
Abstract
Ultrasound-assisted extraction was used to investigate the polyphenolic compounds, particularly anthocyanins, present in myrtle alcoholic extracts. This type of extract is typical in the making of liqueurs obtained from herbs or plants, especially medicinal plants. The leaf extracts were found to contain flavonoids from the quercetin and myricetin families. Besides these, the berry extracts also showed the presence of anthocyanins, hydrolysable tannins and quinic acid. The antioxidant capacity was studied using the ORAC and TEAC methods and the polyphenol content was measured using the Folin–Ciocalteu method. The results showed that the values produced by the ORAC and TEAC methods were in agreement and that the antioxidant capacity correlated with the polyphenol content. The results showed that the leaf extracts exhibited higher antioxidant capacity than the berry extracts. The extraction method was easily implementable, and proved to be a swift method for obtaining bioactive compounds from vegetable matrices.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myrtus communis L., or myrtle, is an evergreen shrub typical of the Mediterranean flora, which grows wild across most of Portugal. It has small white flowers and dark purple berries (Fig. 1). Both the leaves and the flowers are delicately and pleasantly scented. It is a plant that has long been known for its medicinal properties. It is also used in the production of liqueurs, for which the berries or a mixture of leaves and berries is macerated in alcohol for a prescribed amount of time. This process can sometimes take years, as is the case of the myrtle liqueur produced in the Portuguese region of Arrábida, a hilly coastal region not far from Lisbon.

Leaves, flowers and berries of M. communis L.
In this work, we investigated the antioxidant capacity of extracts of M. communis L. obtained by maceration of leaves and berries in a hydroalcoholic solution using ultrasound-assisted extraction (USAE) to obtain extracts with antioxidant capacity (AOC). The high antioxidant capacity of compounds in M. communis L. has already been established in other works (Pereira et al. 2012, 2013, 2016), by using both conventional extraction techniques and supercritical fluid extraction (SFE) techniques. However, some compounds such as anthocyanins seem to be almost absent from SFE extracts. Likewise, the presence of hydrolysable tannins in myrtle extracts (obtained by conventional methods) has been reported by some authors (Aleksic and Knezevic 2014; Wannes et al. 2010; Nassar et al. 2010; Yoshimura et al. 2008) but they are also absent from the SFE extracts. Supercritical fluid extraction methods are known for their high selectivity (Sapkale et al. 2010; Veggi et al. 2011; Mendes et al. 2003). This is because these methods are based on changes of pressure (P) and temperature (T) and, therefore, the number of different extraction conditions is very large, as large as the possible (P, T) combinations. By contrast, ultrasound assisted extraction is a method that can very efficiently extract a large number of compounds from a vegetable matrix, because the ultrasounds cause the cell walls to burst, hence allowing more extensive contact between the vegetable matrix and the solvent. The technique, therefore, produces high extraction yields but is not very selective. To the best of our knowledge the ultrasound assisted extraction method has not been employed for the extraction of polyphenolic compounds of leaves and berries of myrtle. Therefore, the aim of this investigation was to identify as many antioxidant compounds as possible in M. communis L., using the ultrasound-assisted extraction method.
Experimental
Plant material
Samples of myrtle leaves and berries were collected from the Sintra area (central west coast of Portugal—latitude: 38°58′N; longitude: 9°21′W; altitude: 75 m). No soil tillage, fertilization and pest treatments were carried out in this area. After collection, the plant material was identified and deposited in the Herbarium of the Instituto Superior de Agronomia (Institute of Agronomy) of University of Lisbon. The leaves and berries were dried for 2 months out of sunlight, sealed in black bags, and kept at − 20 °C.
Chemicals
Standards of Quercetin-3-β-d-glucoside, myricetin, myricetin-3-O-rhamnoside, malvidin-3-O-glucoside were purchased from Extrasynthese (Genay, France). Quinic acid was purchased from Sigma-Aldrich (Steinheim Germany). Monobasic potassium phosphate was purchased from Merck (Darmstadt, Germany) and dibasic sodium phosphate was obtained from Fluka (Steinheim, Germany). Folin–Ciocalteu (FC) reagent, gallic acid and sodium carbonate were acquired from VWR (Leuven, Belgium). Fluorescein was obtained from Panreac (Barcelona, Spain). 2,2-Azobis(2-methylpropionamidine) dihydrochloride (AAPH) and 6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) were purchased from Acros Organics (Geel, Germany). 2,2-azinobis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) tablets and potassium persulphate were purchased from Sigma-Aldrich (Steinheim, Germany). Absolute ethanol p.a. obtained from Merck (Darmstadt, Germany). Methanol, acetonitrile, formic acid, all of the highest available purity, were purchased from Merck (Darmstadt, Germany).
Ultrasound-assisted extraction (USAE)
The antioxidant compounds were extracted indirectly in a sonication bath (Sonorex Super RK, 510 H, 35 kHz, maximum input power 320 W, Bandelin, Berlin, Germany). A sample of 30 g of plant material was immersed in a solution made of 135 mL of ethanol (96% v/v) and 27 mL of water. The mixture, thus obtained, was then sonicated for 30 min, followed by a stirring period of 24 h, in the dark. At the end of this time, it was again sonicated for 30 min. After the sonication procedure, the mixture was filtrated, to remove the solid residue, first by gravity filtration, followed by a vacuum filtration. The solution obtained was then evaporated in a rotating evaporator at 40 °C and protected from light. The samples were prepared with an extract concentration of 10 mg/mL in methanol.
HPLC–DAD–MS analysis
The extracts were analysed in a LC–MS with a ProStar 410 autosampler, two 212-LC chromatography pumps, a ProStar 335 diode array detector and a 500-MS ion trap mass spectrometer with an electrospray ionisation (ESI) ion source (Varian, Palo Alto, CA, USA). Data acquisition and processing were performed using Varian MS Control 6.9 software. The phenolic compounds were separated in a Polaris column (Varian) RP18-A (150 mm × 2 mm I.D., 5 μm particle size) while the anthocyanins were separated on a Merck Lichrocart RP18 column (250 mm × 4.6 mm ID, 5 µm particle size). In both cases, the columns were kept at a controlled temperature (35 °C). The samples were injected into the column via a Rheodyne injector with a 20 μL loop. The mobile phase, for the phenolic compounds, consisted of 0.1% (v/v) formic acid in water (A) and 0.1% (v/v) formic acid in acetonitrile (B). The gradient adopted, at a flow rate of 0.25 mL/min, was as follows: 0–2 min A:B (93:7) isocratic, 8 min A:B (75:25) isocratic, 35 min A:B (20:80) isocratic, 40 min A:B (0:100) isocratic, 45 min A:B (93:7) linear. For the anthocyanins, the mobile phase consisted of 0.1% (v/v) formic acid in water (A) and 0.1% (v/v) formic acid in methanol (B). The following gradient was used: 0 min A:B (100:0) isocratic, 20 min A:B (10:90) isocratic, 25 min A:B (10:90) isocratic, 30 min A:B (100:0) linear. The flow rate was 1 mL/min, and the LC eluent was post-column split in a ratio of 3:1 before being introduced into the mass spectrometer. The mass spectra were acquired in the ESI negative and positive ion modes, in the range from 100 to 1700 u; the optimized parameters were as follows: ion spray voltage, ± 4.9 kV; capillary voltage, 20 and − 60 V; RF loading, 90%. Nitrogen was used as a nebulising and drying gas, at a pressure of 35 and 10 psi, respectively; drying gas temperature, 350 °C. The multistage MS (MSn) spectra were obtained with an isolation window of 2.0 Da, excitation energy values of 1.0 and 2.5 V and an excitation time of 10 ms.
Identification of individual polyphenols was carried out using their retention times, and both spectroscopic and spectrometric data. Quantification of single polyphenols was directly performed by HPLC–DAD using a six-point regression curve built with the available standards (myricetin-3-O-rhamnoside, malvidin-3-O-glucoside). The regression equations and correlation coefficients were calculated using Microsoft Excel 2007 software with correlation coefficient values greater than 0.997. The UV spectra for the unidentified compounds were compared with the available library of previously recorded UV spectra for a wide range of compounds.
Phenolic content and antioxidant capacity measurement
The total content in polyphenols was obtained by the Folin–Ciocalteu colorimetric method which is based on the number of phenolic groups or other potential oxidisable groups present in compounds from the sample. The antioxidant capacity was evaluated by the TEAC and ORAC methods. These assays were carried out as described by Pereira et al. (2016).
Results and discussion
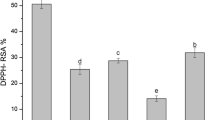
The extraction yield obtained for the extracts of leaves and berries was 10.81 ± 0.02 and 8.52 ± 0.01% w/w, respectively. These results showed that the highest yield was obtained from the leaf extract. There was also a difference in the physical appearance of the solutions. At the end of extraction by sonication, the solution obtained from the berries was reddish in colour, typical of anthocyanins, while the leaf solution was green in colour, characteristic of chlorophylls. Figure 2 shows the results for the total polyphenolic content and for the antioxidant capacity of the extracts. It can be seen that the values obtained for the leaves were significantly higher than those obtained for the berries, similar to the results obtained by other methods of extraction, namely, SFE and conventional extraction in accordance with earlier studies by Pereira et al. (2016), Amensour et al. (2009), Wang and Lin (2000) and Kanoun et al. (2014). Comparing the AOC results with those obtained by SFE (Pereira et al. 2016), the USAE method produced extracts with higher content in polyphenols, 206 μmol GAE/g plant against 30–60 μmol GAE/g plant for the SFE method, for leaves and 97 μmol GAE/g plant against 15–55 μmol GAE/g plant for the SFE method, for the berries. As for the AOC, the USAE method also produced higher values when it was measured by the TEAC method, with a value of 358 μmol Trolox/g plant against 55–130 μmol Trolox/g plant for the SFE method, for the leaves and 179 μmol Trolox/g plant against 25–80 μmol Trolox/g plant for the SFE method, for the berries. As for the AOC determined using the ORAC method, the results were similar for the leaves, 624 μmol Trolox/g plant for the USAE method against 530–759 μmol Trolox/g plant for the SFE method. For the berries, the results are, once again, much higher for the USAE method, 366 μmol Trolox/g plant against 130–250 μmol Trolox/g plant for the SFE method.

Total polyphenol content (TPC) evaluated by the Folin–Ciocalteu (FC) method and antioxidant capacity (AOC) evaluated by TEAC and ORAC methods for the leaves and berries extracts obtained by ultrasound assisted extraction
USAE methods have already been proved more efficient for the extraction of polyphenols, namely in the work reported by Singh et al. (2017) in which USAE was compared with conventional solvent extraction in mung beans.
In order to characterize the main phenolic compounds of the alcoholic extracts of M. communis L., these were further analyzed by HPLC–DAD–ESI–MS/MS. The chromatographic profile shown in Fig. 3 exhibits the separation of 13 compounds which, based on absorption maxima observed in the UV spectrum, fall into three subclasses of phenolic compounds. The first four partially-separated peaks, with a maximum wavelength of around 270 nm, consisted mainly of hydrolizable tannins, principally gallic acid derivatives; the five compounds separated at 525 nm can be assigned to the anthocyanin class; the last components to elute had a maximum absorbance in the region of 350–370 nm, which was characteristic of another group of flavonoids, the flavonols. Identification and assignment of individual polyphenols were established considering the HPLC–DAD analysis together with electrospray mass spectrometry (MS and MSn) data, based on the use of standards when available, and by comparison with published data. MS analysis was preferentially obtained in the ESI negative ion mode which gives higher sensitivity in the detection of the classes of flavonoids (Cuyckens and Claeys 2004), although identification of anthocyanins was achieved in the positive ion mode that provides more confident structural data Oliveira et al. 2001).

HPLC-DAD chromatograms at wavelengths of 268, 525 and 350 nm of berry extract obtained by ultrasound-assisted extraction
Table 1 summarizes the HPLC–DAD and MS data for the berry extract obtained by USAE method. Peak 1 eluting at Rt = 14.1 min displayed in the ESI(−) mass spectrum two main peaks, a double charged ion with m/z 783.5 (base peak), and a less abundant mono charged ion m/z 1568 attributed to the deprotonated molecule of an hydrolyzed tannin. MS/MS analysis of the double charged species originated pairs of monocharged fragment ions, namely ions m/z 935 [C41H27O26]− plus m/z 633 [C27H21O18]−, m/z 783 [C34H23O22]− plus m/z 615 [C27H19O17]− plus loss of a gallic acid unit (170 u), and m/z 765 [C34H21O21]− plus m/z 633 [C27H21O18]− plus loss of a gallic acid unit. Based on literature data (Lee et al. 2005) compound 1 was tentatively assigned as Oenothein B. This compound was identified by Yoshimura et al. (2008) in isolated extracts of M. communis L. leaves.
Peak 2 (Rt = 15.2 min) was attributed to a galloyl-hexahydroxydiphenol-glucose (galloyl-HHDP-glucose). The mass spectrum produced a peak at m/z 633 which gave in the MS2 analysis two product ions, one at m/z 481 due to the loss of a galloyl unit, a second one at m/z 301 attributed to the ellagic acid. Romani et al. (2012) identified this type of compound in M. communis L. extracts of leaves.
Peak 3 (Rt = 16.1 min), showed a deprotonated molecule at m/z 785, was assigned to a diagalloyl-HHDP-glucose (di-galloyl-hexahidroxidifenol-glucose), based on its MS2 fragmentation and by comparison with data reported by Romani et al. (2012).
Peak 4 (Rt = 16.7 min) exhibited a deprotonated molecule at m/z 495 and its MS2 generated fragments at m/z 343 (loss of 152 Da, corresponding to a residue of gallic acid), m/z 325 (loss of H2O), m/z 191 and 169, corresponding to quinic acid and gallic acid, respectively. By comparison with literature data Del Rio et al. (2004), compound 4 was identified as quinic acid 3,5-di-O-gallate. The presence of this acid, in M. communis L. extracts, was reported by Yoshimura et al. (2008).
Peak 5 (Rt = 17.3 min), shown in the ESI(+) spectrum an ion at m/z 465 attributed to a [M]+ cation. Its MS2 spectrum produced a peak at m/z 303 corresponding to the elimination of a glucose residue (loss of 162 Da), and its MS3 spectrum was very similar to that of delphinidin aglycone previously reported by Oliveira et al. (2001). Based on these results, peak 5 was assigned to delphinidin-3-O-monoglucoside (Sun et al. 2007).
As shown in Table 1 two compounds co-eluted in peak 6 at 17.9 min. The MS spectrum displayed two peaks at m/z 449 and 479 which generated in the MS2 analysis product ions at m/z 287 and 317, assigned to the cyanidin and petunidin aglycones, respectively (Oliveira et al. 2001). Peak 6 was attributed to two anthocyanins that eluted together, cyanidin-3-O-monoglucoside and a petunin-3-O-monoglucoside.
Peak 7 (Rt = 18.7 min) also revealed the co-elution of two molecular cations, one major compound with m/z 493, and a minor one with m/z 463. The MS2 spectra of both precursor ions showed loss of a glucose moiety leading to fragment ions with m/z 331 and 301, respectively. MS3 spectrum of the former displayed the characteristic fragments of the malvidin aglycon (Oliveira et al. 2001). The compound with m/z 493 was identified as maldivina-3-O-glucoside by comparing its MS data, UV spectrum and retention time to those of an authentic standard. In addition, the second compound that co-eluted at peak 7 was assigned to a peonidin-3-O-monoglucoside by comparison with published data (Tuberoso et al. 2010; Montoro et al. 2006a, b).
Peak 8 (Rt = 19.3 min) and peak 9 (Rt = 19.8 min) corresponding to cations with m/z 449 and 463, were identified as anthocyanin pentosides. Indeed, both precursor ions showed losses of 132 Da, indicative of a pentose residue, which leads to aglycone ions with m/z 317 and 331, respectively, indicating petunidin and malvidin derivatives. The MS3 spectra of the (Y0 +) ions were compared with literature data (Tuberoso et al. 2010; Montoro et al. 2006a, b) and with a standard of maldivina-3-O-glucoside. As a result, peak 8 was tentatively attributed to petunidin-3-O-pentoside whereas peak 9 was assigned to malvidin-3-O-pentoside.
Peak 10 (Rt = 20.0 min), had a deprotonated molecule at m/z 631 and was identified as myricetin-galactoside-gallate based on its MS data. MS2 spectrum showed only one peak at m/z 479 due to the loss of a gallic acid residue (152 Da), corresponding to the cleavage of the ester of gallic acid. MS3 spectrum exhibited two fragment ions, at m/z 317 and 316, assigned to the aglycone myricetin (Y0 −) and to the aglycone myricetin radical ion (Y0 −–H·), respectively. The formation of the aglycon is associated with the loss of 162 Da indicating the presence of a glucose/galactose in the structure of the molecule. The MS4 spectrum of m/z 317 showed as main fragments ions at m/z 289 (CO loss), 179 (1,2 A −0 ), 151 (1,2 A −0 –CO) and 137 (1,2 B −0 ), the diagnostic fragments of myricetin, and confirmed the presence of a myricetin derivative. Based on data published by Romani et al. (2012) and Romani et al. (1999) for M. communis L., the sugar moiety linked to the myricetin was assigned as galactose.
Peaks 11 and 12 (Rt = 20.7 and 21.3 min) had deprotonated molecules at m/z 479 and 463, similar UV profile (band II—254 nm, band I—362 nm) typical of a flavonol structure and characteristic MS2 fragments at m/z 317 (Y0 −) and m/z 316 (Y0 −–H·) after elimination of a hexose or a deoxyhexose unit (− 162 or 146 Da). The MS3 spectrum obtained for both Y0 − ions showed 1,2 A −0 , 1,2 A −0 –CO, 1,2 B −0 fragment ions, at m/z 179, 151 and 137, respectively, which are diagnostic of the flavonol myricetin. In addition to the i,jA− and i,jB− ions already discussed, the successive losses of 28 Da (CO) and 46 Da (C2H2O) leading to the ions at m/z 289 and 271 were also observed. These data confirmed the presence of myricetin-O-glycosides. Compound 11 was tentatively assigned to the myrcetin-3-O-galactoside, which has been previously reported in myrtle leaves by Romani et al. (1999). Compound 12 was fully identified as myricetin-3-O-rhamnoside by comparison with an authentic standard. This compound was also identified, in myrtle extracts, by Messaoud et al. (2012), Snoussi et al. (2012), Tuberoso et al. (2010), Barboni et al. (2010), Yoshimura et al. (2008) and Romani et al. (1999).
Peak 13 (Rt = 22.4 min) was attributed to quercetin-O-rhamnoside. The negative ESI mass spectrum of 13 displayed an ion with m/z 447, which in the MS2 spectrum produced a peak at m/z 301, due to the loss of 146 Da, indicating the presence of a rhamnose residue linked to the aglycone. In order to confirm the structure of the ion at m/z 301 as that of quercetin, a MS3 experiment was performed. The fragments found at m/z 179 (1,2 A −0 ), 151 (1,2 A −0 –CO) and 121 (1,2 B −0 ) confirmed the diagnostic ions expected for the quercetin aglycon, Fabre et al. (2001). The results were confirmed by comparison with the standard quercetin and with the results of Plazonic et al. (2009), Singh et al. (2009). The compound was also identified in extracts of M. communis L. by Romani et al. (1999, 2012) and Barboni et al. (2010).
Table 2 shows the HPLC–DAD-ESI/MSn results for the leaf extract. Peaks 1, 2 and 4 have already been described as they correspond to peaks 10, 11 and 12 in the berry extract.
Peak 3 (Rt = 14.8 min) was identified as quercetin-galactoside-gallate. The deprotonated molecule at m/z 615 produced a fragment ion at m/z 463 (loss of 152 Da) which, upon MS3 fragmentation, yielded the ion m/z 301 (loss 162 Da) attributed to a quercetin aglycone. This assignment was confirmed through the further dissociation of ion m/z 301 that yielded the typical fragment ions of quercetin. The compound 3 was previously reported by Sannomiya et al. (2005).
Peak 5 (Rt = 16.6 min) was identified as myricetin by comparison with the authentic standard. The MS spectrum showed a deprotonated molecule at m/z 317, which fragmented producing the diagnostic ions of myricetin. This compound was identified on M. communis L. extracts by Tuberoso et al. (2010), Wannes et al. (2010), Barboni et al. (2010) and Montoro et al.(2006a).
It should be mentioned that berry and leaf extracts were analyzed under different chromatographic conditions which explains the differences observed in the retention times of the identified flavonols. However, the order of elution was as expected based on the presence of one more hydroxyl group in the structure of myricetin.
Table 3 shows the quantitative composition obtained for the leaf and berry extracts. It can be observed that they differ in the content of anthocyanins, which are present in the berry extract but not in the leaf extract. This accounts for the different colours of the extracts, a reddish colour for the berry extract and green for the leaf extract.
The fact that the AOC for the extracts obtained by the USAE method is considerably higher than the AOC of the extracts obtained by the SFE method (Pereira et al. 2016) is probably because, as was already mentioned, these methods differ mostly in terms of selectivity. The results obtained here are consistent with this assumption, as the extracts obtained by USAE exhibited a higher number of compounds namely, myricetin-galactoside-gallate, quercetin-galactoside-gallate and myricetin in the leaves and oenothein B, galloyl-HHDP-glucose, digalloyl HHDP-glucose, quinic acid 3,5-di-O-gallate, cyanidin-3-O-glucoside, peonidin-3-O-monoglucoside and myricetin galactoside-gallate in the berries. These compounds are, most probably, the reason for the increase in the AOC found in the extracts obtained by USAE. It is also noticeable, when comparing with the results obtained by the SFE method (Pereira et al. 2016) that, as in the SFE method, extraction by USAE also produces mainly the glycoside form of the polyphenols and produces hardly any of its aglycone forms. The only exception is the myricetin that shows up in the leaf extracts obtained by USAE. Finally, and like other results already mentioned, the leaf extracts obtained here show a higher AOC than that of the extracts obtained from the berries, although in these the anthocyanin content is quite high. This result strengthens the argument that the high AOC observed in myrtle comes from the compounds belonging to the flavonols and flavanols and not from the anthocyanins, unlike what happens in other fruits such as raspberry and blackberry (Sariburun et al. 2010).
Conclusion
Ultrasound-assisted extraction proved to be quite an effective technique in extracting a wide range of polyphenols from a matrix of myrtle leaves and berries, producing extracts with high AOC. The main compounds found were myricetin-galactoside-gallate, quercetin-galactoside-gallate and myricetin in the leaves, and oenothein B, galloyl-HHDP-glucose, digalloyl HHDP-glucose, quinic acid 3,5-di-O-gallate, cyanidin-3-O-glucoside, peonidin-3-O-monoglucoside and myricetin galactoside-gallate in the berries. These compounds were found mainly in their glycoside form. Finally, because of the higher AOC observed in the leaves’ extracts we are led to believe that myrtle’s high AOC derives from the flavonols and flavanol compounds and not from the anthocyanins.
References
Aleksic V, Knezevic P (2014) Antimicrobial and antioxidative activity of extracts and essential oils of Myrtus communis L. Microbiol Res 169:240–254
Amensour M, Sendra E, Abrini J, Bouhdid S, Pérez-Alvarez J, Fernández-López J (2009) Total phenolic content and antioxidant activity of myrtle (Myrtus communis L.) extracts. Nat Prod Commun 4:819–824
Barboni T, Venturini N, Paolini J, Desjobert J-M, Chiaramonti N, Costa J (2010) Characterisation of volatiles and polyphenols for quality assessment of alcoholic beverages prepared from Corsican Myrtus communis L. berries. Food Chem 122:1304–1312
Cuyckens F, Claeys M (2004) Mass spectrometry in the structural analysis of flavonoids. J Mass Spectrom 39:1–15
Del Rio D, Stewart AJ, Mullen W, Burns J, Lean M, Brighenti F, Crozier A (2004) HPLC-MSn analysis of phenolic compounds and purine alkaloids in green and black tea. J Agric Food Chem 52:2807–2815
Fabre N, Rustan I, Hoffmann E, Quetin-Leclercq J (2001) Determination of flavone, flavonol, and flavanone aglycones by negative ion liquid chromatography electrospray ion trap mass spectrometry. J Am Soc Mass Spectrom 12:707–715
Kanoun K, Belyagoubi-Benhammou N, Ghembaza N, Atik Bekkara F (2014) Comparative studies on antioxidant activities of extracts from the leaf, stem and berry of Myrtus communis L. Int Food Res J 21:1957–1962
Lee J-H, Jonhson JV, Talcott ST (2005) Identification of ellagic acid conjugates and other polyphenolics in muscadine grapes by HPLC–ESI–MS. J Agric Food Chem 53:6003–6010
Mendes RL, Nobre BP, Cardoso MT, Pereira AP, Palavra AF (2003) Supercritical carbon dioxide extraction of compounds with pharmaceutical importance from microalgae. Inorg Chim Acta 356:328–334
Messaoud C, Laabidi A, Boussaid M (2012) Myrtus communis L. infusions: the effect of infusion time on phytochemical composition, antioxidant, and antimicrobial activities. J Food Sci 77:941–947
Montoro P, Tuberoso C, Piacente S, Perrone A, De Feo V, Cabras P, Pizza C (2006a) Stability and antioxidant activity of polyphenols in extracts of Myrtus communis L. berries used for the preparation of myrtle liqueur. J Pharm Biomed 41:1614–1619
Montoro P, Tuberoso C, Perrone A, Piacente S, Cabras P, Pizza C (2006b) Characterisation by liquid chromatography–electrospray tandem mass spectrometry of anthocyanins in extracts of Myrtus communis L. berries used for the preparation of myrtle liqueur. J Chromatogr A 1112:232–240
Nassar MI, Aboutabl E-S, Ahmed R, El-Khrisy E-A, Ibrahim K, Sleem A (2010) Secondary metabolites and bioactivities of Myrtus communis L. Pharmacogn Res 2:325–329
Oliveira MC, Esperança P, Ferreira A (2001) Characterisation of anthocyanidins by electrospray ionisation and collision-induced dissociation tandem mass spectrometry. Rapid Commun Mass Spectrom 15:1525–1532
Pereira P, Cebola MJ, Bernardo-Gil MG (2012) Comparison of antioxidant activity in extracts of Myrtus communis L. obtained by SFE vs solvent extraction. J Environ Sci Eng A 1:115–120
Pereira P, Bernardo-Gil MG, Cebola MJ, Maurício E, Romano A (2013) Supercritical fluid extracts with antioxidant and antimicrobial activities from myrtle (Myrtus communis L.) leaves. Response surface optimization. J Supercrit Fluid 83:57–64
Pereira P, Cebola MJ, Oliveira MC, Bernardo-Gil MG (2016) Supercritical fluid extraction vs conventional extraction of myrtle leaves and berries: comparison of antioxidant activity and identification of bioactive compounds. J Supercrit Fluid 113:1–9
Plazonic A, Bucar F, Males Z, Mornar A, Nigovic B, Kujundzic N (2009) Identification and quantification of flavonoids and phenolic acids in burr parsley (Caucalis platycarpos L.), using high-performance liquid chromatography with diode array detection and electrospray ionization mass spectrometry. Molecules 14:2466–2490
Romani A, Pinelli P, Mulinacci N, Vincieri FF, Tattini M (1999) Identification and quantification of polyphenols in leaves of Myrtus communis L. Chromatographia 49:17–22
Romani A, Campo M, Pinelli P (2012) HPLC/DAD/ESI–MS analyses and anti-radical activity of hydrolyzable tannins from different vegetal species. Food Chem 130:214–221
Sannomiya M, Montoro P, Piacente S, Pizza C, Brito AR, Vilegas W (2005) Application of liquid chromatography/electrospray ionization tandem mass spectrometry to the analysis of phenolics compounds from an infusion of Byrsonima crassa Niedenzu. Rapid Commun Mass Spectrom 19:2244–2250
Sapkale GN, Patil SM, Surwase US, Bhatbhage PK (2010) Supercritical fluid extraction—a review. Int J Chem Sci 8:729–743
Sariburun E, Sahin S, Demir C, Turkben C, Uylaser V (2010) Phenolic content and antioxidant activity of raspberry and blackberry cultivars. J Food Sci 75:328–335
Singh AP, Luthria D, Wilson DT, Vorsa N, Singh V, Banuelos GS, Pasakdee S (2009) Polyphenols content and antioxidant capacity of eggplant pulp. Food Chem 114:955–961
Singh B, Singh N, Thakur S, Kaur A (2017) Ultrasound assisted extraction of polyphenols and their distribution in whole mung bean, hull and cotyledon. J Food Sci Technol 54:921–932
Snoussi A, Hayet BH, Essaidi I, Zgoulli S, Moncef CM, Thonart P, Bouzouita N (2012) Improvement of the composition of Tunisian myrtle berries (Myrtus communis L.) alcohol extracts. J Agric Food Chem 60:608–614
Sun J, Liang F, Bin Y, Li P, Duan C (2007) Screening non-colored phenolics in red wines using liquid chromatography/ultraviolet and mass spectrometry/mass spectrometry libraries. Molecules 12:679–693
Tuberoso C, Rosa A, Bifulco E, Melis MP, Atzeri A, Pirisi FM, Dessi MA (2010) Chemical composition and antioxidant activities of Myrtus communis L. berries extracts. Food Chem 123:1242–1251
Veggi PC, Cavalcanti RN, Meireles MAA (2011) Modifier effects on supercritical fluid extraction (SFE) of some Brazilian plants: antioxidant activity and economical evaluation. Proc Food Sci 1:1717–1724
Wang S, Lin H-S (2000) Antioxidant activity in fruits and Leaves of blackberry, raspberry, and strawberry varies with cultivar and developmental stage. J Agric Food Chem 48:140–146
Wannes A, Mhamdi B, Sriti J, Jemia MB, Ouchikh O, Hamdaoui G, Kchouk ME, Marzouk B (2010) Antioxidant activities of the essential oils and methanol extracts from myrtle (Myrtus communis var. italica L.) leaf, stem and flower. Food Chem Toxicol 48:1362–1370
Yoshimura M, Amakura Y, Tokuhara M, Yoshida T (2008) Polyphenolic compounds isolated from the leaves of Myrtus communis L. J Nat Med 63:366–368
Acknowledgements
MCO would like to thank the Fundação para a Ciência e a Tecnologia (FCT), for its financial support from Projects UID/QUI/00100/2013 and REM2013.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Pereira, P., Cebola, MJ., Oliveira, M.C. et al. Antioxidant capacity and identification of bioactive compounds of Myrtus communis L. extract obtained by ultrasound-assisted extraction. J Food Sci Technol 54, 4362–4369 (2017). https://doi.org/10.1007/s13197-017-2907-y
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13197-017-2907-y