
Abstract
In this paper, we provide a phylogenetic overview of Basidiomycota and related phyla in relation to ten years of DNA based phylogenetic studies since the AFTOL publications in 2007. We selected 529 species to address phylogenetic relationships of higher-level taxa using a maximum-likelihood framework and sequence data from six genes traditionally used in fungal molecular systematics (nrLSU, nrSSU, 5.8S, tef1-α, rpb1 and rpb2). These species represent 18 classes, 62 orders, 183 families, and 392 genera from the phyla Basidiomycota (including the newly recognized subphylum Wallemiomycotina) and Entorrhizomycota, and 13 species representing 13 classes of Ascomycota as outgroup taxa. We also conducted a molecular dating analysis based on these six genes for 116 species representing 17 classes and 54 orders of Basidiomycota and Entorrhizomycota. Finally we performed a phyloproteomics analysis from 109 Basidiomycota species and 6 outgroup taxa using amino-acid sequences retrieved from 396 orthologous genes. Recognition of higher taxa follows the criteria in Zhao et al (Fungal Divers 78:239–292, 2016): (i) taxa must be monophyletic and statistically well-supported in molecular dating analyses, (ii) their respective stem ages should be roughly equivalent, and (iii) stem ages of higher taxa must be older than those of lower level taxa. The time-tree indicates that the mean of stem ages of Basidiomycota and Entorrhizomycota are ca. 530 Ma; subphyla of Basidiomycota are 406–490 Ma; most classes are 358–393 Ma for those of Agaricomycotina and 245–356 Ma for those of Pucciniomycotina and Ustilaginomycotina; most orders of those subphyla split 120–290 Ma. Monophyly of most higher-level taxa of Basidiomycota are generally supported, especially those taxa introduced in the recent ten years: phylum Entorrhizomycota, classes Malasseziomycetes, Moniliellomycetes, Spiculogloeomycetes, Tritirachiomycetes and orders Amylocorticiales, Golubeviales, Holtermanniales, Jaapiales, Lepidostromatales, Robbauerales, Stereopsidales and Trichosporonales. However, the younger divergence times of Leucosporidiales (Microbotryomycetes) indicate that its order status is not supported, thus we propose combining it under Microbotryales. On the other hand, the families Buckleyzymaceae and Sakaguchiaceae (Cystobasidiomycetes) are raised to Buckleyzymales and Sakaguchiales due to their older divergence times. Cystofilobasidiales (Tremellomycetes) has an older divergence time and should be amended to a higher rank. We however, do not introduce it as new class here for Cystofilobasidiales, as DNA sequences from these taxa are not from their respective types and thus await further studies. Divergence times for Exobasidiomycetes, Cantharellales, Gomphales and Hysterangiales were obtained based on limited species sequences in molecular dating study. More comprehensive phylogenetic studies on those four taxa are needed in the future because our ML analysis based on wider sampling, shows they are not monophyletic groups. In general, the six-gene phylogenies are in agreement with the phyloproteomics tree except for the placements of Wallemiomycotina, orders Amylocorticiales, Auriculariales, Cantharellales, Geastrales, Sebacinales and Trechisporales from Agaricomycetes. These conflicting placements in the six-gene phylogeny vs the phyloproteomics tree are discussed. This leads to future perspectives for assessing gene orthology and problems in deciphering taxon ranks using divergence times.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The phylum Basidiomycota R.T. Moore (1980) contains 16 classes, 52 orders, 177 families, 1589 genera and more than 30,000 species (Kirk et al. 2008). It is estimated that approximately 32% of the described fungal taxa belong to this phylum (Dai et al. 2015). The Basidiomycota includes the well-known mushrooms, bracket fungi, puffballs, toadstools, but also some microfungi, such as rusts, smuts and yeasts. Although there is an extraordinary diversity within this group, molecular studies indicate there are numerous taxa yet to be described (e.g. Kemler et al. 2009; Wannathes et al. 2009; Zhao et al. 2011; Wu et al. 2014; Li et al. 2016; Hyde et al. 2016). Traditionally Basidiomycota are typically characterized by the presence of basidia and basidiospores. Recent research shows that some variable morphological characters are an expression of a long evolutionary history within the different level taxa of Basidiomycota, such as diversity of cellular constructions in hyphal systems, meiosporangia, the basidiocarps, and ultrastructural characters such as septal pores (Garnica et al. 2007; Van Driel et al. 2009; Yang 2011; Oberwinkler 2012). This indicates that defining higher taxa (e.g. classes, orders) using unique or rather limited morphological characters is subjective.
Basidiomycota species are possibly the main contributors to wood and litter degradation, including degradation of the different components of wood, and they play a key process in carbon recycling (Peláez et al. 1995; Pointing 2001; Pointing et al. 2005; Oberwinkler 2012). Basidiomycota are also an important food source for both humans and animals (Zhang et al. 2015). Mushrooms have been a vital source of food for humans mainly because they are rich in carbohydrates (structural polysaccharides) and proteins, including essential amino acids necessary for humans and contain less fat (Zhang et al. 2015). Basidiomycota can produce a number of biochemical compounds that can be either beneficial or toxic to humans. Thus, they are used as traditional medicines to cure cancer, diabetes and other ailments (De Silva et al. 2012a, b, 2013), but are also the cause of serious food poisoning (Liu 2004; Dai et al. 2009; Chen et al. 2014). Basidiomycota also include plant pathogens, such as rusts and smuts (Sinclair et al. 1987; Wang and Zhuang 1998; Zhuang 2003, 2005, 2012; Dai et al. 2007; Hyde et al. 2014), and also form mutualistic associations with a variety of other organisms, such as ectomycorrhizas with plants (e.g. Bonfante and Genre 2008; Rinaldi et al. 2008), symbioses with liverworts (e.g. Kottke et al. 2003), lichen symbioses (e.g. Lawrey et al. 2007), and fungus-farming (fungiculture) by ants and termites (e.g. Chapela et al. 1994; Mueller et al. 2005).
The phylum Basidiomycota was shown to be a monophyletic group sister to Ascomycota (James et al. 2006; Hibbett et al. 2007). Currently, Basidiomycota includes three major clades—Agaricomycotina (mushrooms, jelly fungi, bracket fungi and others), Pucciniomycotina (rusts), and Ustilaginomycotina (smuts and others) (Hibbett et al. 2007)—as well as Wallemiomycetes (Matheny et al. 2007a; Padamsee et al. 2012). A higher-level classification of Basidiomycota was published around ten years ago, based mainly on a series phylogenetic studies, such as the Deep Hypha and AFTOL projects (Blackwell et al. 2006; James et al. 2006; Hibbett et al. 2007; Lutzoni et al. 2004). However, there has been considerable advances since this time and the classification needs updating. For example, the class Entorrhizomycetes has been split from Basidiomycota and raised to phylum Entorrhizomycota. New classes (Malasseziomycetes, Moniliellomycetes, Spiculogloeomycetes, Tritirachiomycetes), and new orders (Holtermanniales, Trichosporonales, Golubeviales, Robbauerales, Unilacrymales, Amylocorticiales, Jaapiales, Stereopsidales and Lepidostromatales) have been introduced (Binder et al. 2010; Boekhout et al. 2011; Schell et al. 2011; Wuczkowski et al. 2011; Shirouzu et al. 2013; Hodkinson et al. 2014; Sjökvist et al. 2014; Weiss et al. 2014; Wang et al. 2014, 2015c). All of the recently introduced higher-level taxa (hereon refers to orders and above) were studied alongside related taxa. No recent study has combined all high-ranked taxa in a single phylogeny to establish whether they are well-resolved. In this study, we selected 529 taxa across all higher ranks of Basidiomycota in standard phylogenetic and molecular dating analyses, as well as phylogenetic reconstruction with hundreds of proteins, to establish if they are well-supported.
Current systematic and taxonomic research generally focuses on organizing and classifying taxa using a combination of characters, including chemical, molecular, morphological, ecological, physiological and biogeographical data (e.g., Hyde et al. 2013; Kuhnert et al. 2015; Maharachchikumbura et al. 2015, 2016). Taxon distinction is based on the recognition of monophyly (e.g. Vilgalys et al. 1994; Taylor et al. 2000), however, the rank of any taxon at any level is subjective and lacks universal criteria (Avise and Johns 1999; Liu et al. 2016). This is apparent both within (e.g. Ascomycota and Basidiomycota) and outside of the Fungi (e.g. across animals, fungi and plants, Samarakoon et al. 2016). Researchers usually rank monophyletic lineages based on their distinct phylogenetic position, plus a combination of morphological and or other characters, and the level of ranking may thus follow that of their known sister clades.
Researchers have attempted to use other criteria in taxon ranking and a polyphasic approach is generally recommended. This is because the use of morphological, phenotypic or molecular characters may be misleading and differ from group to group. For example, Talavera et al. (2013) have designated an age interval of 4–5 Ma to define genera of blue butterflies using sequence data from nine genes. The arrangement of higher-level taxa has been subjective and has led to many hostile and futile arguments in the past (Liu et al. 2016). The concept of using divergence times as a universal criterion for taxa ranking was first proposed by Hennig (1966). He suggested that a taxonomic rank should reflect its geological age, and divergence time could be used as a universally standardized criterion in the systematics of all known organisms (Hennig 1966). In Hennigs’ day this was not possible, but the estimation of organism divergence times has now become feasible, due to advances of molecular biology techniques and analytical methods. The latter can convert molecular change into evolutionary time (Robinson and Robinson 2001; Drummond et al. 2006, 2012). The first attempt that aimed at reconciling taxonomic ranks with divergence time estimates in fish, arthropods and fruit flies is that of Avise and Johns (1999). They found a notable disparity in the ages of lineages and their taxonomic rank. Since it was established, molecular clock analysis have largely been used in biogeographic and phylogeographic studies (Hickerson et al. 2010; Zhao et al. 2013; Stefani et al. 2014), but not in taxonomic ranking.
Several recent reports have established divergence times in the fungi. For example, Basidiomycota are estimated to have evolved during the Palaeozoic, around 500 million years ago (Ma), and is a sister group to Ascomycota being of similar age (Berbee and Taylor 2010; Oberwinkler 2012; Hibbett 2014). Agaricomycetes diverged ca. 290 Ma (Floudas et al. 2012). The ages of some groups within Agaricomycetes, such as Boletales (Skrede et al. 2011; Wilson et al. 2012), Agaricales (Matheny et al. 2009; Ryberg and Matheny 2012), and brown-rot lineages (Garcia-Sandoval et al. 2011) have been also estimated, as were fungal epiphytes (Hongsanan et al. 2016).
The first attempt at establishing a taxonomic system in the Fungi based on divergence time was a reconstruction of a taxonomic system for Agaricus (Zhao et al. 2016). These authors produced a multigene phylogeny based on combined ITS, nrLSU, tef1-α, and rpb2 sequence data. Divergence times within the multigene phylogeny were calculated and used to standardize the ranking of taxa into subgenera and sections. The following criteria were used to recognize taxa above species level: (i) they must be monophyletic and statistically well-supported in the molecular dating analyses; (ii) their respective stem ages should be roughly equivalent, and higher taxa stem ages must be older than lower level taxa stem ages; and (iii) they should be identifiable phenotypically, whenever possible. Based on those criteria some subgenera or sections were split or rejected, and new ones were discovered and named.
In the present study, we produce a phylogeny of representative species from almost all classes and orders of Basidiomycota and related phyla using six-gene data commonly used in fungal systematics. Divergence times are estimated, and used as a criterion for ranking of higher-level taxa. We also compare this six-gene phylogeny with a tree produced from amino acid data extracted from published genomes (115 taxa and 396 proteins).
Materials and methods
Six-gene sampling
Our dataset comprises nrLSU, nrSSU, 5.8S, tef1-α, rpb1 and rpb2 sequence data from 529 specimens or cultures representing 18 classes, 62 orders, 183 families, 392 genera and 503 species of Basidiomycota and Entorrhizomycota. In addition, 13 species of Ascomycota belonging to 13 different classes, orders, families and genera were chosen as outgroup taxa (Beimforde et al. 2014). Multigene sequence data from 81 specimens are new taxonomic contributions from this study, all of which have been morphologically examined in detail following the methods of Largent (1986a, b). The other sequences were downloaded from GenBank, and their details are listed in Supplementary Table 1.
PCR amplification and sequencing
Genomic DNA was extracted from dried specimens using an E.Z.N.A. Forensic DNA Kit (OMEGA Bio-Tek, Norcross, GA, USA). We used primers ITS4 and ITS5 (White et al. 1990) for the ITS1-5.8S-ITS2 region (ITS) of the nuclear ribosomal DNA repeat, primers LROR and LR5 (Moncalvo et al. 2000) for the nuclear LSU-rDNA region, primers EF1-983F and EF1-1567R (Morehouse et al. 2003) for translation elongation factor alpha (tef1-α), and primers b6F and b7.1R for RNA polymerase II subunit II (rpb2) (Matheny et al. 2007b) and primers RPB1-Ac RPB1-Cr for rpb1 (Matheny et al. 2002). Genes were amplified by polymerase chain reaction (PCR) using the procedures mentioned in those studies (Moncalvo et al. 2000; Morehouse et al. 2003; Matheny et al. 2007b; Zhao et al. 2011). The PCR products were sent to a commercial biotech company (Baimaide Biotechnique Company, Beijing) for sequencing.
Phylogenetic analysis
The sequence data from each gene was aligned using Muscle 3.6 with default settings (Edgar 2004a, b), then manually adjusted in Mesquite 3.2 (Maddison and Maddison 2016). Regions with ambiguous alignment were excluded, then all sequence data from each gene were combined into a single, concatenated datamatrix. Single-gene phylogenies were constructed to detect possible significant conflicts among the single-gene trees. The final alignment has been submitted to TreeBase (submission No. 20837). Maximum-likelihood (ML) analyses were performed using RAxML v7 (Stamatakis 2006) using a GTR + G+I substitution model. To assess the statistical support of clades we ran 1000 fast-bootstrap (BS) replications under the GTR-CAT approximation.
Divergence-time analysis
We selected 116 taxa for dating analysis based on the phylogenetic results from the six-gene phylogeny and sequence data completeness. The selected taxa represent 17 classes and 54 orders of Basidiomycota and two species of Entorrhizomycota as outgroup taxa (Supplementary Table 1).
We used two fossils for calibration: Archaeomarasmius leggetti Hibbett et al., an agaricoid fruiting body preserved in 90 Ma Dominican amber (Hibbett et al. 1997) as representative of the minimum age of Agaricales; and Quatsinoporites cranhamii S.Y. Sm. et al., a poroid fruiting body from Apple Bay on Vancouver Island from 113 Ma (Smith et al. 2004) as representative of the minimum age of Hymenochaetales. In addition, we used a genomic study that concluded that Agaricomycetes are 290 Ma old (Floudas et al. 2012). Divergence times were estimated in BEAST v1.8.4 (Drummond et al. 2012). We first constructed an XML file with BEAUTI v1.8.4 with a combined six-gene alignment. As substitution models, we used the GTR + G as suggested by jModelTest v2 (Darriba et al. 2012). We used the uncorrelated lognormal relaxed clock model (Drummond et al. 2006; Lepage et al. 2007), specifying a gamma distribution prior (offset = 0, scale = 0.001, shape = 1) on the ulcd.mean parameter of each gene partition. For calibration, we specified a gamma distribution prior (scale = 20, shape = 1) on the Agaricales (offset = 90 Ma), and Hymenochaetales (offset = 113 Ma) clades (Sánchez-Ramírez et al. 2015; Zhao et al. 2016). In addition, we constrained the Agaricomycetes to be 230 Ma (mean) by applying a normal distribution prior (SD = 1), based on Floudas et al. (2012). We ran two independent Monte Carlo Markov Chains of 100 million generations, logging states every 10,000 generations. Log files were checked for convergence and mixing in Tracer v1.6 (Rambaut et al. 2013; http://tree.bio.ed.ac.uk/software/tracer/). An ultrametric maximum-clade-credibility (MCC) tree was summarized using TreeAnnotator 1.8.4, discarding 10% of states as burn-in and annotating clades with ≥ 0.8 posterior probability.
Phyloproteomics analysis
One-hundred and fifteen published Basidiomycota and outgroup proteomes were downloaded from JGI and Ensembl (Supplementary Table 2). All-vs-all comparisons were conducted using Diamond BLAST with maximum sensitivity (Buchfink et al. 2015). In-paralogs were identified using a Python script based on the assumption that these should be a better match to another than to any sequence from any other species, and all clusters of in-paralogs were reduced to a single representative sequence. BLAST output was pruned to remove instances of in-paralogs and used for MCL-based identification of orthologs with an inflation parameter of 3.0 (van Dongen 2000). Single-copy orthologs were selected with a minimum taxa occupancy of 70, aligned using Muscle (Edgar 2004a, b), and trimmed using trimAl with the “strict” heuristic option (Capella-Gutiérrez et al. 2009). 396 alignments with trimmed lengths between 100 and 600 sites were selected and used to generate a partitioned super-matrix with 82,536 sites. Maximum likelihood phylogenetic analysis was conducted using ExaML (Kozlov et al. 2015) with 100 bootstraps, gamma rate distribution, and evolutionary models for all partitions determined by maximum likelihood during the analysis.
Results and discussion
Six-gene phylogeny and general dating analysis
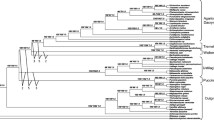
The combined nrLSU, nrSSU, 5.8S, tef1-α, rpb1 and rpb2 dataset comprised 529 species with 3957 bps, after excluding the ambiguous regions. The best scoring RAxML trees with all species and BS support are shown in Fig. 1. (Fig. 1a, b). Figure 1a is a concatenated tree from Fig. 1b and highlights the relationships among classes). The MCC tree is shown in Fig. 2, which is based on 116 taxa with 95% highest posterior density (HPD) of divergence time estimates and PP values at the nodes.







A concatenated tree showing the relationships among classes from the phylogenetic analysis of Basidiomycota and allied phyla generated from maximum likelihood analysis of nrLSU, nrSSU, 5.8S, tef1-α, rpb1 and rpb2 sequence data, rooted with 13 species from Ascomycota. Bootstrap support (BS) values >50% are given at the internodes. The thickened branch indicates the order with a BS values >80% (a A concentred tree from the completely ML tree, in order to show the highlights of the relationships among classes; b The completely ML tree)

Maximium Clade Credibility tree of Basidiomycota and allied phyla based on nrLSU, nrSSU, 5.8S, tef1-α, rpb1 and rpb2 sequence data with the outgroups Entorrhizomycota. Posterior probabilities which are equal or greater than 80% are annotated at the internodes. The 95% highest posterior density of divergence time estimates are marked by horizontal bars. The coloured dots
 refer to the positions of the mean stem age of phyla, subphyls, classes and orders respectively
refer to the positions of the mean stem age of phyla, subphyls, classes and orders respectively
The phylum Basidiomycota is monophyletic with 1.0 PP support in the MCC tree (Fig. 2) and 75% BS support in the ML tree (Fig. 1). Basidiomycota have a stem age of 530 Ma. In this tree, Basidiomycota is composed of four main clades which correspond to Agaricomycotina, Pucciniomycotina, Ustilaginomycotina and Wallemiomycotina with the divergence time period of ca. 406–490 Ma. Entorrhizomycota is sister to Basidiomycota at the divergence time of 530 Ma.
The four subphyla of Basidiomycota, Pucciniomycotina, Ustilaginomycotina and Wallemiomycotina are well-supported in both trees, however, Agaricomycotina has 55% BS support in the ML tree (Fig. 1), but 1 PP support in the MCC tree (Fig. 2). Agaricomycotina comprises three classes: Agaricomycetes, Dacrymycetes and Tremellomycetes. These subphyla were estimated to have diverged 406 to 430 Ma ago. Most classes of Basidiomycota have 99% or 100% BS support, with the exceptions of Agaricostilbomycetes with 61% BS support, while Exobasidiomycetes is polyphyletic in the ML tree (Fig. 1).
Phyloproteomics analysis
Results of our phyloproteomics analysis are depicted in Fig. 3. Overall, there is a high level of congruence with the six-gene phylogeny (Figs. 1, 2). One striking exception, however, is the placement of Wallemiomycotina (represented by its holotype Wallemia), which is sister group to Agaricomycotina in the phyloproteomics tree (Fig. 3) whereas it is sister group to Agaricomycotina + Ustilaginomycotina + Pucciniomycotina in the six-gene phylogeny (Figs. 1, 2).

Phyloproteomic tree of 109 Basidiomycete and six outgroup species, based on 396 protein alignments and a total of 82,536 sites. Statistical support values are based on maximum likelihood, and are shown only for nodes with lower than 100% support
At the order level, this phyloproteomic analysis also reveals inconsistent phylogenetic positions from the six-gene phylogeny for orders Amylocorticiales, Auriculariales, Cantharellales Geastrales, Sebacinales and Trechisporales of Agaricomycetes (Figs. 1, 2, 3).
Taxonomy
From the results depicted in Figs. 1, 2, 3 and the divergence times of higher taxa summarized in Table 1 we propose to treat subphyla, classes and related orders as follows.
Phylum: Basidiomycota R.T. Moore, Bot. Mar. 23(6): 371 (1980)
Subphylum Agaricomycotina Doweld, Prosyllabus Tracheophytorum, Tentamen Systematis Plantarum Vascularium (Tracheophyta) (Moscow): LXXVIII (2001)
Subphylum Pucciniomycotina R. Bauer, Begerow, J.P. Samp., M. Weiss & Oberw., Mycol. Progr. 5(1): 45 (2006)
Subphylum Ustilaginomycotina Doweld, Prosyllabus Tracheophytorum, Tentamen Systematis Plantarum Vascularium (Tracheophyta) (Moscow): LXXVIII (2001)
Subphylum Wallemiomycotina Doweld, Index Fungorum 73 (2014)
1. Subphylum Agaricomycotina
Class Agaricomycetes Doweld, Prosyllabus Tracheophytorum, Tentamen Systematis Plantarum Vascularium (Tracheophyta) (Moscow): LXXVIII (2001)
Class Dacrymycetes Doweld, Prosyllabus Tracheophytorum, Tentamen Systematis Plantarum Vascularium (Tracheophyta) (Moscow): LXXVIII (2001)
Class Tremellomycetes Doweld, Prosyllabus Tracheophytorum, Tentamen Systematis Plantarum Vascularium (Tracheophyta) (Moscow): LXXVIII (2001)
Phylogenetic support
The subphylum Agaricomycotina comprises three lineages: Dacrymycetes, Agaricomycetes and Tremellomycetes (Figs. 1, 2). The classes Agaricomycetes and Dacrymycetes cluster together with 55% BS support, and are sister to Tremellomycetes with poor statistical support in the ML tree (Fig. 1). In the MCC tree (Fig. 2) and phyloproteomic tree (Fig. 3) there is the same topology within this subphylum, but Agaricomycotina has 1 PP/100% BS support (Fig. 2). The mean of stem age of Agaricomycotina is 406 Ma and the classes diverged from 358 Ma to 393 Ma (Table 1).
Discussion
The subphylum Agaricomycotina contains approximately 70% of the known basidiomycetes (Kirk et al. 2008), including mushrooms, shelf fungi, puffballs and others. Historically the subphylum has been named as “Hymenomycetes” using single gene sequences (Swann and Taylor 1993, 1995). This lineage has been well-accepted as a subphylum based on analysis of nrLSU and nrSSU sequence data, and the integrated analysis of ultrastructural features, such as form and behaviour of the spindle pole bodies and types of host–parasite interaction (Bauer et al. 2006).
A phylogenetic overview of Agaricomycotina has been published using sequence data from nuclear rRNA and protein coding genes. The subphylum contains three major clades (Agaricomycetes, Dacrymycetes and Tremellomycetes; Hibbett 2006). In Hibbett’s study, Agaricomycotina is well-supported using combined nuclear rRNA and protein-coding-gene sequence data from 125 taxa when data for Wallemiomycetes and Entorrhizomycetes was not available. However, once sequence data from Wallemiomycetes and Entorrhizomycetes are included, this subphylum is well-supported using nuclear rRNA genes; but lacks strong BS support when using combined nuclear rRNA and protein coding genes (Hibbett 2006).
In our analyses, we used rRNA and protein coding genes and include more than 500 species: Agaricomycotina lacks high BS support in the ML tree (Fig. 1), but has 1 PP/100% BS support in the molecular dating and phyloproteomic analyses (Figs. 2, 3). We therefore accept Agaricomycotina with three classes: Agaricomycetes, Dacrymycetes and Tremellomycetes.
Class Agaricomycetes
Phylogenetic support
This class is monophyletic with 100% BS and 1 PP support (Figs. 1, 2, 3). In the ML tree (Fig. 1), 12 orders (Amylocorticiales, Atheliales, Auriculariales, Boletales, Corticiales, Lepidostromatales, Phallales, Russulales, Sebacinales, Stereopsidales, Thelephorales, Trechisporales) out of 21 orders have more than 85% BS support, and six orders (Agaricales, Cantharellales, Geastrales, Gloeophyllales, Hymenochaetales, and Polyporales) have 62–84% BS support. The problematic orders in this ML study are Gomphales and Hysterangiales. The former lacks statistical support and the latter is polyphyletic. In the molecular dating analysis, all those 21 orders of Agaricomycetes are monophyletic with 0.95 or 1 PP support. In the phyloproteomic analysis, a total of 15 orders are included and those six weak-supported orders (Agaricales, Cantharellales, Geastrales, Gloeophyllales, Hymenochaetales, and Polyporales with 62–84% BS support in six-gene analysis) get 100% BS support (Fig. 3).
Based on the molecular dating analysis (Fig. 2), Agaricomycetes has a mean stem age of 358 Ma. The 21 orders within Agaricomycetes diverged at different times, but mostly within the mean of stem ages ranging from 124 Ma to 290 Ma (Table 1). In that analysis the order Auriculariales is basal (Figs. 1, 2; Table 1). However, the phyloproteomic analysis, the order Cantharellales has the basal position, followed by Sebacinales and Auriculariales (Fig. 3).
Discussion
The members of Agaricomycetes are mostly mushrooms, and include cultivated, saprotrophic and ectomycorrhizal species used as sources of food and medicine (De Silva et al. 2012a, b, 2013; Hibbett et al. 2014). Some species also cause poisoning or psychoactivity when eaten (Chen et al. 2014). Bioactive components from these mushrooms can be used in the modern medical industries (Ruiz-Dueñas and Martínez 2009). This class previously contained 17 orders, more than 100 families, 1000 genera and 21,000 species, which is one-fifth of all known fungi (Hibbett et al. 2007; Kirk et al. 2008).
The early recognized orders have been resolved as monophyletic groups using analysis of rRNA genes, and support for some orders were weak or absent, such as Cantharellales (Binder and Hibbett 2002; Binder et al. 2005; Moncalvo et al. 2006). In part due to elevated rates of evolution in rRNA genes, protein coding genes have been introduced in fungal phylogenetic analyses, such as in Matheny et al. (2006). Several weakly supported orders based on rRNA gene analysis are strongly supported in Matheny et al. (2006). This includes Agaricales, Polyporales, and Cantharellales (excluding species of Tulasnella) and the russuloid and gomphoid-phalloid clades (Binder and Hibbett 2002; Binder et al. 2005; Moncalvo et al. 2006). Since 2010, the orders Amylocorticiales, Jaapiales, Stereopsidales and Lepidostromatales have been introduced within Agaricomycetes (Binder et al. 2010; Hodkinson et al. 2014; Sjökvist et al. 2014).
In this study, we included all 21 orders and in the ML analysis using rRNA and protein coding gene sequences from many more species than in Matheny et al. (2006). When combining the ML and molecular dating analysis (Figs. 1, 2; Table 1) as well as previous studies, the orders Agaricales, Amylocorticiales, Atheliales, Auriculariales, Boletales, Corticiales, Geastrales, Gloeophyllales, Hymenochaetales, Jaapiales, Lepidostromatales, Polyporales, Phallales, Russulales, Sebacinales, Stereopsidales, Thelephorales, and Trechisporales are well-supported. All are monophyletic groups within similar divergence times in our analyses (Figs. 1, 2, 3; Table 1). Within those orders, Jaapiales was established in 2010 based on analysis of a six-locus nuclear dataset of 191 species, and contains a single resupinate genus Jaapia comprising two species (Binder et al. 2010). In that study, Jaapiales is sister to Boletales. However in our study, Jaapia is sister to Gloeophyllales in all our analyses, including the ML (Fig. 1), molecular dating analysis (Fig. 2) and phyloproteomic analysis (Fig. 3).
The main problems in the present taxonomic system of Agaricomycetes are as follows:
-
(i)
Redefinition of Cantharellales. Cantharellales has been thought to be a monophyletic group, represented by members of Cantharellaceae, Clavulinaceae and Hydnaceae (Matheny et al. 2006). Botryobasidiaceae was included in this order by Moncalvo et al. (2006), appearing to be sister to the core cantharelloid clade when using analysis of rRNA genes (Moncalvo et al. 2006). The most recent phylogenetic study on Cantharellales based on ITS sequences from six families, also included Botryobasidiaceae (Veldrea et al. 2013). In our ML analysis, we used rRNA and protein coding gene sequence data from members of Clavulinaceae, Hydnaceae and Botryobasidiaceae. The phylogeny shows this order is monophyletic with 84% BS support. However, this order has a 1.0 PP and 100% BS statistical support in the molecular dating and phyloproteomic analysis, but it does not have a wide taxon sampling. Thus, more comprehensive studies are needed to redefine this order in future work.
-
(ii)
An improved phylogenetic analysis emphasizing on Gomphales is needed. In the ML analysis, this order is paraphyletic as our sample of Phaeoclavulina africana (collection number 39,621) did not cluster with the other twelve taxa of Gomphales, which formed a monophyletic clade, but without statistical support (Fig. 1). However, in the molecular dating analysis this order has 0.97 PP support (Fig. 2) and has a divergence time of 187 Ma. Further studies are needed because this dating analysis is only based on two species sequences.
-
(iii)
A more comprehensive phylogenetic analysis of Hysterangiales is needed. Similar to Gomphales, Hysterangiales is monophyletic with 0.95 PP support, and a divergence time of 133 Ma based on two species sequences. However this order is paraphyletic in the ML analysis when using sequences from 22 species.
Class Dacrymycetes
Phylogenetic support
This class is represented by nine species and is supported by 100% BS (Fig. 1), 1.0 PP in the molecular dating analysis (Fig. 2) and 100% BS in the phyloproteomics analysis (Fig. 3). This class is sister to Agaricomycetes with 57% BS support (Fig. 1). The recently introduced order Unilacrymales, represented by a single species of Unilacryma, nests in the clade comprising Cerinomycetaceae (Dacrymycetales) with 100% BS support (Fig. 1), but is sister to Dacrymycetales in the molecular dating analysis (Fig. 2). The mean stem age for Dacrymycetes is 358 Ma, while Dacrymycetales and Unilacrymales are both 225 Ma (Fig. 2; Table 1).
Discussion
Dacrymycetes comprises the jelly fungi, and traditionally contains a single order Dacrymycetales, one family Dacrymycetaceae, nine genera and more than 100 species (Hibbett 2006; James et al. 2006; Hibbett et al. 2007; Kirk et al. 2008). Members of this class are mainly brown rot and rarely white rot fungi. The divergence time for this class was estimated at 280–360 Ma (Dentinger et al. 2010), so the class is regarded as one of the earliest wood decomposers to diverge in the Basidiomycota as compared to the later-evolved wood decomposers in Agaricomycetes (Shirouzu et al. 2013); such as the order Polyporales (Agaricomycetes), which is about 114 Ma old (Fig. 2). Recent molecular phylogenetic studies support the monophyly of Dacrymycetes and show it has a sister relationship to Agaricomycetes (Hibbett 2006; James et al. 2006; Hibbett et al. 2007).
Cerinomycetaceae (Jülich 1981; Larsson et al. 2004) is a second family of Dacrymycetales introduced by Shirouzu et al. (2009). A new genus Unilacryma was introduced with a single species U. unisporus; this species always occupies a sister position to other species of Dacrymycetes. To accommodate this taxon, a new family Unilacrymaceae and a new order Unilacrymales were introduced (Shirouzu et al. 2013).
However, in our study, Unilacrymales clusters in the clade of Cerinomycetaceae of Dacrymycetales with 100% BS support in ML analysis (Fig. 1), but sister to Dacrymycetales in the molecular dating analysis (Fig. 2). Their divergence times are both 225 Ma (Table 1). Then we accept Unilacrymales as an order in this study, but a more detailed study should be conducted using more genes.
Class Tremellomycetes
Phylogenetic support
In this study, Tremellomycetes as represented by 43 species, is monophyletic with 100% BS support in the ML tree (Fig. 1). Tremellomycetes is sister to the classes Agaricomycetes and Dacrymycetes and they form a larger clade which respect to the subphylum Agaricomycotina with the BS values less than 50% support. However, in the MCC and phyloproteomic trees (Figs. 2, 3), Tremellomycetes is sister to the clade of Agaricomycetes + Dacrymycetes, and all cluster within Agaricomycotina with 1.0 PP/100% BS support.
Both analyses show the same topology within the class Tremellomycetes (Figs. 1, 2): the order Cystofilobasidiales and the orders Filobasidiales, Holtermanniales, Trichosporonales and Tremellales represent a deep dichotomy; Filobasidiales is sister to the clade composed of Holtermanniales, Trichosporonales and Tremellales. All orders have 99–100% BS and 0.99–1.0 PP support.
The mean of stem age of Tremellomycetes is 393 Ma, and the mean of stem ages of its orders mostly range from 153 Ma to 270 Ma with the exception of order Cystofilobasidiales is older (350 Ma, Table 1).
Discussion
Tremellomycetes, the jelly fungi, have gelatinous fruiting bodies. However, the classes also contain yeasts and dimorphic fungi. The phylogenetic position of Tremellomycetes has been well-studied (Fell et al. 2000; Matheny et al. 2006; Hibbett et al. 2007), and three orders Cystofilobasidiales, Filobasidiales and Tremellales have been widely accepted within this class (Kirk et al. 2008). Holtermanniales and Trichosporonales are additional orders (Boekhout et al. 2011; Wuczkowski et al. 2011; Weiss et al. 2014), and a related robust framework and updated taxonomic system of this class has been provided using analysis of combined sequence data from seven genes (Liu et al. 2015a,b).
In our study, Tremellomycetes is strongly supported and it contains five well-supported orders (Figs. 1, 2; Table 1). However, this class is loosely related to the subphylum Agaricomycotina in the ML analysis (Fig. 1), this is similar to the phylogenetic overview of Agaricomycotina (Hibbett 2006): the protein-coding gene sequences reduce the support of Tremellomycetes as a member of subphylum Agaricomycotina. However, in the molecular dating and phyloproteomic analysis, Tremellomycetes is strongly supported as a member of Agaricomycotina. On the other hand, the older age of order Cystofilobasidiales may indicate that it could eventually be raised to a higher taxon.
2. Subphylum Pucciniomycotina R. Bauer, Begerow, J.P. Samp., M. Weiss & Oberw., Mycol. Progr. 5(1): 45 (2006)
Class Agaricostilbomycetes R. Bauer, Begerow, J.P. Samp., M. Weiss & Oberw., Mycol. Progr. 5(1): 45 (2006)
Class Atractiellomycetes R. Bauer, Begerow, J.P. Samp., M. Weiss & Oberw., Mycol. Progr. 5(1): 45 (2006)
Class Classiculomycetes R. Bauer, Begerow, J.P. Samp., M. Weiss & Oberw., Mycol. Progr. 5(1): 46 (2006)
Class Cryptomycocolacomycetes R. Bauer, Begerow, J.P. Samp., M. Weiss & Oberw., Mycol. Progr. 5(1): 46 (2006) (not involved in this study)
Class Cystobasidiomycetes R. Bauer, Begerow, J.P. Samp., M. Weiss & Oberw., Mycol. Progr. 5(1): 46 (2006)
Class Microbotryomycetes R. Bauer, Begerow, J.P. Samp., M. Weiss & Oberw., Mycol. Progr. 5(1): 47 (2006)
Class Mixiomycetes R. Bauer, Begerow, J.P. Samp., M. Weiss & Oberw., Mycol. Progr. 5(1): 47 (2006)
Class Pucciniomycetes R. Bauer, Begerow, J.P. Samp., M. Weiss & Oberw., Mycol. Progr. 5(1): 48 (2006)
Class Spiculogloeomycetes Q.M. Wang, F.Y. Bai, M. Groenew. & Boekhout, Stud. Mycol. 81: 172 (2015)
Class Tritirachiomycetes Aime & Schell, in Schell, Lee & Aime, Mycologia 103(6): 1339 (2011)
Phylogenetic support
This subphylum is a monophyletic clade with 100% BS support (Fig. 1), and sister to subphylum Agaricomycotina. Nine of the ten classes of Pucciniomycotina were included in this study. Cryptomycocolacomycetes is not included as it lacks reliable sequence data. All nine classes are monophyletic with 99% BS or 1.0 PP support, except for Agaricostilbomycetes with 61% BS support in the ML analysis (Fig. 1). Eight classes (Classiculomycetes is excluded due to limited sequence data) are included in the molecular dating analysis. All classes are monophyletic, and have 0.95–1.0 PP support (Fig. 2; Table 1). In the phyloproteomic analysis, this subphylum is fully supported and represented by three classes (Pucciniomycetes, Microbotryomycetes and Mixiomycetes), however it is sister to the Agaricomycotina + Ustilaginomycotina clade (Fig. 3).
The orders Classiculales, Heterogastridiales, Kriegeriales, Mixiales, Naohideales, Sakaguchiales ord. nov. and Septobasidiales are represented by a single species, so they lack statistical support (Fig. 1). The orders Atractiellales, Cystobasidiales, Helicobasidiales, Platygloeales, Pucciniales, Spiculogloeales, Sporidiobolales and Tritirachoales have 93% to 100% BS support; and the orders Agaricostilbales, Erythrobasidiales, Leucosporidiales (proposed as a synonym of Micorbotryales in this study) and Microbotryales have 61, 81, 69 and 71% BS support respectively (Fig. 1). In the class Cystobasidiomycetes, the genera Buckleyzyma, Microsporomyces, Sakaguchia and Symmetrospora are represented by a single type specimen in the ML analysis.
In the molecular dating analysis, the mean of stem age of Pucciniomycotina is 406 Ma, its classes have mean stem ages mostly from ca. 264 Ma to 356 Ma. The mean stem ages of the orders are mainly from 118 Ma to 187 Ma, however Leucosporidiales and Microbotryales have a similar stem age (74 Ma), which is much younger than those of other orders (Fig. 2; Table 1).
Discussion
There are more than 8000 species in Pucciniomycotina, which makes up one third of the described Basidiomycota (Aime et al. 2006, Bauer et al. 2006). Previous systematic studies mainly compared ultrastructural features, such as septal pore apparatus, form and behaviour of the spindle pole bodies, types of host-parasite interaction, presence or absence of colacosomes, symplechosomes, atractosomes and cystosomes as well as analyses of nuclear rRNA gene sequence data. A taxonomic arrangement has been proposed with eight classes: Agaricostilbomycetes, Atractiellomycetes, Classiculomycetes, Cryptomycocolacomycetes, Cystobasidiomycetes, Microbotryomycetes, Mixiomycetes, and Pucciniomycetes (Hamamoto and Nakase 2000; Nakase 2000; Fell et al. 2000; Scorzetti et al. 2002; Aime et al. 2006, 2014; Bauer et al. 2006; Hibbett et al. 2007; Boekhout et al. 2011. Among these classes, Pucciniomycetes includes the most species from this subphylum (around 8000 species, Kirk et al. 2008) and most are plant pathogens. The class Tritirachiomycetes is based on a single genus Tritirachium Limber, which are hyphomycetes recently placed as incertae sedis within the Pezizomycotina (Ascomycota). Now the genus is placed in Tritirachiomycetes and as the ninth class of Pucciniomycotina (Schell et al. 2011). The tenth class, Spiculogloeomycetes Q.M. Wang, F.Y. Bai, M. Groenew. & Boekhout, comprises yeast and yeast-like species which were introduced by Wang et al. (2015a, c).
All classes are monophyletic groups based on the phylogenies of the ML and molecular dating analyses. The recently introduced classes Tritirachiomycetes and Spiculogloeomycetes are well-supported, not only from the phylogenetic analysis, but also have similar divergence times.
Most orders of Pucciniomycotina are well-supported in this study. Exceptions are: (i) In class Microbotryomycetes, Leucosporidiales which is represented by its type species and type specimen Leucosporidium scottii in this study. The molecular dating analysis shows this order has a very young divergence time (74 Ma) and is sister to Microbotryales in molecular dating analysis. In the ML analysis Leucosporidiales is sister to Kriegeriales without statiscal support, then both of them are sister to Microbotryales without statiscal support too. Based on the younger divergence time of Leucosporidiales and its clustering with Microbotryales under 1.0 pp value support, we propose combining Leucosporidiales under Microbotryales. Its related orders (such as Kriegeriales) therefore need further studies. (ii) In class Cystobasidiomycetes, the genera Buckleyzyma, Microsporomyces, Sakaguchia and Symmetrospora are the only genera representing the families Buckleyzymaceae, Microsporomycetaceae, Sakaguchiaceae and Symmetrosporaceae respectively, and are placed in Cystobasidiomycetes incertae sedis at the ordinal level (Wang et al. 2015c). In the ML analysis, the type specimens of those four genera do not cluster with any known orders with good statiscal support. Buckleyzyma armeniaca (type JCM 8977) and Sakaguchia lamellibrachiae (type CBS9598) are included in the molecular dating analysis and are positioned as sister to Erythrobasidiales with divergence times of 136 Ma and 122 Ma respectively. From these results we introduce the new orders Buckleyzymales and Sakaguchiales to accommodate the families Buckleyzymaceae and Sakaguchiaceae. Sequence data for members of the families Microsporomycetaceae and Symmetrosporaceae were not available for molecular dating and phyloproteomic analyses in this study; we therefore keep them in the Cystobasidiomycetes.
Buckleyzymales R.L. Zhao & K. D. Hyde, order nov.
Fungal Names: FN570465
Type Family: Buckleyzymaceae Q.M. Wang, F.Y. Bai, M. Groenew. & Boekhout, Stud. Mycol. 81:176 (2015)
Type genus: Buckleyzyma Q.M. Wang, F.Y. Bai, M. Groenew. & Boekhout. Stud. Mycol. 81:176 (2015)
The diagnosis of the order Buckleyzymales is based on the description of family Buckleyzymaceae, which is based on the description of the genus Buckleyzyma: “Sexual reproduction not known. Colonies brownish-orange or orange and butyrous. Budding cells present. Hyphae and pseudohyphae present or not. Ballistoconidia present or not, ellipsoidal allantoid to amygdaliform” (Wang et al. 2015c).
Sakaguchiales R.L. Zhao & K.D. Hyde, order nov.
Fungal Names: FN570466
Type family: Sakaguchiaceae Q.M. Wang, F.Y. Bai, M. Groenew. & Boekhout, Stud. Mycol. 81:177 (2015)
Type genus: Sakaguchia Y. Yamada et al., Biosc. Biotechn. Biochem. 58:102. 1994. emend. Q.M. Wang, F.Y. Bai, M. Groenew. & Boekhout., Stud. Mycol. 81:177 (2015)
The diagnosis of the order Sakaguchiales is based on the description of the family Sakaguchiaceae, which is based on the genus Sakaguchia: “Sexual reproduction in some species. Clamp connections present. Teliospores laterally or terminally on the hyphae. Teliospores germinate with two- to four-celled metabasidium with lateral and terminal basidiospores. Colonies red or orange-red and butyrous. Budding cells present. Pseudohyphae or true hyphae present or not. Ballistoconidia not produced” (Wang et al. 2015c).
Microbotryales R. Bauer & Oberw., Can. J. Bot. 75:1309 (1997)
= Leucosporidiales J.P. Samp. et al., Mycol. Prog. 2: 61 (2003)
Type family: Microbotryaceae R.T. Moore. Can. J. Bot. 75:1309 (1997)
3. Subphylum Ustilaginomycotina Doweld, Prosyllabus Tracheophytorum, Tentamen Systematis Plantarum Vascularium (Tracheophyta) (Moscow): LXXVIII (2001)
Class Exobasidiomycetes Begerow, M. Stoll & R. Bauer, Mycologia 98(6): 908 (2007) [2006]
Class Malasseziomycetes Denchev & T. Denchev, Index Fungorum 145: 1 (2014)
Class Moniliellomycetes Boekhout, Q.M. Wang & F.Y. Bai, Persoonia 33:46 (2014)
Class Ustilaginomycetes R. Bauer, Oberw. & Vánky, Can. J. Bot. 75: 1311 (1997)
Phylogenetic support
Ustilaginomycotina is a monophyletic group with 100% BS and 1.0 PP support (Figs. 1, 2, 3). The classes Malasseziomycetes, Moliliellomycetes and Ustilaginomycetes have 100% BS and 0.96–1.0 PP support in both analyses, however Exobasidiomycetes is polyphyletic in the molecular dating analysis and lacks good support in the ML analysis. Ustilaginomycotina diverged at 430 Ma, and the classes later diverged between 245 and 265 Ma (Fig. 2; Table 1). The orders of Ustilaginomycetes, Malasseziomycetes and Moliliellomycetes are monophyletic with 100% BS and 1.0 PP support (Figs. 1, 2, 3; Table 1). The orders Exobasidiales and Doassansiales of Exobasidiomycetes do not have high BS support in the ML analysis, but have 1 PP support in the molecular dating analysis, which only includes a small number of species. The mean of stem age of all orders in this subphylum are generally 187–265 Ma (Fig. 2; Table 1). Two recently proposed orders Golubeviales and Robbauerales of Exobasidiomycetes are divergent at 202 Ma (Fig. 2; Table 1).
Discussion
Ustilaginomycotina contains more than 1000 species, and comprises plant pathogenic fungi (smuts), which are mostly dimorphic and have a yeast-like stage during their life cycle. Traditionally Ustilaginomycotina contained two classes: Ustilaginomycetes and Exobasidiomycetes (Begerow et al. 2006; Matheny et al. 2007a; Hibbett et al. 2007). Malassezia grows on skin of animals and Moniliella (syn. Trichosporonoides) is a yeast-like taxon representing Moliliellomycetes. Those two genera and related orders are maintained in Ustilaginomycotina incertae sedis for a long time because their variable phylogenetic positions in different gene trees. They nest into Exobasidiomycetes or in Ustilaginomycetes using rDNA sequence data alone or in combination with protein genes (Begerow et al. 2000, 2006; Bauer et al. 2001; Weiß et al. 2004; Matheny et al. 2007a). Until 2014, different analyses based on the sequences of six-gene confirmed that Malassezia and Moniliella belong to Ustilaginomycotina, and forming deep and well-supported lineages. The classes Malasseziomycetes and Moniliellomycetes were therefore proposed for these lineages, with additional support from phenotypic characters (Wang et al. 2014). In a recent phylogenetic study on Ustilaginomycotina using seven genes, Exobasidiomycetes resulted paraphyletic (Wang et al. 2015b). Two orders Golubeviales and Robbauerales were introduced in Exobasidiomycetes, so that there are eight orders in this class (Wang et al. 2015b).
In our study, the recently introduced classes Malasseziomycetes and Moniliellomycetes along with Ustilaginomycetes are well-supported and their divergence times fall within ranges of 245–330 Ma which is the most classes have. The class Exobasidiomycetes is paraphyletic as in the previous studies (Wang et al. 2014, 2015b). Two recently proposed orders Golubeviales and Robbauerales of Exobasidiomycetes are also supported in our study.
4. Subphylum Wallemiomycotina Doweld, in Index Fungorum no. 73 (2014)
Type class: Wallemiomycetes Zalar, de Hoog et Schroers, Antonie van Leeuwenhoek, 87:322 (2005)
Type order: Wallemiales Zalar, de Hoog & Schroers, Antonie van Leeuwenhoek 87(4): 322 (2005)
Type family: Wallemiaceae R.T. Moore, Rhizoctonia Species, Taxonomy, Molecular Biology, Ecology, Pathology and Disease Control (Dordrecht): 20 (1996)
Type genus: Wallemia Johan-Olsen. designated by Doweld in Index Fungorum no. 73 (2014)
Type species: Wallemia ichthyophaga Johan-Olsen 1887, designated by Zalar et al., Antonie van Leeuwenhoek, 87:322 (2005)
Phylogenetic support
Wallemiomycota is represented by four species in the ML analysis (Fig. 1). Sequences from the type strain of Wallemia tropicalis (stain number EXF_8739, from Jančič et al. 2016), and those of voucher stains for W. ichthyophaga, W. muriae and W. sebi from previous studies (Zalar et al. 2005; Matheny et al. 2007a) are included in the analyses. All Wallemia species cluster together with 100% BS and 1.0 PP support (Figs. 1, 2, 3). Wallemiomycotina is sister to other three subphyla of Basidiomycota in the ML and molecular dating analyses with 100% BS and 1.0 PP support (Figs. 1, 2) respectively. The mean of stem age of Wallemiomycotina is 487 Ma (Fig. 2). However in the phyloproteomics analysis it is sister to subphylum Agaricomycotina, then clade of Agaricomycotina + Wallemiomycotina is sister to the subphylum Ustilaginomycotina (Fig. 3).
Discussion
Subphylum Wallemiomycotina was proposed with a brief morphological description in Index Fungorum (Doweld 2014).
Based on a previous study, Wallemia is classified in the only family Wallemiaceae under the only order Wallemiales and the only class Wallemiomycetes based on phylogenetic analysis of 18S sequence data (Zalar et al. 2005). Four more species have recently been described (Jančič et al. 2016). Species of this class are xerophilic molds and have an unusual mode of asexual reproduction. Matheny et al. (2007a) investigated the phylogenetic placement using six nuclear genes. Their combined gene tree supports this class as being an isolated lineage and its position is basal to the core clade of Pucciniomycotina, Ustilaginomycotina and Agaricomycotina in Basidiomycota (Matheny et al. 2007a).
Our analysis, based on a larger sampling from Basidiomycota and Entorrhizomycota (Ascomycota as outgroups), confirms its distinct phylogenetic position, which is sister to other three subphyla of Basidiomycota with 100% BS and 1.0 PP support (Figs. 1, 2). While in the phyloproteomic analysis it shows up as sister to the Agaricomycotina (Fig. 3), which is in agreement with previous genomic studies (Zajc et al. 2013; Padamsee et al. 2012). In this study, we accept the class Wallemiomycetes to subphylum level and place it under Wallemiomycotina (Doweld 2014) to better fit its evolutionary history.
Phylum Entorrhizomycota R. Bauer, Garnica, Oberw., K. Riess, M. Weiß & Begerow, PLoS ONE 10(7): 10 (2015)
Phylogenetic support
Entorrhizomycota is represented by four species in our analysis, having 100% BS and 1.0 PP support, and is sister to the clade of Basidiomycota + Wallemiomycota (Figs. 1, 2). Entorrhiza and Talbotiomyces represent a deep dichotomy within the Entorrhizomycota in the ML Tree (Fig. 1). The stem age of this phylum is 530 Ma and is represented by two Entorrhiza species (Fig. 2).
Discussion
This phylum was established for a single genus, Entorrhiza, which is a small group of unusual teliosporic root parasites on Juncaceae and Cyperaceae (Begerow et al. 2006). Traditionally, species of Entorrhiza were considered as smut fungi and classified in Tilletiaceae (Zundel 1953). More recently based on ultrastructure and molecular phylogeny, Entorrhiza was accommodated in the distinct family, order and class, Entorrhizaceae, Entorrhizales and Entorrhizomycetes in the subphylum Ustilaginomycotina (Bauer et al. 1997; Begerow et al. 1997, 2006) and phylum Basidiomycota. It was recently separated from Basidiomycota and established as a novel phylum Entorrhizomycota containing the only class Entorrhizomycetes (Bauer et al. 2015). Molecular phylogenetic analyses as well as morphological and ecological data indicate that Talbotiomyces calosporus belongs to Entorrhizomycota, and a novel order Talbotiomycetales K. Riess, R. Bauer, R. Kellner, Kemler, Piątek, Vánky & Begerow and family Talbotiomycetaceae K. Riess, R. Bauer, R. Kellner, Kemler, Piątek, Vánky & Begerow were established based on this genus (Riess et al. 2015). Our study accepts this phylum based on the phylogeny and deep divergence.
Conclusions
Phylogenetic overview of Basidiomycota
In this study, we used sequence data from six-gene and a broad sampling that represents all classes of Basidiomycota except the Cryptomycocolacomycetes which lack proper sequence data. Almost all orders from each class have been included and are represented by all or most of known families. Exceptions are Auriculariales and Cantharellales (Agaricomycetes), Atractiellales (Classiculomycetes) and Ustilaginales (Ustilaginomycetes). We evaluated the relevance of higher-level taxa by considering whether or not they are monophyletic and have similar relative divergence times using the mean of stem age.
In our six-gene phylogenetic analysis (Figs. 1, 2), most higher-level taxa are monophyletic with sufficient statistical support (BS > 50% and PP > 0.8). Taxa that lacked high support included the Agaricomycotina, Gomphales and Exobasidiales (Figs. 1, 2); while some resulted polyphyletic/paraphyletic: Exobasidiomycetes, Cantharellales and Hysterangiales (Agaricomycetes), and Unilacrymales (Dacrymycetes). The recently introduced order Unilacrymales nests in the Cerinomycetaceae clade of Dacrymycetales in ML tree. Some of these unsupported taxa agree with previous studies, such as the polyphyletic groups in Exobasidiomycetes (Wang et al. 2015a), but the others differ from previous studies (Cantharellales in Veldrea et al. 2013; Hysterangiales in Hosaka et al. 2006; Unilacrymales in Shirouzu et al. 2013). Thus, further comprehensive studies are needed to clarify these disagreements.
Our results widely agree with dating estimates reported by previous researchers. For example, the phylum Basidiomycota originated more than 500 Ma according to Floudas et al. (2012), compared to 530 Ma in this study; Agaricomycotina diverged 429 Ma according to Floudas et al. (2012), compared to 406 Ma in this study. The divergence estimates for Agaricomycetidae were 149 Ma in Floudas et al. (2012) and 203–250 Ma in Garcia-Sandoval et al. (2011), versus 173 Ma in our study. The class Dacrymycetes originated 280–360 Ma according to Dentinger et al. (2010), versus 358 Ma in this study. The orders Boletales and Gloeophyllales originated during the early Cretaceous (around 66–146 Ma in Garcia-Sandoval et al. 2011) and in our study, they are divergent at 146 and 124 Ma respectively. However, in our study, Polyporales has a younger divergence time compared to the estimates of Garcia-Sandoval et al. (2011), which estimated the order Polyporales to have evolved during the late Jurassic, about 203–250 Ma.
Based on our study, the phyla and subphyla of Basidiomycota formed ca. 530 Ma and 406–490 Ma respectively, so we accept these divergence times ranges as supplementary criteria in ranking phyla and subphyla. These estimated divergence times of classes within Basidiomycota, were variable in different subphyla. For example, divergence times of classes in Agaricomycotina were estimated in a narrow range of 358–393 Ma. However, divergence times for classes of Pucciniomycotina and Ustilaginomycotina are mostly estimated 245–356 Ma, which are much wider than those classes of Agaricomycotina. However, the estimated divergence times of orders within those different subphyla remain between 120 and 290 Ma (Table 1). We use these divergence time ranges as criteria for the ranking of phyla, subphyla, classes and orders within Basidiomycota in this study.
Based on our relative divergence time estimates and phylogenetic analyses, we conclude that most higher-level taxa in Basidiomycota are well-supported, especially the recently introduced taxa: phylum Entorrhizomycota, classes Malasseziomycetes, Moniliellomycetes, Spiculogloeomycetes, Tritirachiomycetes and orders Holtermanniales, Trichosporonales, Golubeviales, Robbauerales, Amylocorticiales, Jaapiales, Stereopsidales and Lepidostromatales.
However, some taxa are not well-supported. The problems are found at the order level as follows: (1) Unilacrymales (Dacrymycetes) nests in the clade of Cerinomycetaceae of Dacrymycetales with 100% BS in ML tree (Fig. 1), however it is sister to Dacrymycetales with divergence time of 225 Ma in the MCC tree (Fig. 2). In this study we accept Unilacrymales but further scrutiny is needed. (2) Leucosporidiales (Microbotryomycetes) has a divergence of 74 Ma, which is much younger than other orders (120–290 Ma), thus we propose combining Leucosporidiales under the older name of Microbotryales. (3) Two new orders Buckleyzymales and Sakaguchiales are introduced to accommodate the families Buckleyzymaceae and Sakaguchiaceae (Cystobasidiomycetes) due to their older divergence times. (4) The oldest divergence time and distinct phylogenetic position of Cystofilobasidiales of Tremellomycetes indicates it should be raised to a higher rank. We however, do not introduce a new class here, as DNA sequence data from these taxa are not from their respective types, thus we await further studies. (5) Monophyletic evaluation and divergence time estimates for some higher-level taxa (i.e. Exobasidiomycetes, Cantharellales, Gomphales, and Hysterangiales) were based on limited species sequences in this study, therefore, await further scrutiny.
The tree topology of our phyloproteomic analysis largely agrees with the six-gene trees (Figs. 1, 2, 3). The main exceptions are followings: (1) the monophyletic Wallemiomycetes is sister to three subphyla of Basidiomycota (Agaricomycotina + Pucciniomycotina + Ustilaginomycotina) in the six-gene analysis, however, it sister to subphylum Agaricomycotina in phyloproteomic analysis. In this study we recognize this group at the subphylum level (Wallemiomycotina) but further studies are needed. (2) the positions of the Agaricomycetes orders Amylocorticiales, Auriculariales, Cantharellales, Geastrales, Sebacinales and Trechisporales are inconsistent between the six-gene phylogeny and phyloproteomic analyses, therefore, also need further scrutiny (Figs. 1, 2, 3).
Discussion on taxa rank using divergence times
The divergence time of a lineage could be used as a universal criterion for ranking taxa. However, there are still some obstacles to overcome, and one is how to choose proper calibration points in the dating analysis. Fossil isotopic ages aid estimation of molecular divergence time by providing points of calibration (Berbee and Taylor 2010). Several mushroom fossils have been discovered and these are used as calibration points in modern molecular clock analysis within Basidiomycota. Fossils included are Archaeomarasmius leggetti within the order Agaricales (Hibbett et al. 1995, 1997) and Quatsinoporites cranhamii within the order Hymenochaetales (Smith et al. 2004). These fossils calibrations have been used in several studies (Gueidan et al. 2011; Skrede et al. 2011, Floudas et al. 2012; Sánchez-Ramírez et al. 2015; Zhao et al. 2016; see Sánchez-Ramírez et al. 2017 for other potentially useful fossils). However, the fossil record for such a large group as Basidiomycota is relatively poor. Other sources that have the potential to address ages, are vicariant events and fossils of obligate symbionts of ectomycorrhiza hosts (Hibbett 2001; Hibbett and Matheny 2009; Matheny et al. 2009; Wilson et al. 2012; Sánchez-Ramírez et al. 2017), such as the putative suilloid ectomycorrhiza (LePage et al. 1997). The genera Termitomyces and Amylostereum which are associated with termite and sawflies respectively, could be dated through these arthropods (Slippers et al. 2003; Mikheyev et al. 2010; Nobre et al. 2011). Reliably identified fossils are still thought to be too few to establish reliable dates (Berbee and Taylor 2010; Hibbett 2014; Sánchez-Ramírez et al. 2017). Different researchers may also choose different calibration points in their dating analysis, and this may result in inconsistent divergence times for the same group (Sánchez-Ramírez et al. 2017). For example, the divergence estimates for Inocybaceae (Agaricales) was 143 (99–191) Ma according to Matheny et al. (2009), whereas the common ancestor of Inocybaceae and Crepidotaceae evolved ca. 45 (30–60) Ma according to Skrede et al. (2011).
Another difficulty in dating analysis is the diversification rates. In cladistics, a lineage consists of a stem and a crown group. The stem group refers to those of origin, which may include not only the most recent common ancestor of all of its members and their descendants, but also extinct lineages. The crown group comprises the last common ancestor of a living clade, plus all of its descendants (Budd and Jensen 2000; Budd 2001). Reflection of groups in divergence times are stem and crown age respectively. This means that for any given group of taxa the crown age is always younger than the stem age (Stadler et al. 2014). Some researchers have determined that the length of branch between stem ancestor and crown clade depends on factors, such the timescale, the net diversification rate, and the species richness of the clade (McPeek and Brown 2007; Stadler et al. 2014). At the species level, the most recent research has synthesized a global time tree of life from 50,632 species, and examined the pattern and rate of diversification, as well as the timing of speciation using genomic data. The rate of diversification in eukaryotes has been mostly constant (Hedges et al. 2015). In the present and our previous study (Zhao et al. 2016), we use stem ages to reflect divergence times.
Other factors which may affect the reliability of divergence time estimates are the exorbitant data sizes, the difficulty in correctly modelling the rate heterogeneity in highly diverse taxonomic groups, and the uncertain evolutionary rates for most groups of species. Evolutionary rates vary extensively among lineages when following autocorrelated and uncorrelated models (Berbee and Taylor 2010; Tamura et al. 2012). Some researchers have attempted to estimate the dating of evolutionary events using relaxed molecular clock analyses, which could compensate for the rate heterogeneity (Drummond et al. 2006; Garcia-Sandoval et al. 2011).
Tamura et al. (2013) proposed a method of relative time estimates (RelTime) for determining the relative ordering and spacing of evolutionary events, identifying lineages with significantly slower or faster evolutionary rates, diagnosing the effect of selected calibrations on absolute divergence times, and estimating absolute times of divergence when highly reliable calibration points are available. Divergence times from BEAST and RelTime methodologies are becoming universal criteria in taxa ranking. Although challenges in dating analysis are apparent as discussed above, the introduction of divergence times into systematics research under the framework of a robust phylogeny, will provide extra evidence to evaluate the application of higher taxon ranks. Presently, these may not be perfect clocks that reflects perfect lineages, but they provide extra quantitative evidence as compared to the known limitations expressed in morphologies in present modern systematics arrangements.
Phylogenomics perspectives
The examination of more than 400 articles reveals that aprox. 40% of the phylogenetic studies that used more than one gene have reported incongruence among gene phylogenies (Rokas and Chatzimanolis 2010). Thus, the phylogeny based on single-gene data is still insufficient to reconstruct consistent and accurate phylogenetic hypotheses due to the conflicting gene trees. Phylogenetic reconstructions from genome-scale data could eventually solve these problems (Rokas et al. 2003; Delsuc et al. 2005; Bushley and Turgeon 2010; Medina et al. 2011; Capella-Gutiérrez et al. 2012; Shelest and Voigt 2014).
In this study, we used 396 orthologous genes from 109 taxa with published genomes in Basidiomycota to detect the phylogenetic positions of the most higher-level taxa in this phylum. The results largely agree to those of phylogenetic topology based on six-gene sequences. However, there are some inconsistencies, such for the subphylum Wallemiomycotina, orders Amylocorticiales, Auriculariales, Cantharellales, Geastrales, Sebacinales and Trechisporales from Agaricomycetes. The increasing availability of genomic data will enable researchers to overcome those phylogenetic problems in the future. On the other hand, those inconsistent positions would provide the clues to demonstrate potential horizontal gene transfer, introgression, hidden paralogs and lineage sorting throughout the evolution of the Basidiomycota.
References
Aime MC, Matheny PB, Henk DA, Frieders EM et al (2006) An overview of the higher-level classification of Pucciniomycotina based on combined analyses of nuclear large and small subunit rDNA sequences. Mycologia 98:896–905
Aime MC, Toome M, McLaughlin D (2014) The Pucciniomycotina. In: McLaughlin D, Spatafora JW (eds) The mycota VII part A. Systematics and evolution, 2nd edn. Springer, Berlin, pp 271–294
Avise JC, Johns GC (1999) Proposal for a standardized temporal scheme of biological classification for extant species. Proc Natl Acad Sci USA 96:7358–7363
Bauer R, Begerow D, Nagler A, Oberwinkler F (2001) The Georgefischeriales: a phylogenetic hypothesis. Mycol Res 104:416–424
Bauer R, Begerow D, Sampaio JP, Weiß M, Oberwinkler F (2006) The simple-septate basidiomycetes: a synopsis. Mycol Prog 5:41–66
Bauer R, Garnica S, Oberwinkler F, Riess K et al (2015) Entorrhizomycota: A new fungal phylum reveals new perspectives on the evolution of fungi. PLoS ONE 10:e0128183
Bauer R, Oberwinkler F, Vánky K (1997) Ultrastructural markers and systematics in smut fungi and allied taxa. Can J Bot 75:1273–1314
Begerow D, Bauer R, Boekhout T (2000) Phylogenetic placements of ustilaginomycetous anamorphs as deduced from nuclear LSU rDNA sequences. Mycol Res 104:53–60
Begerow D, Bauer R, Oberwinkler F (1997) Phylogenetic studies on nuclear large subunit ribosomal DNA sequences of smut fungi and related taxa. Can J Bot 75:2045–2056
Begerow D, Stoll M, Bauer R (2006) A phylogenetic hypothesis of Ustilaginomycotina based on multiple gene analyses and morphological data. Mycologia 98:906–916
Beimforde C, Feldberg K, Nylinder S, Rikkinen J et al (2014) Estimating the Phanerozoic history of the Ascomycota lineages: combining fossil and molecular data. Mol Phylogenet Evol 78:386–398
Berbee ML, Taylor JW (2010) Dating the molecular clock in fungi—how close are we? Fungal Biol Rev 24:1–16
Binder M, Hibbett DS (2002) Higher-level phylogenetic relationships of homobasidiomycetes (mushroom-forming fungi) inferred from four rDNA regions. Mol Phylogenet Evol 22:76–90
Binder M, Hibbett DS, Larsson KH, Larsson E, Langer E (2005) The phylogenetic distribution of resupinate forms in the homobasidiomycetes. Syst Biodivers 3:113–157
Binder M, Larsson KH, Matheny PB, Hibbett DS (2010) Amylocorticiales ord. nov. and Jaapiales ord. nov.: early diverging clades of Agaricomycetidae dominated by corticioid forms. Mycologia 102:865–880
Blackwell M, Hibbett DS, Taylor JW, Spatafora JW (2006) Research coordination networks: a phylogeny for kingdom Fungi (Deep Hypha). Mycologia 98:829–837
Boekhout T, Fonseca A, Sampaio JP, Bandoni RJ et al (2011) Discussion of teleomorphic and anamorphic basidiomycetous yeasts. In: Kurtzman CP, Fell JW, Boekhout T (eds) The yeasts: a taxonomic study. Elsevier, Amsterdam, pp 1356–1367
Bonfante P, Genre A (2008) Plants and arbuscular mycorrhizal fungi: an evolutionary-developmental perspective. Trends Plant Sci 13:492–498
Buchfink B, Xie C, Huson DH (2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60
Budd GE (2001) Climbing life’s tree. Nature 412:487
Budd GE, Jensen S (2000) A critical reappraisal of the fossil record of the bilaterian phyla. Biol Rev 75:253–295
Bushley KE, Turgeon BG (2010) Phylogenomics reveals subfamilies of fungal nonribosomal peptide synthetases and their evolutionary relationships. BMC Evol Biol 10:26
Capella-Gutiérrez S, Marcet-Houben M, Gabaldón T (2012) Phylogenomics supports microsporidia as the earliest diverging clade of sequenced fungi. BMC Biol 10:47
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973
Chapela IH, Rehner SA, Schultz TR, Mueller UG (1994) Evolutionary history of the symbiosis between fungus-growing ants and their fungi. Science 266:1691–1694
Chen ZH, Zhang P, Zhang ZG (2014) Investigation and analysis of 102 mushroom poisoning cases in Southern China from 1994 to 2012. Fungal Divers 64:123–131
Dai YC, Cui BK, Si J, He SH et al (2015) Dynamics of the worldwide number of fungi with emphasis on fungal diversity in China. Mycol Prog 14:62
Dai YC, Cui BK, Yuan HS, Li BD (2007) Pathogenic wood-decaying fungi in China. For Pathol 37:105–120
Dai YC, Yang ZL, Cui BK, Yu CJ, Zhou LW (2009) Species diversity and utilization of medicinal mushrooms and fungi in China (Review). Int J Med Mushrooms 11:287–302
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772
De Silva DD, Rapior S, Fons F, Bahkali AH, Hyde KD (2012a) Medicinal mushrooms in supportive cancer therapies: an approach to anti-cancer effects and putative mechanisms of action. Fungal Divers 55:1–35
De Silva DD, Rapior S, Hyde KD, Bahkali AH (2012b) Medicinal mushrooms in prevention and control of diabetes mellitus. Fungal Divers 56:1–29
De Silva DD, Rapior S, Sudarman E, Stadler M et al (2013) Bioactive metabolites from macrofungi: ethnopharmacology, biological activities and chemistry. Fungal Divers 62:1–40
Delsuc F, Brinkmann H, Philippe H (2005) Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet 6:361–375
Dentinger BT, Ammirati JF, Both EE, Desjardin DE et al (2010) Molecular phylogenetics of porcini mushrooms (Boletus section Boletus). Mol Phylogenet Evol 57:1276–1292
Doweld AB (2014) Index Fungorum no. 73. http://www.indexfungorum.org/Names/NamesRecord.asp?RecordID=550364
Drummond AJ, Ho SY, Phillips MJ, Rambaut A (2006) Relaxed phylogenetics and dating with confidence. PLoS Biol 4:e88
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Phylogenet Evol 29:1969–1973
Edgar RC (2004a) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Edgar RC (2004b) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform 5:113
Fell JW, Boekhout T, Fonseca A, Scorzetti G, Statzell-Tallman A (2000) Biodiversity and systematics of basidiomycetous yeasts as determined by large-subunit rDNA D1/D2 domain sequence analysis. Int J Syst Evol Microbiol 50:1351–1371
Floudas D, Binder M, Riley R, Barry K et al (2012) The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 336:1715–1719
Garcia-Sandoval R, Wang Z, Binder M, Hibbett DS (2011) Molecular phylogenetics of the Gloeophyllales and relative ages of clades of Agaricomycotina producing a brown rot. Mycologia 103:510–524
Garnica S, Weiss M, Walther G, Oberwinkler F (2007) Reconstructing the evolution of agarics from nuclear gene sequences and basidiospore ultrastructure. Mycol Res 111:1019–1029
Gueidan C, Ruibal C, de Hoog GS, Schneider H (2011) Rock-inhabiting fungi originated during periods of dry climate in the late Devonian and middle Triassic. Fungal Biol 115:987–996
Hamamoto M, Nakase T (2000) Phylogenetic analysis of the ballistoconidium-forming yeast genus Sporobolomyces based on 18S rDNA sequences. Int J Syst Evol Microbiol 50:1373–1380
Hedges S, Marin J, Suleski M, Paymer M, Kumar S (2015) Tree of life reveals clock-like speciation and diversification. Mol Biol Evol. doi:10.1093/molbev/msv037
Hennig W (1966) Phylogenetic systematics. University of Illinois Press, Urbana
Hibbett DS (2001) Shiitake mushrooms and molecular clocks: historical biogeography of Lentinula. J Biogeogr 28:231–241
Hibbett DS (2006) A Phylogenetic overview of the Agaricomycotina. Mycologia 98:917–925
Hibbett DS (2014) Major events in the evolution of the Fungi. In: Losos J (ed) Princeton guide to evolution. Princeton University Press, Princeton, pp 152–158
Hibbett DS, Bauer R, Binder M, Giachini AJ et al (2014) Agaricomycetes. In: McLaughlin DJ, Spatafora JW (eds) The mycota, vol. VII, part A. Systematics and evolution, 2nd edn. Springer, Berlin, pp 373–429
Hibbett DS, Binder M, Bischoff JF, Blackwell M et al (2007) A higher- level phylogenetic classification of the Fungi. Mycol Res 111:509–547
Hibbett DS, Matheny PB (2009) Relative ages of ectomycorrhizal mushrooms and their plant hosts. BMC Biol 7:13
Hibbett DS, Pine EM, Langer E, Langer G, Donoghue MJ (1997) Evolution of gilled mushrooms and puffballs inferred from ribosomal DNA sequences. Proc Natl Acad Sci USA 94:12002–12006
Hibbett DS, Tsuneda A, Fukumasa-Nakai Y, Donoghue MJ (1995) Phylogenetic diversity in shiitake inferred from nuclear ribosomal DNA sequences. Mycologia 87:618–638
Hickerson MJ, Carstens BC, Cavender-Bares J (2010) Phylogeography’s past, present, and future: 10 years after Avise, 2000. Mol Phylogenet Evol 54:291–301
Hodkinson BP, Moncada B, Lücking R (2014) Lepidostromatales, a new order of lichenized fungi (Basidiomycota, Agaricomycetes), with two new genera, Ertzia and Sulzbacheromyces, and one new species, Lepidostroma winklerianum. Fungal Divers 64:165–179
Hongsanan S, Sánchez-Ramírez S, Crous PW, Ariyawansa HA et al (2016) The evolution of fungal epiphytes. Mycosphere 7:1690–1712
Hosaka K, Bates ST, Beever RE, Castellano MA, Colgan W 3rd, Domínguez LS, Nouhra ER, Geml J, Giachini AJ, Kenney SR, Simpson NB, Spatafora JW, Trappe JM (2006) Molecular phylogenetics of the gomphoid-phalloid fungi with an establishment of the new subclass Phallomycetidae and two new orders. Mycologia 98:949–959
Hyde KD, Hongsanan S, Jeewon R, Bhat DJ et al (2016) Fungal Diversity Notes 367–490: taxonomic and phylogenetic contributions to fungal taxa. Fungal Divers. doi:10.1007/s13225-016-0373-x
Hyde KD, Jones EBG, Liu JK, Ariyawansa H et al (2013) Families of Dothideomycetes. Fungal Divers 63:1–313
Hyde KD, Nilsson RHSA, Ariyawansa HA et al (2014) One stop shop: backbones trees for important phytopathogenic genera: I. Fungal Divers 67:21–125
James TY, Kauff F, Schoch CL, Matheny PB et al (2006) Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature 98:829–837
Jančič S, Zalar P, Kocev D, Schroers H-J et al (2016) Halophily reloaded: new insights into the extremophilic life-style of Wallemia with the description of Wallemia hederae sp. nov. Fungal Divers 76:97–118
Jülich W (1981) Higher taxa of Basidiomycetes. Bibliogr Mycol 85:1–845
Kemler M, Lutz M, Göker M, Oberwinkler F, Begerow D (2009) Hidden diversity in the non-caryophyllaceous plant-parasitic members of Microbotryum (Pucciniomycotina: Microbotryales). Syst Biodivers 7:297–306
Kirk PM, Cannon PF, Minter DW, Stalpers JA (2008) Ainsworth & Bisby’s dictionary of the fungi, 10th edn. CABI, Wallingford
Kottke I, Beiter A, Weiss M, Haug I et al (2003) Heterobasidiomycetes form symbiotic associations with hepatics: Jungermanniales have sebacinoid mycobionts while Aneura pinguis (Metzgeriales) is associated with a Tulasnella species. Mycol Res 107:957–968
Kozlov AM, Aberer AJ, Stamatakis A (2015) ExaML version 3: a tool for phylogenomic analyses on supercomputers. Bioinformatics 31:2577–2579
Kuhnert E, Surup F, Sir EB, Lambert C, Hyde KD, Hladki AI, Romero AI, Stadler M (2015) Lenormandins A—G, new azaphilones from Hypoxylon lenormandii and Hypoxylon jaklitschii sp. nov., recognised by chemotaxonomic data. Fungal Divers 71:165–184
Largent DL (1986a) How to identify mushrooms to genus vol. 1. Macroscopic features. Mad River Press, Eureka
Largent DL (1986b) How to identify mushrooms to genus vol. 3. Microscopic features. Mad River Press, Eureka
Larsson KH, Larsson E, Kõljalg U (2004) High phylogenetic diversity among corticioid Homobasidiomycetes. Mycol Res 108:983–1002
Lawrey JD, Binder M, Diederich P, Molina MC et al (2007) Phylogenetic diversity of lichen-associated Homobasidiomycetes. Mol Phylogenet Evol 44:778–789
LePage BA, Currah RS, Stockey RA, Rothwell GW (1997) Fossil ectomycorrhizae from the Middle Eocene. Am J Bot 84:410–412
Lepage T, Bryant D, Philippe H, Lartillot N (2007) A general comparison of relaxed molecular clock models. Mol Biol Evol 24:2669–2680
Li GJ, Hyde KD, Zhao RL, Hongsanan S et al (2016) Fungal Diversity Notes 253–366: taxonomic and phylogenetic contributions to fungal taxa. Fungal Divers 78:1–237
Liu JK (2004) Mycochemistry of high fungi. China Sci and Tech Press, Beijing
Liu NN, Ariyawansa HA, Hyde KD, Maharachchikumbura SSN et al (2016) Mycosphere essays X Perspectives into the value of genera, families and orders in fungal classification. Mycosphere 7:1649–1668
Liu XZ, Wang QM, Göker M, Groenewald M et al (2015a) Towards an integrated phylogenetic classification of the Tremellomycetes. Stud Mycol 81:85–147
Liu XZ, Wang QM, Theelen B, Groenewald M et al (2015b) Phylogeny of tremellomycetous yeasts and related dimorphic and filamentous basidiomycetes reconstructed from multiple gene sequence analyses. Stud Mycol 81:1–26
Lutzoni F, Kauff F, Cox CJ, McLaughlin D et al (2004) Assembling the fungal tree of life: progress, classification, and evolution of subcellular traits. Am J Bot 91:1146–1180
Maddison WP, Maddison DR (2016). Mesquite: a modular system for evolutionary analysis. Version 3.2. http://mesquiteproject.org/mesquite/mesquite.html
Maharachchikumbura SSN, Hyde KD, Jones EBG, McKenzie EHC et al (2015) Towards a natural classification and backbone tree for Sodariomycetes. Fungal Divers 72:199–301
Maharachchikumbura SSN, Hyde KD, Jones EBG, McKenzie EHC et al (2016) Families of Sordariomycetes. Fungal Divers 79:1–317
Matheny PB, Aime MC, Bougher NL, Buyck B et al (2009) Out of the Palaeotropics? Historical biogeography and diversification of the cosmopolitan ectomycorrhizal mushroom family Inocybaceae. J Biogeogr 36:577–592
Matheny PB, Curtis JM, Hofstetter V, Aime MC et al (2006) Major clades of Agaricales: a multilocus phylogenetic overview. Mycologia 98:982–995
Matheny PB, Gossmann JA, Zalar P, Kumar TKA, Hibbett DS (2007a) Resolving the phylogenetic position of the Wallemiomycetes: an enigmatic major lineage of Basidiomycota. Can J Botany 84:1794–1805
Matheny PB, Liu YJ, Ammirati JF, Hall BD (2002) Using RPB1 sequences to improve phylogenetic inference among mushrooms (Inocybe, Agaricales). Am J Bot 89:688–698
Matheny PB, Wang Z, Binder M, Curtis JM et al (2007b) Contributions of rpb2 and tef1 to the phylogeny of mushrooms and allies (Basidiomycota, Fungi). Mol Phylogenet Evol 43:430–451
McPeek MA, Brown JM (2007) Clade age and not diversification rate explains species richness among animal taxa. Am Nat 169:E97–E106
Medina ME, Jones GW, Fitzpatrick DA (2011) Reconstructing the fungal tree of life using phylogenomics and a preliminary investigation of the distribution of yeast prion-like proteins in the fungal kingdom. J Mol Evol 73:116–133
Mikheyev AS, Mueller UG, Abbot P (2010) Comparative dating of attine ant and lepiotaceous cultivar phylogenies reveals coevolutionary synchrony and discord. Am Nat 175(6):E126–E133
Moncalvo J-M, Lutzoni FM, Rehner SA, Johnson J, Vilgalys R (2000) Phylogenetic relationships of agaric fungi based on nuclear large subunit ribosomal DNA sequences. Syst Biol 49:278–305
Moncalvo J-M, Nilsson RH, Koster B, Dunham SM et al (2006) The cantharelloid clade: dealing with incongruent gene trees and phylogenetic reconstruction methods. Mycologia 98:937–948
Moore RT (1980) Taxonomic proposals for the classification of marine yeasts and other yeast-like fungi including the smuts. Bot Mar 23:361–373
Morehouse EA, James TY, Ganley ARD, Vilgalys R et al (2003) Multilocus sequence typing suggests the chytrid pathogen of amphibians is a recently emerged clone. Mol Ecol 12:395–403
Mueller UG, Gerardo NM, Aanen DK, Six DL, Schultz TR (2005) The evolution of agriculture in insects. Annu Rev Ecol Evol S 36:563–569
Nakase T (2000) Expanding world of ballistosporous yeasts: distribution in the phyllosphere, systematics and phylogeny. J Gen Appl Microbiol 46:189–216
Nobre T, Fernandes C, Boomsma JJ, Korb J, Aanen DK (2011) Farming termites determine the genetic population structure of Termitomyces fungal symbionts. Mol Ecol 20:2023–2033
Oberwinkler F (2012) Evolutionary trends in Basidiomycota. Stapfia 96:45–104
Padamsee M, Kumar TK, Riley R, Binder M et al (2012) The genome of the xerotolerant mold Wallemia sebi reveals adaptations to osmotic stress and suggests cryptic sexual reproduction. Fungal Genet Biol 49:217–226
Peláez F, Martínez MJ, Martínez AT (1995) Screening of 68 species of Basidiomycetes for enzymes involved in lignin degradation. Mycol Res 99:37–42
Pointing S (2001) Feasibility of bioremediation by white-rot fungi. Appl Microbiol Biotechnol 57:20–33
Pointing SB, Pelling AL, Smith GJD, Hyde KD, Reddy CA (2005) Screening of basidiomycetes and xylariaceous fungi for lignin peroxidase and laccase gene-specific sequences. Mycol Res 109:115–124
Rambaut A, Suchard M, Drummond A (2013) Tracer. http://tree.bio.ed.ac.uk/software/tracer/. Accessed 15 Sept 2016
Riess K, Bauer R, Kellner R, Kemler M et al (2015) Identification of a new order of root-colonising fungi in the Entorrhizomycota: Talbotiomycetales ord. nov. on eudicotyledons. IMA Fungus 6:129–133
Rinaldi AC, Comandini O, Kuyper TW (2008) Ectomycorrhizal fungal diversity: Separating the wheat from the chaff. Fungal Divers 33:1–45
Robinson NE, Robinson AB (2001) Molecular clocks. Proc Natl Acad Sci USA 98:944–949
Rokas A, Chatzimanolis S (2010) From gene-scale to genome-scale phylogenetics: the data flood in, but the challenges remain. In: Murphy WJ (ed) Phylogenomics. Humana Press, Totowa
Rokas A, Williams BL, King N, Carroll SB (2003) Genome-scale approachs to resolving incongruence in molecular phylogenies. Nature 425:798–804
Ruiz-Dueñas FJ, Martínez AT (2009) Microbial degradation of lignin: how a bulky recalcitrant polymer is efficiently recycled in nature and how we can take advantage of this. Microbiol Biotechnol 2:164–167
Ryberg M, Matheny PB (2012) Asynchronous origins of ectomycorrhizal clades of Agaricales). Proc R Soc B Biol Sci 279:2003–2011
Samarakoon MC, Hyde KD, Ariyawansa HA, Hongsanan S (2016) Mycosphere Essays X: divergence and ranking of taxa across the kingdoms Animalia, Fungi and Plantae. Mycosphere 7:1678–1689
Sánchez-Ramírez S, Wilson AW, Ryberg M (2017) An overview of phylogenetic and historical approaches to mycorrhizal biogeography, diversity, and evolution. In: Tedersoo L (ed) Mycorrhizal biogeography, ecological studies 230. Springer, Berlin
Sánchez-Ramírez S, Tulloss RE, Amalfi M, Moncalvo J-M (2015) Palaeotropical origins, boreotropical distribution and increased rates of diversification in a clade of edible ectomycorrhizal mushrooms (Amanita section Caesareae). J Biogeogr 42:351–363
Schell WA, Lee AG, Aime MC (2011) A new lineage in Pucciniomycotina: class Tritirachiomycetes, order Tritirachiales, family Tritirachiaceae. Mycologia 103:1331–1340
Scorzetti G, Fell JW, Fonseca A, Statzell-Tallman A (2002) Systematics of basidiomycetous yeasts: a comparison of large subunit D1/D2 and internal transcribed spacer rDNA regions. FEMS Yeast Res 2:495–517
Shelest E, Voigt K (2014) Genomics to study basal lineage fungal biology: phylogenomics suggests a common origin. Fungal genomics. In: Nowrousian M (ed) The mycota XIII, 2nd edn. Springer, Berlin
Shirouzu T, Hirose D, Oberwinkler F, Shimomura N et al (2013) Combined molecular and morphological data for improving phylogenetic hypothesis in Dacrymycetes. Mycologia 105:1110–1125
Shirouzu T, Hirose D, Tokumasu S (2009) Taxonomic study of the Japanese Dacrymycetes. Persoonia 23:16–34
Sinclair WA, Lyon HH, Johnson WT (1987) Diseases of trees and shrubs. Cornell University Press, Ithaca, p 574
Sjökvist E, Pfeil BE, Larsson E, Larsson KH (2014) Stereopsidales—a new order of mushroom-forming fungi. PLoS ONE 9:e95227
Skrede I, Engh IB, Binder M, Carlsen T et al (2011) Evolutionary history of Serpulaceae (Basidiomycota): molecular phylogeny, historical biogeography and evidence for a single transition of nutritional mode. BMC Evol Biol 11:230
Slippers B, Coutinho TA, Wingfield BD, Wingfield MJ (2003) A review of the genus Amylostereum and its association with woodwasps. S Afr J Sci 99:70–74
Smith SY, Currah RS, Stockey RA (2004) Cretaceous and Eocene poroid hymenophores from Vancouver Island, British Columbia. Mycologia 96:180–186
Stadler T, Rabosky DL, Ricklefs R, Bokma F (2014) On age and species richness of higher taxa. Am Nat 184:447–455
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690
Stefani FOP, Jones RH, May TW (2014) Concordance of seven gene genealogies compared to phenotypic data reveals multiple cryptic species in Australian dermocyboid Cortinarius (Agaricales). Mol Phylogenet Evol 71:249–260
Swann EC, Taylor JW (1993) Higher taxa of Basidiomycetes: an 18S rRNA gene perspective. Mycologia 85:923–936
Swann EC, Taylor JW (1995) Phylogenetic perspectives on basidiomycete systematics: evidence from the 18S rRNA gene. Can J Bot 73:S862–S868
Talavera G, Lukhtanov VA, Pierce NE, Vila R (2013) Establishing criteria for higher-level classification using molecular data: the systematics of Polyommatus blue butterflies (Lepidoptera, Lycaenidae). Cladistics 29:166–192
Tamura K, Battistuzzib FU, Billing-Rossb P, Murillob O et al (2012) Estimating divergence times in large molecular phylogenies. Proc Natl Acad Sci USA 109(47):19333–19338
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12):2725–2729
Taylor JW, Jacobson DJ, Kroken S, Kasuga T et al (2000) Phylogenetic species recognition and species concepts in fungi. Fungal Gen Biol 31:21–32
van Dongen S (2000) Graph clustering by flow simulation. PhD thesis, University of Utrecht, Netherlands
Van Driel GA, Humbel BM, Verkleu AJ, Stalpers J et al (2009) Septal pore complex morphology on the Agaricomycotina (Basidiomycota) with emphasis on the Cantharellales and Hymenochaetales. Mycol Res 113:559–576
Veldrea V, Abarenkova K, Bahrama M, Martosc F et al (2013) Evolution of nutritional modes of Ceratobasidiaceae (Cantharellales, Basidiomycota) as revealed from publicly available ITS sequences. Fungal Ecol 6:256–268
Vilgalys R, Hopple JS, Hibbett DS (1994) Phylogenetic implications of generic concepts in fungi: the impact of molecular systematic studies. Mycol Helv 6:73–91
Wang QM, Begerow D, Groenewald M, Liu XZ et al (2015a) Multigene phylogeny and taxonomic revision of yeasts and related fungi in the Ustilaginomycotina. Stud Mycol 81:55–83
Wang QM, Groenewald M, Takashima M, Theelen B et al (2015b) Phylogeny of yeasts and related filamentous fungi within Pucciniomycotina determined from multigene sequence analyses. Stud Mycol 81:27–53
Wang QM, Theelen B, Groenewald M, Bai FY, Boekhout T (2014) Moniliellomycetes and Malasseziomycetes, two new classes in Ustilaginomycotina. Persoonia 33:41–47
Wang QM, Yurkov AM, Göker M, Lumbsch HT et al (2015c) Phylogenetic classification of yeasts and related taxa within Pucciniomycotina. Stud Mycol 81:146–189
Wang YZ, Zhuang JY (1998) Flora fungorum sinicorum vol. 10 Pucciniales 1. Scinece Press, Beijing (in Chinese)
Wannathes N, Desjardin DE, Hyde KD, Perry BA, Lumyong S (2009) A monograph of Marasmius (Basidiomycota) from Northern Thailand based on morphological and molecular (ITS sequences) data. Fungal Divers 37:209–306
Weiß M, Bauer R, Begerow D (2004) Spotlights on heterobasidiomycetes. In: Agerer R, Blanz P, Piepen-bring M (eds) Frontiers in Basidiomycete mycology. Germany, IHW-Verlag, München, pp 7–48
Weiss M, Bauer R, Sampaio JP, Oberwinkler F (2014) Tremellomycetes and related groups. In: McLaughlin DJ, Spatafora JW (eds) Systematics and evolution, the mycota VII part A. Springer, Berlin, pp 349–350
White TJ, Bruns T, Lee S, Taylor JW (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic Press, New York, pp 315–322
Wilson AW, Binder M, Hibbett DS (2012) Diversity and evolution of ectomycorrhizal host associations in the Sclerodermatineae (Boletales, Basidiomycota). New Phytol 194:1079–1095
Wu G, Feng B, Xu J, Zhu XT et al (2014) Molecular phylogenetic analyses redefine seven major clades and reveal 22 new generic lineages in the fungal family Boletaceae. Fungal Divers 69:93–115
Wuczkowski M, Passoth V, Turchetti B, Andersson AC et al (2011) Description of Holtermanniella gen. nov., including Holtermanniella takashimae sp. nov. and four new combinations, and proposal of the order Holtermanniales to accommodate tremellomycetous yeasts of the Holtermannia clade. Int J Syst Evol Microbiol 61:680–689
Yang ZL (2011) Molecular techniques revolutionize knowledge of basidiomycete evolution. Fungal Divers 50:47–58
Zajc J, Liu Y, Dai W, Yang Z, Hu J, Gostinčar C, Gunde-Cimerman N (2013) Genome and transcriptome sequencing of the halophilic fungus Wallemia ichthyophaga: haloadaptations present and absent. BMC Genom 14:617
Zalar P, de Hoog GS, Schroers H-J, Frank JM, Gunde-Cimerman N (2005) Taxonomy and phylogeny of the xerophilic genus Wallemia (Wallemiomycetes and Wallemiales, cl. et ord. nov.). Antonie Van Leeuwenhoek J Microb 87:311–328
Zhang JX, Chen Q, Huang CY, Gao W, Qu JB (2015) History, current situation and trend of edible mushroom industry development. Mycosystema 34:524–540
Zhao Q, Feng B, Yang ZL, Dai YC et al (2013) New species and distinctive geographical divergences of the genus Sparassis (Basidiomycota): evidence from morphological and molecular data. Mycol Prog 12:445–454
Zhao RL, Karunarathna SC, Raspé O, Parra LA et al (2011) Major clades in tropical Agaricus. Fungal Divers 51:279–296
Zhao RL, Zhou JL, Chen J, Margaritescu S et al (2016) Towards standardizing taxonomic ranks using divergence times—a case study for reconstruction of the Agaricus taxonomic system. Fungal Divers 78:239–292
Zhuang JY (2003) Flora Fungorum Sinicorum Vol. 19 Pucciniales 2. Scinece Press, Beijing, 324 pp (In Chinese)
Zhuang JY (2005) Flora fungorum sinicorum vol. 25 Pucciniales 3. Scinece Press, Beijing (in Chinese)
Zhuang JY (2012) Flora fungorum sinicorum vol. 41 Pucciniales 4. Scinece Press, Beijing (in Chinese)
Zundel GL (1953) The Ustilaginales of the world, vol 176. Contributions from the Department of Botany Pennsylvania State College, pp. 1–410
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China to R.-L. Zhao (Project IDs 31470152 and 31360014) and G.-J. Li (Project ID 31500013), the Innovative Group of Edible Mushrooms Industry of Beijing (Project ID: BAIC05-2017) and the Key Research and Development Program from Government of Guangxi Zhuang Autonomous Region (Project ID: 2016AB05317) to R.-L. Zhao, the Thailand Research Fund to K.D. Hyde (Grant BRG 5580009), and the Natural Sciences and Engineering Research Council of Canada and the ROM Governors to J.-M. Moncalvo.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhao, RL., Li, GJ., Sánchez-Ramírez, S. et al. A six-gene phylogenetic overview of Basidiomycota and allied phyla with estimated divergence times of higher taxa and a phyloproteomics perspective. Fungal Diversity 84, 43–74 (2017). https://doi.org/10.1007/s13225-017-0381-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13225-017-0381-5