
Abstract
Lignin peroxidase was purified (72-fold) from Acinetobacter calcoaceticus NCIM 2890. The purified lignin peroxidase (55–65 kDa) showed dimeric nature. The maximum enzyme activity was observed at pH 1.0, between a broad temperature range of 50 and 70°C, at H2O2 concentration (40 mM) and the substrate concentration (n-propanol, 100 mM). Purified lignin peroxidase was able to oxidize a variety of substrates including Mn2+, tryptophan, mimosine, l-Dopa, hydroquinone, xylidine, n-propanol, veratryl alcohol, and ten textile dyes of various groups indicating as a versatile peroxidase. Most of the dyes decolorized up to 90%. Tryptophan stabilizes the lignin peroxidase activity during decolorization of dyes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
More than 80,000 tonnes of reactive dyes are produced and consumed every year [1]. Among these, azo dyes are a prominent class of colorants used in tattooing, cosmetics, printing, and consumer’s products as well as in textile dying because of their chemical stability and versatility. Their durability, however, causes pollution once the dyes are released into the environment [2]. Synthetic textile dyes belong to the most dangerous pollutants, which as a part of the industrial effluents contaminate steadily higher amounts of wastewater. Based on the chemical structure of the chromophoric groups, dyes can be classified as azo, triphenylmethane, phthalocyanine, and anthraquinone dyes. Most of them are mutagenic and carcinogenic, and their removal is very difficult [3]. Processes based on physical and chemical treatment for decolorization of textile wastewater have a number of operational problems, secondary pollution problems, and high cost [4]. Currently, bioremediation is becoming important because of cost effectiveness, environmental friendliness, and production of less sludge as compare to the chemical and physical decomposition processes [5]. Lignin peroxidase (LiP) was the first discovered enzyme that catalyzes the partial depolymerization of methylated lignin in vitro [6]. Peroxidase catalyzes a variety of oxidation reactions [7]. LiP is capable of oxidizing a variety of xenobiotic compounds including polycyclic aromatic hydrocarbons, polychlorinated phenols, nitroaromatics, and azo dyes [8].
A majority of the work on degradation of dyestuffs by whole cultures, crude enzyme preparations, and purified ligninolytic enzymes have been carried out for the decolorization of the different dyes using Phanerochaete chrysosporium [9, 10], whereas the ligninolytic enzyme such as LiP from different bacterial sources involved in decolorization of dyes has been reported recently [11–13]. Versatile peroxidase (VP) has been recently described as a new family of ligninolytic peroxidases, together with LiP and manganese peroxidase (MnP) in P. chrysosporium for the first time [14]. The use of several Acinetobacter strains as a biocatalyst to remediate various environmental contaminants and other biotechnological applications has been reported recently [15]. Effluent-adapted strains of Acinetobacter, Bacillus, and Legionella have potentials for color removal, and strains of Acinetobacter, Bacillus, and Pseudomonas show chemical oxygen demand removal activities [16].
This study persuades us to study LiP, so the attempt was made for the purification of LiP from A. calcoaceticus and its characteristics like temperature, pH, substrate (n-propanol), and H2O2 optima. In addition, the role of tryptophan (TRP) as a stabilizer for the oxidation of four structurally different groups such as azo, thiazin, heterocyclic, and polymeric dyes were studied.
Materials and Methods
Dyes and Chemicals
Molecular weight markers were obtained from Bangalore Genei. Textile dyes such as Disperse red DK, Reactive red M5B, Direct blue GLL, Direct red 5B, Direct brown MR, and Direct brown T4LL were a generous gift from Manpasand Textile Processors, Ichalkaranji, India. Methyl orange and n-propanol were obtained from E-Merk (India). Methyl red and H2O2 were purchased from Qualigens. Ethylene blue and Congo red were purchased from BDH, India.
Microorganism, Media, and Culture Conditions
Acinetobacter calcoaceticus NCIM 2890 was obtained from the National Collection of Industrial Microorganisms (NCIM), National Chemical Laboratory, Pune, India. Enzyme production was studied in a batch culture nutrient medium containing NaCl 0.5%, bacteriological peptone1.0%, and beef extract 1.0% in 100 mL placed in 250-mL Erlenmeyer flasks at 30 °C.
Purification
Cells harvested after 24 h were grown in 250-mL Erlenmeyer flasks containing 100 mL nutrient medium after centrifugation at 7,000 rpm for 15 min at 4 °C and suspended (75 mg/mL) in 50 mM potassium phosphate buffer (pH 7.4). Cells were gently homogenized and disrupted by using sonication (Sonics vibracell, ultrasonic processor) at 4 °C giving ten strokes each of amplitude of 40 for 40 s with a 2-min interval. The crude enzyme was centrifuged at 10,000 rpm for 15 min at 4 °C. The crude enzyme preparation (80 mL) containing 0.138 U was applied on a diethylaminoethyl (DEAE) cellulose column (1 × 16 cm; Bio-Rad) pre-equilibrated with 50 mM sodium phosphate buffer (pH 7.0). The column was washed with 100 mL with the same buffer and eluted using a linear gradient of NaCl (0.0–1.0 M) with 50 mM sodium phosphate buffer (pH 7.0) containing 10 mM TRP as the stabilizing agent for LiP. The flow rate was maintained at 0.5 mL/min, and a total of 80 fractions each of 2.0 mL were collected. Each fraction was assayed for protein (A280 nm) and LiP activity with n-propanol (method mentioned below). The pooled active fractions were dialyzed against 50 mM sodium phosphate buffer (pH 7.0) and concentrated using a freeze dryer (Operon), stored at 4 °C, and used for the characterization and dye degradation experiments [17].
Enzyme Assays
The n-propanol oxidase activity was measured by monitoring the formation of propanaldehyde at 300 nm in a reaction mixture of 2.5 mL containing 100 mM n-propanol, 250 mM tartaric acid, and 10 mM H2O2 [18]. The enzyme assay was carried out at 30 °C, where the reference blank contained all components except the enzyme. One unit of enzyme activity was defined as a change in the absorbance U min−1 mg−1 of enzyme. The protein content of the crude LiP preparation was estimated by using the Biuret method. Absorbance at 280 nm was used to monitor the protein content of fractions.
MnP activity was determined by the modified method [19]. The 2.5-mL assay mixture contained 0.05 M sodium tartarate buffer (pH 4.5) and 1 mM MnSO4, and the reaction was started by the addition of 10 mM H2O2 and monitored at 238 nm.
Native and SDS-PAGE
Native and sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE) was performed on the vertical slab gel electrophoresis using acrylamide gel (10%) [20]. The samples were prepared by incubation at room temperature for 15 min in Tris–HCl buffer pH 6.8 (containing 50% sucrose, 5.0% SDS, and 0.5% bromophenol blue). Boiling of the sample was avoided because it causes aggregation of protein and loss of enzyme activity. After electrophoresis, the gels were stained with 0.1% coomossie brilliant blue (R-250) solution prepared in methanol–water–acetic acid (4:5:1) and destained in the same solution without dye. LiP bands on native and SDS-PAGE gels were developed by activity staining with l-Dopa [21, 22]. The assessments of purity of the enzyme after DEAE cellulose column chromatography, molecular weight, and the dimeric nature of LiP were analyzed by electrophoresis.
pH and Temperature Tolerance of Lignin Peroxidase
The pH optima of LiP were studied in the reaction mixture (2.5 mL) containing 100 mM n-propanol, 40 mM H2O2, HCl–KCl buffer 0.2 M (pH 1.0–2.2), and citrate phosphate buffer 0.1 M (pH 2.6–7.0), which was carried out at 30 °C. The temperature optima were studied in the reaction mixture of 2.5 mL containing 100 mM n-propanol, 40 mM H2O2, and 0.2 M HCl–KCl buffer, pH 1.0, which was first preincubated until required temperatures were reached (20–80 °C). The n-propanol oxidase activity was initiated by adding 0.2 mL enzyme.
H2O2 and Substrate Optima of Lignin Peroxidase
To study the H2O2 optima of LiP, a reaction mixture (2.5 mL) containing 100 mM n-propanol, 0.2 M HCl–KCl buffer pH 1.0, and H2O2 (2.5–100 mM) at 30 °C was used, and the substrate optima for n-propanol oxidation were studied in a reaction mixture (2.5 mL) containing n-propanol (10–140 mM) and 40 mM H2O2 in 0.2 M HCl–KCl buffer pH 1.0 at 30°C. The n-propanol oxidase activity was initiated by adding 0.2 mL enzyme.
Substrate Specificity for Lignin Peroxidase
The activity of LiP was determined by taking a reaction mixture of 2.5 mL containing 40 mM H2O2 in 0.2 M HCl–KCl buffer pH 1.0 at 30 °C, and the substrates, each of concentration 10 mM, include l-Dopa, n-propanol, TRP, mimosine, 8-hydroxyquinone, and veratryl alcohol (VA; lignin model compound); the absorbance change was measured at 300, 300, 305, 303, 310, and 300 nm, respectively. Substrate specificity of LiP was studied for different substrates [23–26].
Decolorization of Textile Dyes of Different Classes by Purified Lignin Peroxidase
The decolorization of ten different textile dyes by purified LiP was studied using 2.0 mL of the assay mixture containing 50 mM potassium phosphate buffer (pH 7.0), 40 mM H2O2, 2.5 mM TRP as the stabilizing agent, and 0.24 U of purified LiP. Dye concentration (µg) used for decolorization assay was Methyl red (200), Methyl orange (25), Ethylene blue (25), Disperse red DK (150), Congo red (50), Direct blue GLL (100), Direct red 5B (50), Direct brown MR (50), Direct brown T4LL (50), and Reactive red M5B (100). After incubation at 30 °C, decolorization was assayed by measuring the absorbance at the respective wavelength of the dye, and percent decolorization was determined as follows:
Results and Discussion
Purification of Lignin Peroxidase
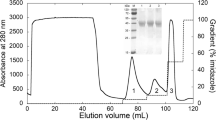
The fractionation of enzyme proteins on the basis of charge was achieved by the DEAE cellulose ion exchange column chromatography technique. The elution was performed by using the linear gradient system of NaCl (0.0–1.0 M) with 50 mM sodium phosphate buffer (pH 7.0). Two major protein peaks were observed; the first peak does not showed LiP activity, while the second peak (fraction no. 71–77) showed the presence of LiP activity (Fig. 1). The fractions containing LiP activity were pooled, concentrated, and used for further studies. The purification fold obtained after DEAE cellulose column chromatography was 72 along with 6.0 U (Table 1). Protein staining as well as l-Dopa activity staining (for the identification of LiP) of SDS-PAGE revealed the purity of LiP.

Elution profile of DEAE cellulose chromatography of lignin peroxidase from A. calcoaceticus protein (filled squares) and lignin peroxidase activity (empty squares)
Properties of Lignin Peroxidase
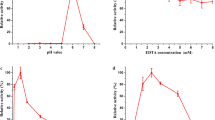
The properties of the H2O2-dependent LiP that catalyzed the oxidation of n-propanol were determined from initial velocity studies in which the pH and temperature variations were carried out. The pH optimum of LiP that catalyzed n-propanol oxidation was found to be 1.0 (Fig. 2). The LiP activity was observed at broad temperature range of 40–70 °C (Fig. 3). LiP uses H2O2 for its catalysis, but it is also known that it gets inactivated in the presence of higher concentrations of H2O2 [27]. The H2O2 optimum for LiP was 40 mM, while the substrate optimum for n-propanol oxidation was 100 mM (Fig. 4).

pH optima of lignin peroxidase

Temperature optima of lignin peroxidase

H2O2 (filled squares) and n-propanol (empty squares) optima of lignin peroxidase
LiP is characterized by its ability to oxidize high-redox potential aromatic compounds including VA and by Mn2+ oxidation. VP has been recently described as a new family of ligninolytic peroxidases, together with LiP and MnP in P. chrysosporium [14].
Purified LiP from A. calcoaceticus was proficient to oxidize a variety of (high- and low-redox potential) substrates including Mn2+, TRP, mimosine, l-Dopa, hydroquinone, and n-propanol (Table 2) and different types of dyes indicating VP. LiP when applied on PAGE showed two bands for LiP activity at 55–65 and 110–130 kDa. However, SDS-PAGE of the same sample showed only one band at 55–65 kDa, indicating that the band obtained at 110–130 kDa in PAGE is due to the dimeric nature of the protein. The molecular weight of purified LiP (intracellular and extracellular) estimated by native as well as SDS-PAGE was 55–65 kDa for the monomer, while the dimer showed the molecular weight 110–130 kDa, which is always present in the purified sample. The results also showed a similar nature of intracellular and extracellular LiP (Fig. 5). The molecular weights of these two major proteins (H2 and H8) from P. chrysosporium as determined by SDS-PAGE were 38,500 (H2) and 42,000 (H8). The pH optimum for VA oxidase activity was pH 2.5 at 25 °C [28]. These characteristics showed a significant difference with well-studied LiP from P. chrysosporium [29].

Polyacrylamide gel electrophoresis: a Native page activity staining by l-Dopa (10 mM); b native page protein staining. Lane 1, purified LiP; lane 2 molecular weight markers in order of increasing molecular weight; lane 3, cell-free extract. c SDS-PAGE protein staining. Lane 1, molecular weight markers; lane 2, purified LiP. d SDS-PAGE activity staining by l-Dopa (10 mM)
Dye Decolorization by Lignin Peroxidase
We have determined the pH optimum for the decolorization of azo-dye Methyl orange and Ethylene blue, which was found to be at pH 7.0. Hence, we have studied the decolorization of dyes at pH 7. However, the pH optimum was 1 when n-propanol is used as a substrate. This also indicates that the pH optimum is depending on the substrate.
Purified LiP from A. calcoaceticus degraded ten different dyes at various extents tested from different classes, indicating that it contributes extensive decolorization ability in the presence of 2.5 mM TRP. TRP enhances the oxidation of azo-dyes in the same way that VA does by LiP [30]. A novel peroxidase (DyP) studied from Geotrichum candidum degraded 7 of the 18 dyes, which was reported by Seong and Makoto [10]. When the decolorization of different dyes was tested, purified LiP showed a higher decolorization rate for monoazo-dyes than for thiazin and heterocyclic textile dyes (Fig. 6). A high decolorization rate was observed for the azo-dye such as Methyl red 98% and Methyl orange 96% within 24 h, while Direct blue GLL, Direct red 5B, Direct brown MR, Reactive red 2, and Ethylene blue decolorized up to 87%, 79%, 75%, 73%, and 84%, within 48 h, respectively. Direct brown T4LL, Disperse red DK, and Congo red showed comparatively less decolorization than the others (63.2%, 63.3%, and 67.9%, within 48 h, respectively). These results were compatible with the decolorization activity obtained for the azo-dye RB182 [10] and the decolorization of Congo red, Poly R-478, and Poly T-128, by LiP isoenzymes from P. chrysosporium [9].

Percent decolorization of textile dyes by purified lignin peroxidase from A. calcoaceticus at pH 7.0
Conclusions
The DEAE cellulose ion exchange column chromatography technique was used to fractionate enzyme proteins on the basis of charge up to 72-fold. The properties of LiP showed the pH optimum at 1.0, broad temperature optimum between 50 and 70 °C, H2O2 optimum at 40 mM, and substrate optimum at 100 mM for n-propanol oxidation. Purified LiP from A. calcoaceticus shows wide substrate specificity and ability to oxidize a variety of substrates (high and low redox potential) including Mn2+, mimosine, TRP, l-Dopa, xylidine, hydroquinone, n-propanol, VA, and ten dyes from the azo, heterocyclic, thiazin, and polymeric groups.
References
Hessel, C. C., Allegre, M., Maisseu, F., & Charbit, P. (2007). Review Journal of Environmental Management, 83, 171–180.
Jadhav, J. P., & Govindwar, S. P. (2006). Yeast, 23, 315–323.
Eichlerova, I., Homoika, L., & Nerud, F. (2006). Journal of Industrial Microbiology & Biotechnology, 33, 759–766.
Marco, S. L., Carla, A., Ana, S., Jose, A. P., & Albino, A. D. (2005). Enzyme and Microbial Technology, 39, 151–155.
Verma, P., & Madamwar, D. (2003). World Journal of Microbiology & Biotechnology, 19, 615–618.
Tien, M., & Kirk, T. K. (1983). Science, 221, 661–663.
Kohler, H., & Jenzer, H. (1989). Free Radical Biology & Medicine, 6, 323–339.
Hammel, K. E. (2005). Organopollutant degradation by ligninolytic fungi. In L. Y. Young & K. E. Cerniglia (Eds), Microbial transformation and degradation of toxic organic chemicals (pp. 331–346). Wiley Series in Ecological and Applied Microbiology. New York: Wiley-Liss Inc.
Ollikka, P., Kirsi, A., Veli-Matti, L., Tuomo, G., Timo, R., & Ilari, S. (1993). Applied and Environmental Microbiology, 59, 4010–4016.
Seong, J. K., & Makoto, S. (1999). Applied and Environmental Microbiology, 65, 1029–1035.
Kalme, S. D., Parshetti, G. K., Jadhav, S. U., & Govindwar, S. P. (2007). Bioresource Technology, 98, 1405–1410.
Parshetti, G. K., Kalme, S. D., Saratale, G. D., & Govindwar, S. P. (2006). Acta Chimica Slovenica, 53, 492–498.
Kalyani, D. C., Patil, P. S., Jadhav, J. P., & Govindwar, S. P. (2008). Bioresource Technology, 99, 4635–4641.
Martinez, A. T. (2002). Enzyme and Microbial Technology, 30, 425–444.
Desouky, A. E. H. (2003). African Journal and Biotechnology, 2, 71–74.
Olukanni, O. D., Osuntoki, A. A., & Gbenle, G. O. (2006). African Journal and Biotechnology, 5, 1980–1984.
Jayraman, J. (1881). Desalting of proteins by dialysis. In J. Jayraman (Ed.), Laboratory manual of biochemistry (pp. 1–180, 1st ed.). New Delhi: Wiley.
Shanmugam, V., Kumari, M., & Yadav, K. D. (1999). Indian Journal of Biochemistry & Biophysics, 36, 39–43.
Hatvani, N., & Mecs, I. (2001). Process Biochemistry, 37, 491–496.
Laemmli, U. K. (1970). Nature, 227, 680–685.
Levin, L., Forchiassin, F., & Ramos, A. M. (2002). Mycolo, 94, 377–383.
Michael, A. B. T., & Rivers, D. B. (2007). Journal of Invertebrate Pathology, 94, 108–118.
Perez-Boada, M., Ruiz-Duen, F. J., Pogni, R., Basosi, R., Choinowski, T., Martinez, M. J., et al. (2005). Journal of Molecular Biology, 354, 385–402.
Ruiz-Duenas, F. J., Camarero, S., Perez-Boada, M., Martinez, M. J., & Martinez, A. T. (2001). Biochemical Society Transactions, 29, 117–121.
Victor, G. T., Martinez, A. T., Martinez, M. J., & Francisco, G. (2001). FEBS European Journal of Biochemistry, 268, 4787–4793.
Ferapontova, E. E., Castillo, J., & Gorton, L. (2006). Biochimica et Biophysica Acta, 1760, 1343–1354.
Rimko, T. H., Sybe, H., Pauline, J. M. T., & Jim, A. F. (1998). FEBS Letters, 422, 391–394.
Tuisel, H., Sinclair, R., Bumpus, J. A., Ashbaugh, W., Brock, B. J., & Aust, S. D. (1990). Archives of Biochemistry and Biophysics, 279, 158–166.
Helfried, T., Robert, S., John, A., Bumpus, W. A., Barry, J. B., & Steven, D. A. (1990). Archives of Biochemistry and Biophysics, 279, 158–166.
Collins, P. J., Field, J. A., Teunissen, P., & Dobson, A. D. W. (1997). Applied and Environmental Microbiology, 63, 2543–2548.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ghodake, G.S., Kalme, S.D., Jadhav, J.P. et al. Purification and Partial Characterization of Lignin Peroxidase from Acinetobacter calcoaceticus NCIM 2890 and Its Application in Decolorization of Textile Dyes. Appl Biochem Biotechnol 152, 6–14 (2009). https://doi.org/10.1007/s12010-008-8258-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-008-8258-4