour growth and metastatisation: implications for the resistance to anti-angiogenic therapy. Tumor Biol. 36, 6603–6614 (2015). https://doi.org/10.1007/s13277-015-3823-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-3823-2