
Abstract
Lignin peroxidase (LiP) enzyme was purified from Kocuria rosea MTCC 1532. The enzyme was produced and subjected to purification by ion exchange chromatography followed by gel filtration chromatography; 79-fold purity was obtained. The molecular weight of LiP was determined to be 66 kDa. The pH optimum of the purified enzyme was 3.0, and the temperature optimum was found to be around 50°C. Purified LiP showed a much higher activity towards n-propanol than towards L-Dopa, 8-hydroquinone, mimosine, veratryl alcohol, and xylidine. The effect of inhibitor and metal ion on LiP activity was analyzed. Purified LiP was able to decolorize synthetic dyes from various groups, indicating that it is a versatile peroxidase.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
Dyes are released into the environment as industrial effluents from two major industrial sources, textile and dyestuff processing. About 10–15% of all dyes are directly lost to wastewater in the dyeing process (Robinson et al. 2001; Chen et al. 2008); consequently, the wastewater must be treated before being released back into the natural environment. The main methods currently used to treat textile wastewater involve physical and/or chemical processes, which are often very costly and, although the dyes are removed, the accumulation of concentrated sludge creates a disposal problem (Alam et al. 2009). There is also the possibility that a secondary pollution problem will arise due to excessive chemical use and that any subsequent treatment is economically unfeasible to solve. Biological processes provide an alternative to existing remediation technologies because they are more cost effective and environmentally friendly, and they do not produce large quantities of sludge (Robinson et al. 2001; Chen et al. 2010). The interest in new biocatalyst (enzyme) usage has been growing over the last two decades due to the increasing usage of xenobiotics, the degradation of which is not effective and efficient by means of conventional chemical and biological processes (Alam et al. 2009). The biodegradation ability of the bacteria is assumed to be associated with the production of lignolytic (LiP, laccase) and other biotransformation enzymes (Kalme et al. 2008; Kalyani et al. 2010; Parshetti et al. 2010). Ligninolytic enzymes have wide applications and are currently used in the removal of dyes from industrial (Ferreira-Leitặo et al. 2007) and bio-bleaching effluents (Silva et al. 2005) and for the treatment of hazardous waste (Huang et al. 2003). In nature, lignin peroxidase can oxidize both phenolic and non-phenolic lignin-related compounds, resulting in the cleavage of the Cα–Cβ bond and the aryl Cα bond, aromatic ring opening, phenolic oxidation, and demethoxylation (Christian et al. 2005). Due to its high redox potential and enlarged substrate range in the presence of specific mediators, LiP has great potential for application in various industrial processes.
Much of the work carried out to date on the degradation of dyestuffs by whole cultures, crude enzyme preparations, and purified ligninolytic enzymes has been carried out to effect the decolorization of different dyes using Phanerochaete chrysosporium (Podgornik et al. 1999). In recent years, a great deal of research has focused on developing processes in which purified enzymes are employed to remove/transform dyes from polluted wastewater (Telke et al. 2009). The demand for removal of dyes from the textile industrial waste using microbial peroxidase is steadily increasing (Ghodake et al. 2009; Gomare et al. 2008; Dawkar et al. 2009). In the study reported here, we purified and characterized the LiP from Kocuria rosea MTCC 1532 and then tested it for its ability to decolorize different groups of dyes.
Materials and methods
Microorganism and chemicals
Kocuria rosea MTCC 1532 was obtained from the Microbial Type Culture Collection (MTCC) Chandighar, India. The pure culture was maintained on nutrient agar (g l−1: NaCl, 5; peptone, 5; beef extract, 3) slants at 4°C with sub-culture once per month. DEAE (diethyl aminoethyl) cellulose was purchased from Sisco Research Laboratories (Mumbai, India), Biogel P100 was purchased from Bio-Rad (Hercules, CA), and 2, 2-azinobis-(3-ethylbenzthiazoline-6-sulfonate) (ABTS) was purchased from Sigma–Aldrich (St. Louis, MO). Methyl orange, cotton blue, and methyl violet were obtained from Sd. Fine Chemicals Limited (Biosar, India), and the textile dyes, amido black, orange HE2R, reactive blue 25, direct blue 6, reactive yellow 81, red HE4B, green HE4B, and reactive green 19 A were the generous gift of the local textile industry (Solapur, India). All chemicals were of the highest purity available and of analytical grade.
LiP production
The time course of LiP production by K. rosea MTCC 1532 was studied in 100 ml of nutrient broth at 30°C in the static condition. LiP activity (described in detail in following section) was measured in the crude cell extract of K. rosea MTCC 1532 cells grown at different time intervals. The dry weight of cells was measured according to Parshetti et al. 2010). For higher LiP production, 10% inoculum of 24-h (OD600nm: 1.0) culture of K. rosea MTCC 1532 was inoculated into 2 l of nutrient medium and incubated for 24 h at 30°C. The cells were collected by centrifugation at 8000 rpm for 15 min and suspended (100 mg ml−1) in 50 mM sodium phosphate buffer (pH 7.0; buffer A). These cells were then suspended in potassium phosphate buffer (50 mM, pH 7.4) for sonication (Model Vibra-cell; Sonics, Newtown, CT; sonifier output 40% amp; temperature 4°C; 7 strokes, each of 1-s duration with a 1-min interval between strokes; Parshetti et al. 2010). The cell lysate obtained was centrifuged twice at 10,000 rpm for 20 min at 4°C, and the clear supernatant used immediately or stored at 4°C until used to purify LiP.
Purification of LiP
All purification steps were carried out using a Bio-Rad protein purification system (EP 1-Econo pump) at 4°C. The clear supernatant obtained after centrifugation was loaded on a DEAE cellulose fast flow column (15 × 120 mm), equilibrated with buffer A (Ghodake et al. 2009). The column was washed with the same buffer (twofold the column volume), and the enzyme was eluted with a linear gradient of 0–1.0 M NaCl. Each fraction was assayed for protein and LiP activity. Fractions containing LiP activity were pooled and dialyzed against 1 mM sodium phosphate buffer (pH 6.0). The dialyzed sample was concentrated (1–2 ml) by ultrafiltration using a YM10 cut-off membrane (Amicon, Boston, MA) and loaded on a Biogel P100 column (10 × 500 mm) equilibrated with 50 mM sodium phosphate buffer (pH 6.0, buffer B). The protein elution was carried with the same buffer at a flow rate of 5 ml h−1. Fractions containing LiP activity were pooled and stored at 4°C until use.
Protein determination and enzyme activity
The protein concentration of each fraction was monitored by absorbance at 280 nm or by the Lowry method with bovine serum albumin as the standard (Lowry et al. 1951). Lignin peroxidase activity was determined by monitoring the propanaldehyde that formed at 300 nm in a reaction mixture of 2.5 ml containing 100 mM n-propanol, 250 mM tartaric acid, and 10 mM H2O2 (Shanmugam et al. 1999). The enzyme assay was carried out at 30°C; reference blanks contained all components except the assayed enzyme. One unit of enzyme activity was defined as the amount of enzyme required to oxidize 1 μmol of substrate.
Characterization of purified LiP
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out following the protocol of Laemmli (Laemmli 1970), with a 4% (w/v) stacking and 10% (w/v) resolving gel and run on a slab gel unit (Genetech Laboratories, Hyderabad, India). Protein bands were stained with 0.1% Coomassie brilliant blue R-250 (w/v) in methanol/acetic acid/water (v/v/v) (4:1:5) for 1 h at room temperature, followed by destaining using same solution without the stain. The molecular mass of the purified LiP was determined by calculating the relative mobility of standard protein markers (Genie, India) run on the same gel slab (205,000-Da myosin rabbit muscle, 0.5 mg ml−1; 97,400-Da phosphorylase b. 0.5 mg ml−1; 66,000-Da bovine serum albumin, 0.5 mg ml−1; 43,000-Da ovalbumin, 0.75 mg ml−1; 29,000-Da carbonic anhydrase, 0.5 mg ml−1; 20,100-Da soyabean trypsin inhibitor, 2.0 mg ml−1; 14,300-Da lysozyme, 0.75 mg ml−1). Zymogram analysis for LiP activity was performed on native-PAGE using 1 mM L-Dopa (L-3,4-dihydroxyphenylalanine) in 0.1 M acetate buffer (pH 4.9), after the gels had been washed for 1 h with the same buffer. The optimum pH for the purified LiP was examined in the pH range 1.0–7.0 (0.1 M KCl–HCl buffer, pH 1.0–2.0; glycine-HCl buffer, pH 3.0; sodium-acetate buffer, pH 4.0–5.0; sodium-phosphate buffer, pH 6.0-7.0) with 100 mM – propanol as a substrate in a 2.5-ml reaction mixture. The temperature optima for the purified LiP were studied in 2.5 ml of reaction mixture containing 100 mM n-propanol, 10 mM H2O2, and 0.2 M HCl–KCl buffer, pH 1.0. For the temperature study (20–70°C), the reaction mixture was incubated at each temperature for 10 min before the enzyme was added.
Substrate specificity and effect of inhibitors
Substrate specificity of the purified LiP was determined spectrophotometrically at 30°C in a 2.5-ml reaction mixture containing 10 mM H2O2 in 0.2 M HCl–KCl buffer, pH 3.0, at 30°C; the substrates (concentration 100 mM) included n-propanol, L-Dopa, 8-hydroxyquinone, mimosine, veratryl alcohol, and xylidine. The activity of peroxidase was tested in the presence of several metal ions (Cu2+, Cd2+, and Hg2+), supplied at a concentration of 100 mM. The activities were determined using n-propanol as substrate.
Decolorization dyes of different classes by purified lignin peroxidase
The decolorization of 11 different dyes by purified LiP was studied using 2.5 ml of the assay mixture containing 50 mM potassium phosphate buffer (pH 7.0), 40 mM H2O2, and 0.25 U of purified LiP. The dyes (50 μg) used in the decolorization assay were methyl orange, cotton blue, methyl violet, amido black, orange HE2R, reactive blue 25, direct blue 6, reactive yellow 81, red HE4B, green HE4B, and reactive green 19 A. After incubation at 30°C, decolorization was assayed by measuring the absorbance at the respective wavelength maximum of the dye; the percentage decolorization was determined as follows:
where A i is the initial absorbance and A t is the absorbance at incubation time t.
Results and discussion
Purification of LiP
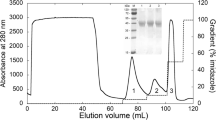
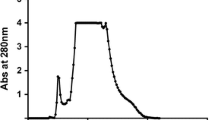
Peak intracellular activity of LiP in the crude cell-free extract was 0.197 ± 0.004 U after 24 h of cell culture (Fig. 1), with maximum LiP production occurring at 24 h of incubation. Although the dry cell weight increased up to 36 h of incubation, this was accompanied by a significant decline in LiP activity. The purification of LiP from K. rosea MTCC 1532 consisted of three steps, namely, isolation of crude LiP, anion exchange, and molecular sieve chromatography. The specific activity of LiP increased from 0.197 U in the crude cell lyzate to 70.0 U protein after passage through anion-exchange chromatography on a DEAE cellulose column. Figure 2 shows the elution pattern of LiP on the DEAE cellulose column. The anion-exchange resin (DEAE) served further to separate LiP activity from that of other proteins present in the fraction. The purification fold of LiP obtained through DEAE cellulose column was 33.14. A further increase in LiP activity and purification fold, up to 168.33 U and 79.70 fold, respectively, occurred following passage through a Biogel P100 column (gel filtration chromatography). Figure 3 shows the elution pattern of LiP on a Biogel P100 column. The purification steps, LiP activity, specific activity, and fold purification are summarized in Table 1. K. rosea MTCC 1532 is able to degrade dyes (Parshetti et al. 2010) and to induce the dye-degrading enzymes peroxidase, laccase, and reductase during the degradation of dyes. We required two chromatography steps to purify peroxidase from this species; in comparison; using Posocarpus decumbens P6, Yang et al. (2005) required four steps. The unique dye-decolorizing peroxidase, DyP, purified from Geotrichum candidum Dec 1 also required three steps for purification (Sugano et al. 2000).

Time course evolution of Kocuria rosea MTCC 1532 expressed as growth (dry weight) and lignin peroxidase (LiP) production, in the static condition at 30°C in 100 ml nutrient broth

Purification of K. rosea MTCC 1532 LiP by anion-exchange chromatography on a DEAE-cellulose column

Purification of K. rosea MTCC 1532 LiP by gel filtration chromatography on a Biogel P100 column
Characterization of peroxidase
Following molecular sieve chromatography of the enzyme, a single protein band appeared on the SDS–PAGE gel, with molecular mass of 66 kDa (Fig. 4, lane B), which is similar to an earlier report (Ghodake et al. 2009). The molecular weight of peroxidase varies with the bacterial species. The molecular weight of Lip obtained from Pseudomonas sp. SUK1 was estimated to be about 86 kDa (Kalyani et al. 2010), whereas the LiP obtained from Brevibacillus laterosporus MTCC 2298 was estimated to be about 205 kDa (Gomare et al. 2008). When the native gel was stained with L-Dopa, the band turned dark brown, indicating the presence of LiP (Fig. 4, lane C); the similar molecular mass also revealed the monomeric nature of peroxidase.

SDS–PAGE analysis of purified LiP. Lanes: A Molecular size markers, B protein staining of LiP purified by passage through a Biogel P100 column, C zymogram analysis for LiP performed on native- AGE using L-Dopa staining
Substrate specificity of LiP
The conventional substrates of LiP, such as n-propanol, L-Dopa, 8-hydroxyquinone, mimosine veratryl alcohol, and xylidine, were oxidized, and their relative activities are as given in Table 2. Of the different substrates tested, n-propanol was the substrate that was subjected to maximum oxidation by LiP.. In relation to n-propanol, the substrate specificity of LiP was, in descending order, veratryl alcohol 8-hydroxyquinone > L-Dopa > mimosine > xylidine. The peroxidase enzyme produced by the various microorganisms has wide substrate specificity; oxidization of hydroxyl- and methoxy-substituted phenols has been reported (Kim and Shoda 1999; Dawkar et al. 2009; Ghodake et al. 2009).
pH, temperature stability
The intact enzyme may contain both positively and negatively charged groups at any given pH. Such ionizable groups are often part of the active site (Gomare et al. 2008). Variation in the pH of the medium can result in changes in both the ionic forms of the active site and the activity of enzyme and, consequently, the reaction rate (Hossain and Anantharaman 2006). The effect of pH on enzyme activity was examined using n-propanol as substrate. Our resultr revealed that the optimum pH for this enzyme was 3.0 (Fig. 5a), which indicates that the LiP under study differs from that of Phanerochaete chrysosporium which had no appreciable activity at pH values close to 3.0. In two studies, LiP activity for n-propanol was maximal near pH 3.0 (Dawkar et al. 2009; Kalyani et al. 2010). The effect of temperature on LiP activity showed that it was active in the temperature range 40–70°C, with maximum activity at 50°C (Fig. 5b). The temperature range for the active enzyme is remarkably wide. Enzymatic activity declined with an increase in the temperature above 50°C.

pH (a) and temperature (b) optima for purified LiP from K. rosea MTCC 1532 with n-propanol as substrate
Effect various substrates and inhibitors of LiP activity
The substrate specificity of the purified peroxidase was examined using various substrates (Table 2). The peroxidase of K. rosea MTCC 1532 was able to oxidize n-propanol and various phenolic compounds, including veratryl alcohol, 3-(3, 4-dihydroxyphenyl) L-alanine (L-Dopa), 8-hydroxyquinone, xylidine, and mimosine. This peroxidase oxidized n-propanol much faster than the other substrates (hydroxyquinone, L-Dopa, and veratrole), which are phenolic and aromatic compounds. Typically, LiP produced by the microorganisms has wide substrate specificity, oxidizing hydroxyl- and methoxy-substituted phenols (Gomare et al. 2008; Dawkar et al. 2009; Kalyani et al. 2010). The peroxidases isolated from lignocellulose cultures were found to be able to oxidize different substituted phenols, such as guaiacol, syringol, and methoxyhydroquinone and also able to oxidize nonphenolic aromatic molecules, such as veratryl alcohol (Camarero et al. 1999). Similar results were also observed with the peroxidase we purified from K. rosea MTCC 1532, which can be used for degradation of different types of textile dyes.
The effect of peroxidase inhibitors was determined with n-propanol as a substrate at room temperature. Peroxidase activity was completely inhibited by NaN3 but only partially inhibited by EDTA. The effect of several metal ions, including Cu2+, Hg2+, and Cd2+, were tested on the activity of peroxidase. Peroxidase activity was inhibited by Cu2+, Cd2+, and Hg2+ (70, 40, and 35%, respectively) when present at a concentration of 100 mM. In terms of industrial applications, the activity of peroxidase in the presence of metal ions is an important property. Peroxidase purified from Lentinula edodes was found to be strongly inhibited in the presence of low concentrations of metal ions, while that from K. rosea MTCC 1532 is active at higher concentrations (Schwartz et al. 2001).
Decolorization of dyes
In our study, purified LiP under the long incubation conditions decolorized all dyes, but the heat-denatured enzyme did not decolorize the tested dyes. These results indicate that LiP is involved in color removal and corroborates recent observations (López et al. 2006). Purified LiP from K. rosea MTCC 1532 decolorized 11 different dyes to various extents, including azo, triphenylmethane, heterocyclic, polymeric, and metal complexes at neutral pH, indicating that the enzyme contributes decolorization ability at neutral pH and that peroxidases play a vital role in dye decolorization. Figure 6 shows the decolorization of dyes by purified lignin peroxidase observed after 5 h. The purified K. rosea MTCC 1532 MTCC 1532 LiP decolorized methyl orange (100%), cotton blue (60%), methyl violet (80%), amido black (60%), orange HE2R (90%), reactive blue 25 (100%), direct blue 6 (70%), reactive yellow 81 (80%), red HE4B (90%), green HE4B (100%), and reactive green 19 A (60%). The variation in the extent of the decolorization is possibly due to structural variations in these dyes (Murugesan et al. 2006)

Percentage decolorization of textile dyes by purified lignin peroxidase from K. rosea MTCC 1532 at pH 7.0
Conclusions
The ion exchange and gel filtration column chromatography technique were used to purify LiP enzyme from K. rosea MTCC 1532 up to 79-fold. The molecular weight of purified LiP was determined to be 66 kDa from SDS–PAGE analysis. Analysis of the properties of LiP showed the pH optimum to be 3.0 and that there was a wide range in the temperature optimum (40–70°C). Based on our results, purified LiP from K. rosea MTCC 1532 has a wide substrate specificity in terms of oxidation, including n-propanol, L-Dopa, 8-hydroxyquinone, mimosine, veratryl alcohol, and xylidine. We conclude that purified LiP from K. rosea MTCC 1532 is a versatile degrader of synthetic dyes of various groups.
References
Alam MZ, Mansor MF, Jalal KCA (2009) Optimization of decolorization of methylene blue by lignin peroxidase enzyme produced from sewage sludge with Phanerocheate chrysosporium. J Hazard Mater 162:708–715
Camarero SS, Sarkar FJ, Duenas R, Martınez MJ, Martınez AT (1999) Description of a versatile peroxidase involved in the natural degradation of lignin that has both manganese peroxidase and lignin peroxidase substrate interaction sites. J Biol Chem 274:10324–10330
Chen C, Chang C, Ho C, Tsai T, Liu S (2008) Biodegradation of crystal violet by a Shewanella sp. NTOU1. Chemosphere 72:1712–20
Chen C, Chang C, Liu S (2010) Partial degradation mechanisms of malachite green and methyl violet B by Shewanella decolorationis NTOU1 under anaerobic conditions. J Hazard Mater 177:281–89
Christian V, Shrivastavaa R, Shukla D, Modi H, Vyas BRM (2005) Mediator role of veratryl alcohol in the lignin peroxidase-catalyzed oxidative decolorization of Remazol Brilliant Blue R. Enzyme Microb Technol 36:327–332
Dawkar VV, Jadhav UU, Telke AA, Govindwar SP (2009) Peroxidase from Bacillus sp.VUS and its role in the decolorization of textile dyes. Biotechnol Bioprod Eng 14:361–368
Ferreira-Leitặo VS, de Carvalho MEA, Bon EPS (2007) Lignin peroxidase efficiency for methylene blue decolouration: comparison to reported methods. Dyes Pigments 74:230–236
Ghodake GS, Kalme SD, Jadhav JP, Govindwar SP (2009) Purification and partial characterization of lignin peroxides from Acinetobacter calcoaceticus NCIM 2890 and its application in decolourization of textile dyes. Appl Biochem Biotechnol 152:6–14
Gomare SS, Jadhav JP, Govindwar SP (2008) Degradation of sulfonated azo dyes by the purified lignin peroxidase from Brevibacillus laterosporus MTCC 2298. Biotechnol Bioprod Eng 13:136–143
Huang X, Wang D, Liu C, Hu M, Qu Y, Gao P (2003) The roles of veratryl alcohol and nonionic surfactant in the oxidation of phenolic compounds by lignin peroxidase. Biochem Biophys Res Commun 311:491–494
Hossain SM, AnantharamanN (2006) Activity enhancement of ligninolytic enzymes ofTrametes versicolor with bagasse powder. Afr J Biotechnol 5:189–194
Kalme S, Parshetti G, Gomare S, Govindwar S (2008) Diesel and kerosene degradation by diesel and kerosene degradation by Pseudomonas desmolyticum NCIM 2112 and Nocardia hydrocarbonoxydans NCIM 2386. Curr Microbiol 56:581–86
Kalyani DC, Phugare SS, Shedbalkar UU, Jadhav JP (2010) Purification and characterization of a bacterial peroxidase from the isolated strain Pseudomonas sp. SUK1 and its application for textile dye decolorization. Ann Microbiol. doi:10.1007/s13213-010-0162-9
Kim SJ, Shoda M (1999). Purification and characterization of anovel peroxidase from Geotrichum candidum Dec. 1 involved indecolourization of dyes. Appl Environ Microbiol 65:1029–1035
Laemmli U (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–85
López M, Guisado G, Vargas-García M, Suárez-Estrella F, Moreno J (2006) Decolorization of industrial dyes by ligninolytic microorganisms isolated from composting environment. Enzyme Microb Technol 40:42–45
Lowry O, Rosebrough N, Farr A, Randall R (1951) Protein measurement with the folin phenol reagent. J Biol Chem 193:265–75
Murugesan K, Arulmani M, Nam IH, Kim YM, Chang YS, Kalaichelvan PT (2006) Purification and characterization of laccase produced by a white rot fungus Pleurotus sajor-caju under submerged culture condition and its potential in decolorization of azo dyes. Appl Microbiol Biotechnol 72:939–946
Parshetti G, Telke A, Kalyani D, Govindwar S (2010) Decolorization and detoxification of sulfonated azo dye methyl orange by Kocuria rosea MTCC 1532. J Hazard Mater 176:503–509
Podgornik H, Grgiimag I, Perdih A (1999) Decolorization rate of dyes using lignin peroxidases of Phanerochaete chrysosporium. Chemosphere 38:1353–1359
Robinson T, McMullan G, Marchant R, Nigam P (2001) Remediation of dyes in textile effluent: a critical review on current treatment technologies with a proposed alternative. Bioresour Technol 77:247–55
Schwartz B, Olgin A, Klinman J (2001) The role of cooper in topa quinone biogenesis and catalysis, as probed by azide inhibiton of a copper amine oxidase from yeast. Biochemistry 40:2954–63
Shanmugam V, Kumari M, Yadav K (1999) n-Propanol as a substrate for assaying the lignin peroxidase activity of Phanerochaete chrysoporium. Ind J Biochem Biophys 36:39–43
Silva CMMS, Melo IS, Oliveira PB (2005) Ligninolytic enzyme production by Ganoderma spp. Enzyme Microb Technol 37:324–329
Sugano Y, Nakano R, Sasaki K, Shoda M (2000) Efficient heterologous expression in Aspergillus oryzae of a unique dye-decolorizing peroxidase, DyP, of Geotrichum candidum Dec 1. Appl Environ Microbiol 66:1754–1758
Telke A, Kalyani D, Jadhav U, Parshetti G, Govindwar S (2009) Purification and characterization of an extracellular laccase from a Pseudomonas sp. LBC1 and its application for the removal of bisphenol A. J Mol Catal B–Enzymes 61:252–60
Yang J, Yuan H, Wang HH, Chen W (2005) Purification and characterization of lignin peroxidases from Penicillium decumbens P6. World J Microbiol Biotechnol 21:435–440
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Parshetti, G.K., Parshetti, S., Kalyani, D.C. et al. Industrial dye decolorizing lignin peroxidase from Kocuria rosea MTCC 1532. Ann Microbiol 62, 217–223 (2012). https://doi.org/10.1007/s13213-011-0249-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13213-011-0249-y