
Abstract
Hepatocellular carcinoma (HCC) has very poor prognosis. Astemizole has gained great interest as a potential anticancer drug because it targets several proteins involved in cancer including the Eag1 (ether à-go-go-1) potassium channel that is overexpressed in human HCC. Eag1 channels are regulated by cancer etiological factors and have been proposed as early tumor markers. Here, we found that HepG2 and HuH-7 HCC cells displayed Eag1 messenger RNA (mRNA) and protein expression, determined by real-time RT-PCR and immunochemistry, respectively. Astemizole inhibited human HCC cell proliferation (assessed by metabolic activity assay) and induced apoptosis (studied with flow cytometry) in both cell lines. The subcellular Eag1 protein localization was modified by astemizole in the HepG2 cells. The treatment with astemizole prevented diethylnitrosamine (DEN)-induced rat HCC development in vivo (followed by studying γ-glutamyl transpeptidase (GGT) activity). The Eag1 mRNA and protein levels were increased in most DEN-treated groups but decreased after astemizole treatment. GGT activity was decreased by astemizole. The Eag1 protein was detected in cirrhotic and dysplastic rat livers. Astemizole might have clinical utility for HCC prevention and treatment, and Eag1 channels may be potential early HCC biomarkers. These data provide significant basis to include astemizole in HCC clinical trials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary liver cancer is the sixth most common cancer worldwide [1] and the third most common cause of cancer mortality [1, 2]. Hepatocellular carcinoma (HCC) accounts for up to 90 % of primary liver cancers [3]. The major risk factors associated with the development of HCC are chronic infection with hepatitis B or C virus and cirrhosis [3, 4]. HCC has a poor prognosis and is often not detected until advanced stages, at which point patients do not respond to the existing chemotherapy and only palliative treatments are provided [5]. Thus, alternative tumor markers and therapeutic options for HCC patients are urgently needed.
Astemizole is an antihistamine and has been suggested as an anticancer treatment [6–15]. Interestingly, astemizole targets different proteins involved in cancer, including H1 histamine receptors, P-glycoproteins, and potassium channels [6, 7, 10–16]. Some H1 and H2 histamine receptor antagonists inhibit the proliferative effect of histamine in malignant cells [8], and astemizole greatly potentiates the growth inhibitory activity of doxorubicin in doxorubicin-resistant human leukemia cells by inhibiting P-glycoprotein [16]. More recently, the inhibitory effect of astemizole on cancer cells has also been attributed to its blocking effects on potassium channels of the ether à-go-go family [7, 9, 12, 15].
Ether à-go-go-1 (Eag1, KCNH1, Kv10.1) is a voltage-dependent potassium channel that displays oncogenic properties and has gained enormous interest in cancer research [13, 15, 17, 18]. Eag1 transcripts are normally detected in healthy brain, but low amounts can be detected in placenta, testes, and adrenal glands and transiently in myoblasts [17, 19, 20]. In contrast, Eag1 is overexpressed in most human tumors, including liver, cervical, lung, breast, colon, and prostate cancer [20–22]. Moreover, Eag1 is expressed in several human tumor cell lines, including those from breast, lung, cervical, gastric, and ovarian cancer, as well as neuroblastoma, melanoma, and leukemia [11, 17, 22–27]. The Eag1 channels have been proposed as early tumor biomarkers and therapeutic targets for different types of tumors [11, 20–22, 28, 29]. Inhibition of either Eag1 gene expression or channel activity with antisense oligonucleotides, siRNA, monoclonal antibodies, or nonspecific Eag1 channel inhibitors reduces tumor cell proliferation in vitro and in vivo [10, 11, 13, 14, 17, 23, 25, 28]. Astemizole inhibits Eag1 channels and decreases cell proliferation of Eag1-expressing tumor cells; it is hypothesized that this inhibition may be due to astemizole’s blockage of the Eag1 channel [10–12]. In vivo studies showed that astemizole administration reduced the growth rate of xenograft tumors in mice implanted with Eag1-expressing cells [12]. Thus, Eag1 channels are inhibited by astemizole and may be potential targets for cancer therapy [7, 13, 15].
Due to the poor prognosis of HCC, we examined if astemizole inhibits the proliferation of human HCC cells in vitro and tumor development in vivo. We also investigated if Eag1 channels could serve as early markers for HCC by studying Eag1 expression during tumor development in vivo.
Material and methods
Cell line
The human liver cancer HepG2 and HuH-7 cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and cultured according to manufacturer’s instructions. This cell line was authenticated at the Laboratory for Genomic Diagnosis (National Institute of Genomic Medicine, Mexico) by multiplex PCR and capillary electrophoresis. Short tandem repeats (STRs) of several markers were studied. The gene profile obtained shared the alleles reported by ATCC for the human cell lines HepG2 and HuH-7 and was not contaminated with other cells.
Metabolic activity and apoptosis assays
Cell proliferation (assessed by metabolic activity) and apoptosis were studied as previously described [11]. Cell proliferation was assayed by a colorimetric method with the [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell proliferation kit I, Boehringer Mannheim GmbH]. Cells (3000 cells per well) were seeded in 96-well plates and incubated for 48, 72, or 96 h in culture medium either alone or in the presence of astemizole (2 and 5 μmol/L) (kindly provided by Liomont Laboratories Mexico), imipramine (10 μmol/L), or DMSO as vehicle (reagents were purchased from Sigma Chemical Co., St. Louis, MO, USA). MTT (0.5 mg/mL) was added 4 h before completing the whole incubation time. Absorbance data were obtained with a microplate photometer (Sunrise Touchscreen). Apoptosis was determined with the Annexin V-FITC kit (Invitrogen Co.) binding to phosphatidylserine and DNA staining by propidium iodide (PI). Cells (40,000 cells per dish) were seeded and incubated for 48, 72, or 96 h. Camptothecin (which induces apoptosis) and methanol (which induces necrosis) were used as controls. Experiments were carried out with the flow cytometer CYAN ADP (DAKO). Percentages of viable (FITC and PI negative), apoptotic (FITC positive and PI negative), and late apoptotic (FITC and PI positive) cells were obtained by quadrant analysis using the Summit 4.3 software.
Animal experimentation
Diethylnitrosamine (DEN) was used to induce liver injury in rats (Fig. 1). This model was chosen because it recapitulates the sequential change from cirrhosis to HCC, as it occurs in humans. Animal care and procedures conformed to the Institutional Animal Care and Use Committee Guidelines. All animals received humane care. Rats were fed a Purina chow rat diet and water ad libitum. Figure 1 schematically shows the different groups studied and the treatments administered. Male Wistar rats weighing 200 g (UPEAL-Cinvestav, Mexico) received intraperitoneal injections of DEN (50 mg/kg) (Sigma Chemical Co., St. Louis, MO, USA) once a week for either 12 or 16 weeks, following the procedure described by Schiffer et al. [30]. Oral administration of astemizole (50 mg/kg) was given daily for 4, 6, 12, or 18 weeks. The control group received PBS. Animals were weighed weekly. At the end of treatments, animals were sacrificed by decapitation. Livers were rapidly removed, weighed, and photographed. Liver tissues were frozen and stored at −80 °C and homogenized in TRIzol or fixed in a buffered 4 % formaldehyde solution.

Dosage regimen and groups studied in the HCC in vivo model. Group A received only the vehicle PBS. DEN was administered for either 12 (groups B and C) or 16 weeks (groups D–I). Astemizole was administered either from the beginning (groups C and F), from week 12 (group G), or after DEN treatment (group I)
Histology and immunochemistry
Representative 20-μm thick sections from frozen liver slices were stained for γ-glutamyl transpeptidase (GGT) activity, as previously described [31]. Image analysis to determine the percentage of GGT-positive tissue was performed with the ImageJ software 1.45 g (NIH, USA). Liver tissue was prepared for histology and embedded in paraffin. Immunohistochemistry was performed as previously described [11, 29]. The anti-Eag1 monoclonal antibody was used at a 1:500 dilution (Novus International Inc., USA) overnight at −4 °C. The specific staining reaction was completed by incubating the slides in the presence of 3,3′-diaminobenzidine in buffer reaction solution (Bio SB) and observed as a brown staining. Sections were counterstained with hematoxylin (Dako, Denmark). HepG2 cells were seeded on charged glass slides, boiled for antigen retrieval, and processed as above described for tissues. The slides were observed using an Olympus IX51 microscope, and photos were taken with an Olympus DP70 camera.
Real-time RT-PCR
Total RNA was extracted from either liver tissue homogenates or cell cultures with TRIzol reagent. Five micrograms of total RNA was reverse-transcribed using the Moloney Murine Leukemia Virus Reverse transcriptase (M-MuLV) (BioLabs Inc.). Real-time PCR was performed with 2 μL of cDNA using the TaqMan™ detection system (Applied Biosystem, Foster City, CA, USA) and the Universal PCR Master Mix reagents kit (Life Technologies). The following primer sequences were used: GATCTTCGCCGTAGCCATCAT (forward), TCCCAAAGATGGTGGCATACAG (reverse), probe: AAGGGAGCCAATCATC for rat-Eag1, and GTCCATTCCTATGACTGTAGATTTTATCAGA (forward), AATAACTTTTATGTCCCCCGTTGACT (reverse), probe: CTGAAGAGCTACTGTAATGAC for rat hypoxanthine-guanine phosphoribosyl transferase (HPRT) which was used as constitutive gene. For human genes, the following primer sequences were used: GATCTTTGCAGTGGCCATCATG (forward), CATTCCCGAAGATGGTGGCATA (reverse), probe: ATGATTGGCTCACTTCTC for Eag1 while the predeveloped Taq Man Assays Reagents h-HPRT Part Number 4326321E was used for HPRT. The PCR reaction protocol was 95 °C for 15 s and 60 °C for 1 min (40 cycles). The 2−∆∆Ct method was used to analyze the Ct ss.
Statistical analysis
Statistical analysis was performed with either ANOVA followed by Tukey-Kramer test or Student’s t test using GraphPad Prism software version 5.0. P values <0.05 were considered to be statistically significant.
Results
Astemizole decreases proliferation and induces apoptosis in Eag1-expressing human liver cancer cells
First, we looked for Eag1 expression in the human HCC cell lines HepG2 and HuH-7. Real-time PCR experiments showed that both cell lines express Eag1 mRNA (data not shown). In accordance, immunocytochemistry assays revealed the presence of the Eag1 protein in both cell lines (Fig. 2a, f). The Eag1 protein was localized in the cytoplasm in most of the cells but perinuclear membrane staining was also observed. Thus, we wondered whether two widely used nonspecific Eag1 inhibitors, astemizole and imipramine, might affect the proliferation and apoptosis of HepG2 cells. Incubation of HepG2 cells with astemizole (5 μM) for 48 or 72 h decreased cell proliferation by 20 %, while incubation for 72 h increased apoptosis (data not shown). However, astemizole decreased HepG2 cell proliferation in up to 50 % (Fig. 2d), and imipramine showed a modest but statistically significant effect at 96 h of incubation with the drug. Both astemizole and imipramine had pronounced effects on apoptosis in HepG2 cells (Fig. 2e). In the case of HUH-7 cells, astemizole (5 μM) decreased cell proliferation by 50 % and increased apoptosis after 72 h of treatment (data not shown). Interestingly, astemizole (5 μM) decreased HuH-7 cell proliferation in more than 60 % (Fig. 2i) and increased apoptosis more than tenfold (Fig. 2j) at 96 h of treatment. Eag1 mRNA levels were not detected in the presence of astemizole (data not shown). Interestingly, the localization of the Eag1 protein was mainly cytoplasmic in HepG2 cells incubated with 2 μM astemizole (Fig. 2b) but mainly perinuclear with 5 μM of astemizole (Fig. 2c). The localization of the protein was more homogeneous in the absence or presence of astemizole in the HuH-7 cells. The remarkable effects of astemizole in vitro prompted us to study its effects in vivo.

Astemizole decreases proliferation and induces apoptosis in human HCC cells. Eag1 protein expression was detected mainly in the cytoplasm of human HCC HepG2 and HuH-7 cells (a, f, respectively, brown immunostaining). No signal was observed in the absence of the primary antibody (data not shown). Astemizole treatment (2 μM) preserved mainly cytoplasmic Eag1 protein expression (b) but 5 μM of astemizole induced mainly perinuclear protein localization in HepG2 cells (c). Protein localization was not changed by astemizole in HuH-7 cells (g, h). Cell proliferation (assayed by metabolic activity) was decreased by 50 % and more than 60 % due to astemizole in HepG2 and HuH-7 cells, respectively (d, i). Astemizole increased apoptosis several fold in both cell lines (e, j). A representative flow cytometry plot for each cell type incubated with astemizole (5 μM) is shown in the middle. The experiments were performed three times using sextuplicates (cell proliferation assay) or triplicates (apoptosis) each time. *p < 0.05 versus control. Magnification ×400×
Preventive and therapeutic effects of astemizole in a HCC in vivo model
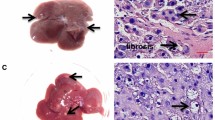
We examined the potential preventive and/or therapeutic effect of astemizole on HCC development in rats. Tumor development was followed by histopathology and by quantitation of GGT activity, a commonly used liver tumor marker. The histological changes observed for each group are summarized in Table 1. Macroscopically, the vehicle-treated group showed normal liver architecture (Fig. 3a and Table 1), whereas DEN treatment clearly induced the appearance of cirrhosis and macroscopic nodules (Fig. 3b, d, e, h and Table 1). The potential preventive effect of astemizole was studied by administering the drug to DEN-treated rats either from the beginning (day 1) or from week 12 of DEN treatment. Astemizole clearly improved liver appearance and histological characteristics in all groups (Fig. 3c, f, g and Table 1). However, the most dramatic preventive effect of the drug was observed in the group treated with DEN for 16 weeks then treated for an additional 2 weeks with no DEN plus astemizole from day 1 (Fig. 3f), compared with the corresponding group without astemizole treatment (Fig. 3e) in which we can observe two representative livers from different rats with numerous neoplastic nodules and necrotic regions. Importantly, astemizole, given simultaneously with DEN from the initiation of treatment, almost completely preserved the normal liver appearance (Fig. 3f). Astemizole also had a very important protective effect when given from week 12 in this group (Fig. 3g).

In vivo chemopreventive and antitumoral effects of astemizole on HCC. Representative livers are shown for the indicated groups (a–i). Two livers from different animals are shown for (e, h) to emphasize the dramatic effect of astemizole in these groups (f, i, respectively). j Percentage of GGT-positive tissue (from the groups from a to i); this liver tumor marker was increased in DEN-treated alone groups and decreased in DEN + astemizole-treated groups. Ast astemizole, wk weeks. *p < 0.05 versus vehicle (a), **p < 0.05 versus DEN 16 weeks + 2 weeks DEN-free (e). Bars represent mean ± s.d. n = 4–8 rats in each group
To study the potential antineoplastic effect of astemizole, the control group was treated with DEN for 16 weeks and untreated for an additional 6 weeks (Fig. 3h, two livers from different rats are shown). Astemizole was administered for 4 weeks after the 16 weeks with DEN plus 2 weeks DEN-free treatment; both groups were studied for a total of 22 weeks. While rats not receiving astemizole developed large tumors, astemizole treatment clearly prevented the appearance of large tumors (Fig. 3h, i, respectively). As expected, GGT activity in liver sections increased during HCC development (DEN-treated rats). Notably, astemizole treatment significantly decreased the presence of GGT-positive lesions especially when administered from the beginning of the study to rats treated with DEN for 16 weeks plus 2 weeks DEN-free (Fig. 3j). It is also worth mentioning that in the group receiving astemizole for 4 weeks after the complete DEN treatment (Fig. 3i, j), the histological appearance was improved and GGT activity was lower than in the group treated for 16 weeks with DEN + 2 weeks DEN-free but that was not treated with astemizole (Fig. 3e, j and Table 1). Thus, astemizole might also be used to prevent the growth of already established liver tumors.
Eag1 channels as potential early markers of HCC
Because of the clear effects of astemizole on HCC prevention, we determined if Eag1 protein could be expressed at early stages of HCC. Eag1 mRNA expression showed an increasing trend during HCC development in most of the DEN-treated groups (Fig. 4a). Eag1 mRNA reached its highest level in the 16 weeks plus 2 weeks DEN-free group; interestingly, Eag1 mRNA expression was lower in the corresponding group treated with astemizole from week 12 (Fig. 4a). Weak Eag1 protein expression was observed in the vehicle-treated group (Fig. 4b) while strong Eag1 protein expression was found in livers with tumors, cirrhosis, or preneoplastic lesions (brown immunostaining in Fig. 4c, e and Table 2). Eag1 protein expression was lower in the corresponding groups treated with astemizole (Fig. 4d, f and Table 2). Interestingly, Eag1 subcellular localization in the vehicle-treated groups was only observed in the cytoplasm and plasma membrane, while Eag1 protein expression in most of the DEN-treated groups was also found in the nucleus. Intriguingly, Eag1 protein expression was almost lost in livers displaying HCC (Fig. 4g) but was slightly higher in the corresponding group treated with astemizole (Fig. 4h and Table 2). These results suggest Eag1 protein channels are potential early markers of HCC.

Eag1 as a potential HCC early biomarker. Real-time PCR and immunohistochemistry experiments were preformed to study Eag1 mRNA and protein expression, respectively, in the in vivo HCC model. a Relative Eag1 mRNA expression was increased in most DEN-treated groups. This increase was partially prevented in some groups treated with astemizole. b–h Eag1 immunohistochemistry; b vehicle-treated group showed weak Eag1 expression (membrane and cytoplasm); c DEN 12 week group showed strong Eag1 expression (cytoplasm and nucleus), but Eag1 expression was lower when astemizole was administered from day 1 (d). e Strong Eag1 expression was found in the cytoplasm and nucleus, but the expression was also lower when astemizole was administered from week 12 (f). g No Eag1 expression was found under the indicated condition, but Eag1 expression was detected in the corresponding group treated with astemizole (h). *p < 0.05 versus vehicle, **p < 0.05 versus DEN 16 weeks + 2 weeks DEN-free. Bars represent mean ± s.d. n = 4–8 rats in each group
Discussion
HCC is a leading cause of cancer mortality and has a very poor prognosis. Our results strongly suggest that the antihistamine astemizole might have clinical utility as a novel therapeutic approach for HCC treatment and prevention. In addition, our findings in rat liver tissues suggest that the Eag1 protein may serve as a potential early marker of HCC.
Identifying novel antitumor properties in “old” drugs may offer a viable and cost-effective alternative to fight cancer. For instance, thalidomide had been withdrawn from the market due to its teratogenic effects but has gained great relevance in recent years because of its potential anticancer effect in multiple myeloma [32]. Likewise, the old antihistamine astemizole was withdrawn from the market in some countries due to its adverse effects on cardiac function but has gained great interest as a novel anticancer drug [6–15]. Astemizole is very attractive as a potential anticancer molecule because it targets several proteins involved in cancer, including H1 histamine receptors, P-glycoproteins, and potassium channels [6, 7, 10–16].
Here, we show that astemizole inhibits proliferation and increases apoptosis of human HepG2 and HuH-7 HCC cells. Although astemizole has different targets, this drug’s inhibition of Eag1 channels is a potential mechanism of its antiproliferative and proapoptotic effect on human HCC cells, as Eag1 mRNA and protein are expressed in these cells. Interestingly, the treatment with astemizole modified the protein localization in HepG2 cells from mainly cytoplasmic to mainly perinuclear. This nuclear Eag1 protein localization has been reported for other cancer cell types and is expected because of the nuclear localization signal found in the Eag1 channel [11–14, 20, 33]. Actually, ionic currents resembling Eag1 channel activity in the nuclear membrane have been already reported and a potential interaction between Eag1 and the heterochromatin has been suggested [34]. However, the mechanism of Eag1 translocation after astemizole treatment and the precise role of Eag1 under these conditions remain elusive. Because Eag1 expression has been reported in human biopsies from liver tumors [20], astemizole might be used as an anticancer therapy in patients with liver cancer.
We observed that astemizole clearly prevented the development of HCC in vivo, assessed by the histological presence of GGT activity (a commonly used liver tumor marker [35]), histopathological changes, and macroscopic liver appearance. Astemizole prevented tumor formation when administered either from the start of DEN treatment or from week 12 of DEN treatment, when cirrhosis is already present. These results suggest that astemizole might be used as a chemopreventive agent in patients at risk of developing liver cancer, namely, patients with liver cirrhosis or hepatitis virus infection. This latter case remains to be studied in the corresponding in vivo model.
The antineoplastic effects of astemizole were studied in rats treated with DEN during 16 weeks and then untreated for an additional 2 or 6 weeks. Astemizole-treated rats displayed fewer GGT-positive lesions and improved histological features and liver appearance. It is worth mentioning that liver histological features were improved and GGT activity was even lower in the astemizole-treated group (total treatment 22 weeks) than in the group treated for 16 weeks with DEN + 2 weeks DEN-free. Thus, astemizole might also be used to avoid the growth of already established liver tumors. Occasionally, liver tumors are not resectable because of their size, so astemizole treatment represents a potential alternative to decrease tumor size and make liver tumors resectable.
Astemizole targets the P-glycoprotein, which confers drug resistance to some cancers, including HCC [16, 36]. Thus, cancer therapy might be improved if anticancer drugs transported by the P-glycoprotein are used in combination with astemizole. Accordingly, astemizole greatly potentiates the growth inhibitory activity of doxorubicin in doxorubicin-resistant human leukemia cells [16]. This effect is thought to be mediated through the inhibition of the P-glycoprotein efflux pump activity by astemizole. Therefore, the combined use of astemizole with conventional chemotherapeutic drugs to improve the anticancer effects is also a potential approach for HCC treatment.
In this study, we did not observe changes in body weight or appetite loss in the animals treated with astemizole. Other in vivo studies using astemizole at the same dosage (50 mg/kg/day) showed that astemizole inhibits the tumor growth of breast cancer cells implanted in mice without producing obvious side effects [12]. This dosage is much lower than the 2052 mg/kg astemizole lethal dose (LD50) indicated by the Drug Bank [7]. Because the severe adverse effects of astemizole were exclusively cases of overdose [7], astemizole use might be very safe if recommended doses are administered and if cardiac function is properly followed.
Eag1 channels have been suggested as potential tumor markers and therapeutic targets for different types of cancers [13, 15, 17, 18]. In addition, Eag1 has been proposed as a potential early tumor marker for cervical, breast, and colon cancers [20–22]. Here, we observed Eag1 mRNA and protein expression in the in vivo HCC model. Interestingly, high Eag1 mRNA expression was detected in most of the DEN-treated groups, and strong Eag1 immunostaining was observed at early stages of HCC development. Thus, our results suggest Eag1 may have potential as an early biomarker for HCC. It may be possible to track Eag1 protein expression using sensitive imaging techniques in human livers. In one study, Eag1 protein expression was detected in vivo in nonpalpable tumors by using dye-tagged antibodies and imaging techniques in the near-infrared spectral range [33]. This type of approach might also be used to screen Eag1 expression in the liver of patients at risk of developing HCC and in the animal model used here. We observed an unexpected low Eag1 expression in the livers presenting HCC. Eag1 expression in the corresponding group treated with astemizole was slightly higher. It is likely that Eag1 expression is more related to early events in HCC development. The precise role of Eag1 channels in HCC development remains to be elucidated. This might be accomplished in different manners, such as the use of mouse models. Recently, Eag1 knockout mice were generated, and interestingly, the animals did not show any major abnormalities in the central nervous system level [37]. It would be very interesting to investigate whether these knockout mice are resistant to the development of liver cirrhosis and HCC.
Because of the poor prognosis of HCC, it is imperative to find novel therapeutic alternatives. Our results suggest astemizole may be a very promising preventive option for patients at risk of developing HCC, as well as a hopeful therapeutic option for HCC patients. We also propose that Eag1 may be a potential early biomarker of HCC. Astemizole-based prevention and therapy and early Eag1 detection in the liver should help to reduce mortality from this disease. These data provide supportive basis to include astemizole in HCC clinical trials.
References
Globocan 2008. International Agency for Research on Cancer. https://globocan.iarc.fr Accessed 22 Jun 2013.
Parkin DM. Global cancer statistics in the year 2000. Lancet Oncol. 2001;2(9):533–43.
El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–76.
Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol. 2009;27(9):1485–91.
Befeler AS, Di Bisceglie AM. Hepatocellular carcinoma: diagnosis and treatment. Gastroenterology. 2002;122(6):1609–19.
Parsons ME, Ganellin CR. Histamine and its receptors. Br J Pharmacol. 2006;147 Suppl 1:S127–35.
Garcia-Quiroz J, Camacho J. Astemizole: an old anti-histamine as a new promising anti-cancer drug. Anti Cancer Agents Med Chem. 2011;11(3):307–14.
Reynolds JL, Akhter J, Morris DL. In vitro effect of histamine and histamine H1 and H2 receptor antagonists on cellular proliferation of human malignant melanoma cell lines. Melanoma Res. 1996;6(2):95–9.
Garcia-Ferreiro RE, Kerschensteiner D, Major F, Monje F, Stuhmer W, Pardo LA. Mechanism of block of hEag1 K+ channels by imipramine and astemizole. J Gen Physiol. 2004;124(4):301–17.
Ouadid-Ahidouch H, Le Bourhis X, Roudbaraki M, Toillon RA, Delcourt P, Prevarskaya N. Changes in the K+ current-density of MCF-7 cells during progression through the cell cycle: possible involvement of a h-ether.a-gogo K+ channel. Recept Channels. 2001;7(5):345–56.
Diaz L, Ceja-Ochoa I, Restrepo-Angulo I, et al. Estrogens and human papilloma virus oncogenes regulate human ether-a-go-go-1 potassium channel expression. Cancer Res. 2009;69(8):3300–7.
Downie BR, Sanchez A, Knotgen H, et al. Eag1 expression interferes with hypoxia homeostasis and induces angiogenesis in tumors. J Biol Chem. 2008;283(52):36234–40.
Pardo LA, Stuhmer W. Eag1: an emerging oncological target. Cancer Res. 2008;68(6):1611–3.
Weber C, Mello de Queiroz F, Downie BR, Suckow A, Stuhmer W, Pardo LA. Silencing the activity and proliferative properties of the human EagI Potassium Channel by RNA Interference. J Biol Chem. 2006;281(19):13030–7.
Wulff H, Castle NA, Pardo LA. Voltage-gated potassium channels as therapeutic targets. Nat Rev Drug Discov. 2009;8(12):982–1001. doi:10.1038/nrd2983.
Ishikawa M, Fujita R, Takayanagi M, Takayanagi Y, Sasaki K. Reversal of acquired resistance to doxorubicin in K562 human leukemia cells by astemizole. Biol Pharm Bull. 2000;23(1):112–5.
Pardo LA, del Camino D, Sánchez A, et al. Oncogenic potential of EAG K(+) channels. EMBO J. 1999;18(20):5540–7.
Rodriguez-Rasgado JA, Acuna-Macias I, Camacho J. Eag1 channels as potential cancer biomarkers. Sensors (Basel). 2012;12(5):5986–95.
Occhiodoro T, Bernheim L, Liu JH, et al. Cloning of a human ether-a-go-go potassium channel expressed in myoblasts at the onset of fusion. FEBS Lett. 1998;434(1–2):177–82.
Hemmerlein B, Weseloh RM, Mello de Queiroz F, et al. Overexpression of Eag1 potassium channels in clinical tumours. Mol Cancer. 2006;5:41.
Farias LM, Ocana DB, Diaz L, et al. Ether a go-go potassium channels as human cervical cancer markers. Cancer Res. 2004;64(19):6996–7001.
Ousingsawat J, Spitzner M, Puntheeranurak S, et al. Expression of voltage-gated potassium channels in human and mouse colonic carcinoma. Clin Cancer Res. 2007;13(3):824–31.
Garcia-Becerra R, Diaz L, Camacho J, et al. Calcitriol inhibits Ether-a go-go potassium channel expression and cell proliferation in human breast cancer cells. Exp Cell Res. 2010;316(3):433–42.
Meyer R, Schonherr R, Gavrilova-Ruch O, Wohlrab W, Heinemann SH. Identification of ether a go-go and calcium-activated potassium channels in human melanoma cells. J Membr Biol. 1999;171(2):107–15.
Gavrilova-Ruch O, Schonherr K, Gessner G, et al. Effects of imipramine on ion channels and proliferation of IGR1 melanoma cells. J Membr Biol. 2002;188(2):137–49.
Asher V, Khan R, Warren A, et al. The Eag potassium channel as a new prognostic marker in ovarian cancer. Diagn Pathol. 2010;5:78. doi:10.1186/1746-1596-5-78.
Agarwal JR, Griesinger F, Stuhmer W, Pardo LA. The potassium channel Ether a go-go is a novel prognostic factor with functional relevance in acute myeloid leukemia. Mol Cancer. 2010;9:18. doi:10.1186/1476-4598-9-18.
Gomez-Varela D, Zwick-Wallasch E, Knotgen H, et al. Monoclonal antibody blockade of the human Eag1 potassium channel function exerts antitumor activity. Cancer Res. 2007;67(15):7343–9.
Ortiz CS, Montante-Montes D, Saqui-Salces M, et al. Eag1 potassium channels as markers of cervical dysplasia. Oncol Rep. 2011;26(6):1377–83.
Schiffer E, Housset C, Cacheux W, et al. Gefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosis. Hepatology. 2005;41(2):307–14.
Rutenburg AM, Kim H, Fischbein JW, Hanker JS, Wasserkrug HL, Seligman AM. Histochemical and ultrastructural demonstration of gamma-glutamyl transpeptidase activity. J Histochem Cytochem. 1969;17(8):517–26.
Suzuki K. Current therapeutic strategy for multiple myeloma. Jpn J Clin Oncol. 2013;43(2):116–24.
Pardo LA, Contreras-Jurado C, Zientkowska M, Alves F, Stuhmer W. Role of voltage-gated potassium channels in cancer. J Membr Biol. 2005;205(3):115–24.
Chen Y, Sánchez A, Rubio ME, Kohl T, Pardo LA, Stühmer W. Functional K(v)10.1 channels localize to the inner nuclear membrane. PLoS One. 2011;6(5):e19257. doi:10.1371/journal.pone.0019257.
Hanigan MH, Pitot HC. Gamma-glutamyl transpeptidase—its role in hepatocarcinogenesis. Carcinogenesis. 1985;6(2):165–72.
Breier A, Gibalova L, Seres M, Barancik M, Sulova Z. New insight into p-glycoprotein as a drug target. Anti Cancer Agents Med Chem. 2013;13(1):159–70.
Ufartes R, Schneider T, Mortensen LS, et al. Behavioural and functional characterization of Kv10.1 (Eag1) knockout mice. Hum Mol Genet. 2013;22(11):2247–62.
Acknowledgments
We thank Beatriz Alcántara for her secretarial work and Guadalupe Montiel and Eduardo García Osornio for their technical assistance. This work was partially supported by the Consejo Nacional de Ciencia y Tecnología (Conacyt) grant number 168102 to JC.
Compliance with Ethical Standards
ᅟ
Conflicts of interest
None
Ethical approval
This article does not contain any studies with human participants performed by any of the authors. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
de Guadalupe Chávez-López, M., Pérez-Carreón, J.I., Zuñiga-García, V. et al. Astemizole-based anticancer therapy for hepatocellular carcinoma (HCC), and Eag1 channels as potential early-stage markers of HCC. Tumor Biol. 36, 6149–6158 (2015). https://doi.org/10.1007/s13277-015-3299-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-3299-0