
Abstract
Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis are neurodegenerative disorders that are characterized by a progressive degeneration of nerve cells eventually leading to dementia. While these diseases affect different neuronal populations and present distinct clinical features, they share in common several features and signaling pathways. In particular, energy metabolism defects, oxidative stress, and excitotoxicity are commonly described and might be correlated with AMP-activated protein kinase (AMPK) deregulation. AMPK is a master energy sensor which was reported to be overactivated in the brain of patients affected by these neurodegenerative disorders. While the exact role played by AMPK in these diseases remains to be clearly established, several studies reported the implication of AMPK in various signaling pathways that are involved in these diseases’ progression. In this chapter, we review the current literature regarding the involvement of AMPK in the development of these diseases and discuss the common pathways involved.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- AMPK
- Neurodegeneration
- Alzheimer’s disease
- Parkinson’s disease
- Huntington’s disease
- Amyotrophic lateral sclerosis
1 Introduction
Neurodegenerative diseases including Alzheimer’s (AD), Parkinson’s (PD), Huntington’s (HD), and amyotrophic lateral sclerosis (ALS) are characterized by the progressive degeneration of nerve cells eventually leading to dementia. While these disorders affect different neuronal populations, they share in common several features. For instance, they are characterized by the presence of protein aggregates in degenerating neurons that likely result from defective clearance mechanisms including proteasomal dysfunction and lysosomal clearance. In addition, metabolic alterations, excitotoxicity, and oxidative stress are often described. All of the latter could participate in the deregulation of AMP-activated protein kinase (AMPK) that was reported to occur in these diseases (Fig. 7.1). AMPK is a heterotrimer composed of one α, β, and γ subunits, often referred to as a master energy sensor. Indeed, AMPK possesses on its regulatory γ subunit four CBS (cystathionine-beta-synthase) domains which are binding sites for adenine nucleotides. Three of these sites can bind AMP, ADP, and ATP (Sanders et al. 2007; Gowans et al. 2013; Xiao et al. 2011). Metabolic stresses that increase the AMP:ATP ratio will allow the preferential binding of AMP to the γ subunit, thereby inducing a conformational change and favoring the phosphorylation of the residue Thr172 located on the catalytic α subunit by upstream AMPKs (Sanders et al. 2007; Gowans et al. 2013). The liver kinase B1 (LKB1) seems to be mostly responsible for AMPK phosphorylation in these conditions (Hawley et al. 2003; Woods et al. 2003; Shaw et al. 2004). At least two other kinases were reported to phosphorylate AMPK on Thr172, the calcium/calmodulin-dependent protein kinase kinase II (CamKKII) that is regulated by an increase in intracellular calcium levels (Woods et al. 2005; Hawley et al. 2005; Hurley et al. 2005; Connolly et al. 2014) and the transforming growth factor β-activated kinase 1 (TAK1) that was reported to phosphorylate AMPK under oxidative stress conditions (Momcilovic et al. 2006; Chen et al. 2013). While not much is known about AMPK function in neuronal cells, studies realized in other cell types demonstrated that AMPK is a very important hub involved in the regulation of many intracellular pathways. In order to preserve energy levels, AMPK was described to downregulate many energy-consuming pathways. These include protein synthesis in particular through the regulation of mTORC1-mediated translational control (Inoki et al. 2003; Gwinn et al. 2008) and eukaryotic elongation factor 2 (eEF2)-mediated translation (Browne et al. 2004; Horman et al. 2002) and fatty acid synthesis through the direct phosphorylation of acetyl CoA carboxylase 1 (ACC1) and the expression of enzymes involved in fatty acid synthesis by inhibition of the lipogenic transcription factor sterol regulatory element-binding protein C1 [SREBP1C; Li et al. (2011)]. On the opposite, AMPK upregulates energy-producing pathways such as mitochondrial biogenesis through the activation of the PGC-1α (peroxisome proliferator-activated receptor-γ coactivator 1α) pathway (Jager et al. 2007); glucose uptake through the regulation of glucose transporters expression (Zheng et al. 2001) and cell surface localization (Russell et al. 1999; Abbud et al. 2000; Weisova et al. 2009); glucose utilization through the direct phosphorylation of enzymes involved in the glycolytic pathway including hexokinase (Abnous and Storey 2008), 6-phosphofructo-2-kinase [PFK-2, Marsin et al. (2000)], and pyruvate dehydrogenase kinase [PDK, Wu et al. (2013)]; and autophagy through the inhibition of ULK1 (Egan et al. 2011; Kim et al. 2011) and mTORC1 complex [review in Shaw (2009)].

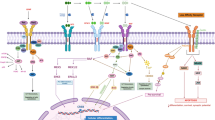
Regulation of AMPK. AMPK is a metabolic sensor which is activated by different stresses. Excitotoxic and oxidative stresses promote, respectively, the activation of CamKKII and TAK1 that phosphorylate AMPK on its residue Thr172 which is necessary for its activation. Metabolic stress induces an increase of the AMP/ATP ratio that promotes AMP binding to the γ subunit of AMPK. This induces a conformational change that allows the phosphorylation of AMPK by LKB1. Once activated, AMPK triggers catabolic pathways and represses anabolic pathways in order to maintain energetic homeostasis. ROS reactive oxygen species, CamKKII Calcium/calmodulin kinase kinase II, LKB1 liver kinase B1, TAK1 transforming growth factor β-activated kinase 1, NMDAR N-methyl d-aspartate receptor. Figure was produced in part using Servier MedicalArt
In this chapter, we review the current literature regarding AMPK involvement in the development of main neurodegenerative diseases that include Alzheimer’s, Parkinson’s, Huntington’s, and amyotrophic lateral sclerosis and discuss the possible common pathological mechanisms involved. It is also important to note that AMPK is also studied in the context of ischemic stroke in animal models. While ischemic stroke can be considered as a neurodegenerative disease, the involvement of AMPK in this context has already been the subject of many reviews (Manwani and McCullough 2013; Weisova et al. 2011) and will not be discussed here.
2 AMPK in Neurodegenerative Diseases
2.1 Alzheimer’s Disease
AD is a progressive neurodegenerative disorder characterized by memory loss and behavioral abnormalities that are correlated with neuronal and synaptic degeneration in specific brain areas. Brain regions are sequentially affected by the pathology starting from the entorhinal cortex to the hippocampus and whole neocortex following cortico-cortical connections. At the histological level, AD is characterized by the presence of senile plaques and neurofibrillary tangles in the brain. Senile plaques result from the extracellular aggregation of a peptide called Amyloid-β (Aβ). Aβ peptides are produced upon the sequential proteolytic processing of its precursor protein (APP) by β- and γ-secretases. Neurofibrillary tangles are composed of paired helical filaments that result from the intracellular aggregation of hyper- and abnormally phosphorylated tau proteins. Tau is a microtubule-associated protein whose main function which is regulated by phosphorylation consists of the regulation of microtubule dynamics. While the exact role of APP remains to be clearly established, there are some rare familial forms of AD which present mutations in APP, Presenilin-1 or Presenilin-2 genes; the latter two being the core components of the γ-secretase complex. However, the vast majorities of AD cases are of sporadic origin and are likely driven by a combination of genetic and environmental factors. The main genetic risk factor is the allele ε4 of APOE (coding for Apolipoprotein E). In addition, other risks factors have been identified following genome-wide association studies and include CLU (coding for clusterin), CR1 (coding for the complement component receptor 1), PICALM, and BIN1 (Lambert et al. 2009; Harold et al. 2009; Seshadri et al. 2010). Environmental factors include age, arterial hypertension, obesity, diabetes, and metabolic syndrome [review in Barberger-Gateau et al. (2013)].
Besides senile plaques and neurofibrillary tangles, perturbations in calcium homeostasis, oxidative stress, and energy metabolism defects are observed in the brain of AD patients (Bezprozvanny and Mattson 2008; Green and LaFerla 2008; Mattson 2007; Sayre et al. 2008). For instance, positron emission tomography (PET) imaging with the 2-[18F]-fluorodeoxyglucose (FDG) tracer is used as a diagnostic marker in AD where reduced glucose energy metabolism can be observed even at early stages of the disease (Mosconi 2005; Ferreira et al. 2010). Additionally, mitochondrial dysfunctions are also commonly described to be associated with AD [for a review, see Cabezas-Opazo et al. (2015)]. These include mitochondrial morphology, dynamics, and bioenergetics defects (DuBoff et al. 2013; Bubber et al. 2005; Garcia-Escudero et al. 2013). Interestingly, these mitochondrial abnormalities were found to be restricted to vulnerable neurons and to occur in neurons lacking neurofibrillary tangles, thus suggesting that they could represent an early event in AD (Hirai et al. 2001). Additionally, mitochondrial axonal transport is also impaired (Wang et al. 2015; Sheng 2014). Both amyloid and tau proteins have been shown to induce mitochondrial dysfunctions (Grimm et al. 2016). Conversely, studies also report that mitochondrial complexes I and III dysfunctions associated with reactive oxygen species (ROS) generation enhance Aβ production both in vitro and in vivo (Leuner et al. 2012).
AMPK was found to be deregulated in AD brains where immunohistochemistry studies revealed that activated AMPK co-localized with hyper-phosphorylated tau in pre-tangle and tangle-bearing neurons (Vingtdeux et al. 2011b). In addition, AMPK activation in AD was also demonstrated by Western blotting where phosphorylated AMPK was significantly upregulated in AD brains as well as in APPSWE,IND(J20) and APPSWE/PS1dE9 mice models of the disease (Ma et al. 2014; Mairet-Coello et al. 2013; Son et al. 2012). AMPK deregulation was also observed in Tauopathies, a subset of neurodegenerative disorders characterized by the presence of abnormally and hyper-phosphorylated tau proteins, including tangle-predominant dementia, Guam Parkinson dementia complex, Pick’s disease, frontotemporal dementia with Parkinsonism linked to chromosome 17, corticobasal degeneration, progressive supranuclear palsy, and argyrophilic grain disease (Vingtdeux et al. 2011b).
The exact role played by AMPK in AD remains controversial. The fact that AMPK co-localizes with hyper-phosphorylated tau in AD led to the hypothesis that AMPK could represent a new tau kinase. Indeed, in vitro studies using recombinant proteins showed that AMPK could phosphorylate tau at several epitopes including Thr231, Ser262, Ser356, and Ser396/404 (Thornton et al. 2011; Vingtdeux et al. 2011b). In cellular models, AMPK was also found to phosphorylate tau under stress conditions (Domise et al. 2016; Thornton et al. 2011). More particularly, Aβ oligomers were found to induce specifically AMPK α1 subunit activation by increasing intracellular calcium concentration and subsequent CamKKII activation. This Aβ oligomer-mediated AMPK activation was suggested to induce tau phosphorylation at epitopes Ser262 and Ser396/404 in primary neuronal cultures (Thornton et al. 2011). In addition, it was postulated that this pathway was responsible for the toxic effects induced by Aβ oligomers on translational block (Yoon et al. 2012), dendritic spines (Mairet-Coello et al. 2013), and synaptic plasticity (Ma et al. 2014). Indeed, AMPK activation following 2-deoxy-d-glucose (2-DG) or Aβ oligomers treatment was found to impair long-term potentiation (LTP) in ex vivo hippocampal slices (Potter et al. 2010; Ma et al. 2014). These results were corroborated in APPSWE/PS1dE9 transgenic animals where AMPK inhibition was found to rescue the LTP impairments mediated by Aβ (Ma et al. 2014). In these studies, AMPK negative effects on synaptic plasticity were found to be the result of decreased protein synthesis through mTORC1 and eEF2 pathways, respectively (Potter et al. 2010; Ma et al. 2014). In addition, AMPK was recently found to modulate tau pathology in vivo (Domise et al. 2016). On the contrary, other studies reported that AMPK activation induced by leptin or metformin reduced tau phosphorylation (Greco et al. 2009, 2011; Kickstein et al. 2010). The effect of metformin might, however, be AMPK independent. Indeed, metformin was suggested to induce protein phosphatase 2A (PP2A) activation, thereby leading to tau dephosphorylation (Kickstein et al. 2010). In a recent study, AMPK modulation was also related to tau dephosphorylation and rather correlated to AMPK phosphorylation at Ser485, which is thought to be an inhibitory AMPK phosphorylation site prohibiting further phosphorylation at epitope Thr172 (Horman et al. 2006). In conditions of metabolic syndrome, insulin resistance or glucose depletion, tau phosphorylation might be differently regulated either because AMPK activation status could differ or because other tau kinases and phosphatases might be involved (Kim et al. 2015). While these findings are somehow controversial, it is clear that tau is an AMPK target either direct or indirect depending on the environmental conditions. Tau epitopes regulated by AMPK include Ser262 and Ser356 which are KXGS domains located in tau microtubules binding repeat regions. Phosphorylation of these particular epitopes regulates tau affinity for microtubules (Fischer et al. 2009). As a consequence, AMPK-mediated tau phosphorylation might control tau binding with microtubules and thereby axonal transport of cargos including mitochondria (Sato-Harada et al. 1996; Reddy 2011). Tau Thr231 is another central epitope since it was reported to serve as a priming site for GSK3β, a very important tau kinase participating to tau hyper-phosphorylation and aggregation (Lin et al. 2007).
AMPK was also found to be involved in APP metabolism. A decrease of Aβ production was reported in primary neurons after AICAR (5-aminoimidazole-4-carboxamide ribonucleotide)-dependent AMPK stimulation; conversely, Aβ levels were increased in primary neurons lacking the AMPK α2 subunit (Won et al. 2010). Opposite results have also been obtained, and for instance, AMPK activation following metformin treatment was reported to increase the transcription of BACE1, one of the enzymes involved in Aβ production and hence to be associated with increased Aβ levels (Chen et al. 2009). The effect of AMPK on Aβ production and/or degradation is likely to be controlled by energy status given that depending on the extracellular glucose concentrations opposite results are obtained (Yang et al. 2015). As a master regulator of autophagy, AMPK activation following resveratrol or AICAR treatment was found to reduce Aβ secretion by increasing its degradation through the autophagic/lysosomal pathway (Vingtdeux et al. 2010, 2011a). In general, AMPK activation might be beneficial by helping clearing protein aggregates through autophagy induction. However, in latter stages of the disease, lysosomal-mediated degradation is impaired (Nixon and Yang 2011), consequently, increasing autophagosomes production without increasing autophagic flux might have deleterious consequences. Indeed, inhibition of autophagic flux will decrease the degradation of misfolded proteins including Aβ and tau (Pickford et al. 2008; Wang et al. 2010) as well as dysfunctional mitochondria. In addition, autophagosomes accumulation might be a source for Aβ production (Yu et al. 2005), thereby inducing a vicious circle.
In conclusion, these data support a role for AMPK in AD as an upstream player in the pathology development. Overall, AMPK could play a role in AD by participating in Aβ production and/or clearance as well as on tau phosphorylation, the two hallmarks of AD. Additionally, AMPK was found to mediate the toxic effects of Aβ on synapses number and synaptic plasticity. These detrimental effects of AMPK in the latter stages of AD are summarized in Fig. 7.2.

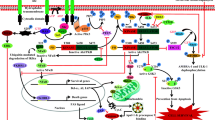
Harmful roles of AMPK in the late stages of Alzheimer’s disease. Alzheimer’s disease is characterized by excitotoxicity as well as metabolic and oxidative stresses. Mitochondrial dysfunction eventually leads to the production of ROS and to the increase of the AMP/ATP ratio that correspond, respectively, to oxidative and metabolic stresses. These two events activate AMPK which in turn decreases protein synthesis ultimately leading to synaptic loss and LTP impairments that contribute to memory loss. AMPK is also involved in tau and amyloid pathologies. On one side, AMPK phosphorylates tau protein thereby altering microtubules assembly and as a result axonal transport of vesicles and mitochondria. On the other side, AMPK plays a part in the production and degradation of Aβ peptides. Finally, Aβ and tau might contribute to the chronic activation of AMPK by inducing mitochondrial impairments and excitotoxicity. LTP long-term potentiation, ROS reactive oxygen species. Figure was produced in part using Servier Medical Art
2.2 Parkinson’s Disease
PD is characterized by resting tremor, rigidity, bradykinesia, gait disturbance, and postural instability. Pathological features include loss of dopaminergic neurons in the substantia nigra associated with Lewy bodies inclusions (Beitz 2014). These Lewy bodies are mainly composed of aggregated α-synuclein. PD etiology involves many genetic and environmental factors (Olanow and Tatton 1999; Verstraeten et al. 2015). While the majority of cases are sporadic, mutations in a number of genes were identified to be responsible for rare familial forms of the disease. These genes include SNCA (coding for α-synuclein), Park2 (coding for the cytosolic E3 ubiquitin ligase Parkin), and PINK1 (coding for PTEN-induced kinase 1). In addition, genetic variants have been identified as PD risk alleles in LRRK2 (leucine-rich repeat kinase 2), SNCA, H1 haplotype of microtubule-associated protein tau, and GBA (coding for beta acid glucosidase) [for a review, see Verstraeten et al. (2015)]. Environmental factors include exposure to environmental toxins (pesticides, herbicides, and industrial chemicals) and drugs of abuse (Olanow and Tatton 1999).
Interestingly, many of these genetic and environmental factors are linked to mitochondrial function. For example, PINK1 is localized to the mitochondria where it exerts a protective role that is abolished by mutations, overall resulting in a cellular increased susceptibility to stress (Valente et al. 2004). Parkin is a protein that was found to be recruited specifically to dysfunctional mitochondria to promote their degradation by the autophagic pathway (Narendra et al. 2008), referred to as mitophagy [for a review, see Youle and Narendra (2011)]. In addition, PINK1 was found to activate Parkin on impaired mitochondria (Narendra et al. 2010). Therefore, it was proposed that Parkin might be involved in mitochondrial quality control as a way to remove damaged mitochondria. Additionally, α-synuclein itself was also reported to induce mitochondrial alterations in neuronal cells and transgenic mice (Hsu et al. 2000; Martin et al. 2006). As for sporadic cases, a decrease in the activity of mitochondrial respiratory chain complex I was found in the substantia nigra of PD patients brain (Schapira et al. 1990). Complex I was found to be functionally impaired, i.e., oxidatively damaged and misassembled (Keeney et al. 2006). In addition, regarding environmental risk factors, many pesticides and 1-methyl-4-1,2,3,6-tetrahydropyridine (MPTP) share the common mechanism of causing mitochondrial dysfunction (Sherer et al. 2002). Finally, FDG–PET studies also demonstrated marked reductions in glucose metabolism in the brain of PD patients (Eckert et al. 2005).
AMPK deregulation was observed in the brain of PD patients where activated AMPK was found near the rim of Lewy bodies in the cytoplasm as opposed to control individuals where AMPK was mainly nuclear (Jiang et al. 2013). AMPK activation was also reported in animal models of PD induced by intra-striatal injection of 6-hydroxydopamine (6-OHDA) or MPTP (Kim et al. 2013; Choi et al. 2010). On the contrary, α-synuclein expression in cell models was reported to downregulate AMPK activation (Dulovic et al. 2014). Whether AMPK activation is beneficial or detrimental in PD remains controversial. AMPK activation was reported in some instance to be detrimental given that further activation of AMPK, for example, following metformin administration significantly enhanced dopaminergic neuron degeneration induced by 6-OHDA, whereas overexpression of a dominant-negative AMPK in the striatum reduced dopaminergic neuron degeneration following 6-OHDA (Kim et al. 2013). In cellular models, PD toxins (6-OHDA, MPP+, or rotenone) induced AMPK activation and Akt inactivation that cooperatively contributed to the downregulation of mTOR-mediated S6K1 (ribosomal p70 S6 kinase) and 4E-BP1 (eukaryotic initiation factor 4E binding protein 1), thereby leading to neuronal cell death (Xu et al. 2014). AMPK might also participate in Lewy bodies’ accumulation through direct phosphorylation of α-synuclein (Jiang et al. 2013) that could impair the clearance of its aggregates (Tenreiro et al. 2014). On the opposite, AMPK activation using AICAR or metformin was reported to reduce the toxicity mediated by α-synuclein (Dulovic et al. 2014). AMPK also protected cells against rotenone toxicity by enhancing autophagy (Hou et al. 2015). This AMPK-induced autophagic pathway also regulates α-synuclein degradation following resveratrol treatment (Wu et al. 2011). AMPK might also participate in mitochondrial function regulation in PD. Results obtained in Drosophila melanogaster models suggest that AMPK activation could be beneficial for familial forms of PD that present mutations in Parkin or LRRK2. Indeed, genetic inactivation of AMPK was reported to reduce the beneficial effects of epigallocatechin gallate (EGCG), an antioxidant found in green tea, in mutant LRRK2 and Parkin-null flies (Ng et al. 2012). In addition, results obtained from patient’s primary fibroblasts presenting Park2 mutations also suggest that the beneficial effects on mitochondrial function and autophagy induced by resveratrol were due to AMPK activation (Ferretta et al. 2014).
Altogether, these studies highlight the potential double role that can be played by AMPK in PD (Fig. 7.3). On one side, AMPK could be neuroprotective by participating, for example, in mitochondrial quality control; yet under other circumstances, AMPK could participate in neurodegeneration.

Dual role of AMPK in Parkinson’s disease. Environmental and genetic risk factors are involved in the buildup of mitochondrial alterations. These alterations eventually lead to oxidative stress through the production of ROS and metabolic stress via an increase of the AMP/ATP ratio. These stresses induce the activation of AMPK which phosphorylates α-synuclein, the latter promoting its aggregation and ultimately neurodegeneration. Neurodegeneration might also result from decreased protein synthesis triggered by AMPK activation. On the contrary, AMPK could also exert a neuroprotective effect in particular by inducing the degradation of damaged mitochondria and α-synuclein aggregates via autophagy. ROS reactive oxygen species. Figure was produced in part using Servier Medical Art
2.3 Huntington’s Disease
Clinical manifestations of HD include motor disturbances comprising chorea and dystonia and cognitive and behavioral dysfunctions. HD is characterized by the loss of medium spiny neurons in the striatum and eventually more widespread loss of cortical, thalamic, hippocampal, and hypothalamic neurons. Another characteristic of the disease is the appearance of nuclear and cytoplasmic inclusions that contain mutant huntingtin and polyglutamine (Walker 2007). HD is an autosomal dominant genetic disease that is induced by the repetition of a polyglutamine CAG triplet repeat in the exon 1 of the huntingtin (Htt) gene with 41 or more polyQ repeats being fully penetrant. (The Huntington’s Disease Collaborative Research Group 1993.) These repeats might confer a toxic gain of function for mutant Htt (mHtt) or a loss of normal Htt function (Zuccato et al. 2010). The physiological role of Htt remains poorly understood; however, it was suggested to be involved in axonal, vesicular, and mitochondrial transport (Smith et al. 2009; Tian et al. 2014).
In HD, mitochondrial dynamics, fusion and fission mechanisms as well as the activity of enzymes involved in oxidative phosphorylation are disturbed (Shirendeb et al. 2011; Song et al. 2011; Browne et al. 1997; Gu et al. 1996). These perturbations have for consequence to increase the accumulation of fragmented and damaged mitochondria eventually leading to oxidative stress. Additionally, mitophagy defects were also proposed to participate in the disease progression (Wong and Holzbaur 2014). A selective impairment of glycolytic metabolism in the striatum of HD patient early in the course of their disease was observed by in vivo PET measurements (Powers et al. 2007). This glucose hypometabolism in the early stages of the disease was also reported in the cerebral cortex and in the brain caudate (Shin et al. 2013; Ciarmiello et al. 2012). Deficits in glycolysis have also been reported in striatal neurons in a rat model of the disease (Gouarne et al. 2013). Huntingtin itself might play a role in glycolysis. Indeed, Htt was found to interact with the glycolytic enzyme GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (Burke et al. 1996). However, studies of HD patients’ brains did not conclusively find an alteration of GAPDH activity (Browne et al. 1997; Tabrizi et al. 1999; Kish et al. 1998; Olah et al. 2008). GAPDH was described to bear additional functions unrelated to its energetic role. GAPDH might act in concert with the ubiquitin-E3-ligase Siah1 to induce mHtt neurotoxicity by assisting its nuclear translocation (Bae et al. 2006). Huntingtin could also be involved in fast axonal transport by scaffolding GAPDH on vesicles, thereby providing onboard energy (Zala et al. 2013). Finally, a recent study demonstrated that mHtt interfered with mitophagy. Indeed, mHtt was found to affect GAPDH-driven mitophagy, thereby leading to the accumulation of damaged mitochondria (Hwang et al. 2015).
The α1 subunit of AMPK seems to be particularly involved in HD pathogenesis. Indeed, it was found to be activated in the nucleus of striatal neurons where it was suggested to downregulate the antiapoptotic protein Bcl2, thus inducing cell death (Ju et al. 2011) (Fig. 7.4). Accumulation of activated AMPK was also reported in the striatum of transgenic mouse models of HD, R6/2 mice harboring exon 1 of the human Htt gene with 144 CAG repeats (Chou et al. 2005; Mochel et al. 2012; Ju et al. 2014). This overactivation of AMPK could be reversed by activating A2A receptors using an agonist, additionally diminishing the HD-like pathology in these animals (Chou et al. 2005; Ju et al. 2011). A2A receptors signaling pathway involves PKA activation. PKA was reported to phosphorylate AMPK α1 at residue Ser173, thereby preventing the activating phosphorylation at Thr172 (Djouder et al. 2010). Additionally, AMPK activation in this mouse model might also result from increased oxidative stress (Ju et al. 2014). On the contrary, in cellular models, AMPK activation through viniferin treatment was reported to provide neuroprotection against mHtt (Fu et al. 2012). Finally, metformin, which can activate AMPK was reported to be beneficial in male R6/2 mice (Ma et al. 2007). However, the exact mechanism behind metformin’s beneficial effects remains to be determined.

Model of AMPK-mediated apoptosis in Huntington’s disease. Mutant Huntingtin induces mitochondrial alterations that lead to oxidative stress and hypometabolism. These participate in the activation of AMPK and its translocation from the cytoplasm to the nucleus where AMPK downregulates the antiapoptotic protein Bcl2. This pathway promotes apoptosis and thereby neurodegeneration. ROS reactive oxygen species, Htt Huntingtin. Schematic is adapted from Ju et al. (2011). Figure was produced in part using Servier Medical Art
Overall, these studies also highlight AMPK signaling pathway as a potential player in the pathology of HD.
2.4 Amyotrophic Lateral Sclerosis
Amyotrophic Lateral Sclerosis (ALS) is characterized by the progressive loss of upper and lower motor neurons at the spinal or bulbar level (Rowland and Shneider 2001). The most common symptoms of ALS are muscle weakness, muscular atrophy, spasticity, and eventually paralysis. While the exact cause of the disease is unknown, around 10 % of familial forms exist involving, for example, the SOD1 gene (superoxide dismutase 1), TARDBP (encoding TAR DNA-binding protein 43), FUS (fused in sarcoma), and hexanucleotide repeat expansion in C9ORF72 (Zarei et al. 2015; Renton et al. 2014). The sporadic forms of the disease might be driven by genetic and lifestyle risk factors (Ingre et al. 2015). At the histological level, ALS is characterized by the aggregation of ubiquitinated proteins that can include TDP43, p62, and FUS in affected neurons (Blokhuis et al. 2013). ALS is associated with defects in energy metabolism comprising weight loss, increased resting energy expenditure (hypermetabolism), and hyperlipidemia (Dupuis et al. 2011). The precise origin of these metabolic dysfunctions remains unclear.
AMPK activation was found to be deregulated in motor neurons of ALS patients (Liu et al. 2015b). In cells and mouse models of the disease, AMPK regulation differs according to the model used. In the mSOD1G93A mouse model, AMPK activity is increased in spinal cords from symptom onset (Lim et al. 2012; Perera et al. 2014; Zhao et al. 2015). Similar results were obtained in vitro, in spinal cord cultures, in motor neuron cell lines expressing mutant SOD1, and in embryonic neural stem cells derived from SOD1G93A mice (Lim et al. 2012; Perera et al. 2014; Sui et al. 2014). On the opposite, AMPK activation was reported to be downregulated in mutant TDP43A315T mouse models of spinal cord and brain (Perera et al. 2014). Similar results were also obtained in motor neuronal cell lines expressing mutant TDP-43, probably as a consequence of increased PP2A activity (Perera et al. 2014). On the contrary, AMPK activity was reported to be increased in the spinal cord of a mouse model overexpressing wild-type TDP43 (Liu et al. 2015b). AMPK was also suggested to be involved in TDP-43 mislocalization from the nucleus to the cytoplasm (Liu et al. 2015a). Similarly, AMPK activation was described to induce the human antigen R [HuR, a major mRNA stabilizer recently shown to regulate TDP-43 and FUS (Lu et al. 2014)] delocalization by directly phosphorylating importin-α1 (Liu et al. 2015b). The impact of AMPK activation in this disease remains a matter of debate. Indeed, modulation of AMPK activity in these various models has given conflicting data. Metformin administration in SOD1G93A mice accelerated disease onset and progression in females only (Davis and Lin 2011), while resveratrol was found to provide beneficial effects (Mancuso et al. 2014; Song et al. 2014). The beneficial effect of resveratrol could act in part through an increase of Sirtuin 1 expression, normalization of autophagic flux, and reduced oxidative stress (Mancuso et al. 2014; Song et al. 2014). Similarly, preconditioning with latrepirdine, a small molecule shown to activate AMPK (Weisova et al. 2013), was reported to delay symptoms onset and increase the lifespan of SODG93A mice (Coughlan et al. 2015). Decreasing AMPK activity in cell cultures or in Caenorhabditis elegans expressing mutant SOD1 or TDP43 was reported to be beneficial (Mancuso et al. 2014) and to rescue TDP43 mislocalization in motor neuronal cells and to delay disease progression in TDP43 wild-type mice (Liu et al. 2015b). Finally, AMPK α2-deficient mice were recently described to present gait abnormalities resembling early stages of ALS supporting a key role for AMPK in the development of this disease (Vergouts et al. 2015).
In conclusion, the role played by AMPK in ALS might vary according to the nature of the disease as mutations in SOD1 and TDP43 were reported to affect differently the kinase.
3 General Considerations
The specific vulnerability of the neuronal populations affected in each of these diseases is likely to be driven by both genetic and environmental factors. Interestingly, many of these factors converge to an impairment of cellular energy metabolism. This is the case, for instance, of mutations in genes that are directly involved in mitochondrial function or clearance (such as SOD1, PINK1, and Parkin). These mitochondrial dysfunctions might contribute to the increase in neuronal excitotoxicity and AMPK deregulation. In addition to impairing energy metabolism, mitochondrial insults can cause an imbalance between ROS production and removal, thereby participating in oxidative stress, another common factor of these diseases (Sayre et al. 2008). This oxidative stress through the activation of TAK1 might also contribute to the chronic activation of AMPK. Conversely, given its role on mitochondria function, biogenesis, and degradation, it is also possible that AMPK participates in the establishment of mitochondrial dysfunctions that are observed in these diseases. Whether AMPK deregulation is triggered by these metabolic perturbations or could be involved in their development will be an important issue to investigate.
While AMPK is highly expressed in neurons, its physiological function remains poorly studied. Nonetheless, AMPK is vital for neuronal survival. Indeed, results obtained in Drosophila demonstrated that genetic ablation of AMPK subunits γ [lochrig mutant, Tschape et al. (2002)] or β [alicorn mutant, Spasic et al. (2008)] induces progressive neurodegeneration. Although it is becoming increasingly evident that AMPK might participate in these neurodegenerative diseases development, whether this activation is beneficial or detrimental remains matter of debate. In general, the contradictory results that have been obtained in vivo regarding the beneficial or detrimental role of AMPK could also be due to peripheral AMPK activity. Several papers reported beneficial effects of peripheral AMPK activation on cognition. For instance, it was shown that AMPK activation following AICAR administration in mice enhanced endurance and spatial memory in the Morris water maze (Kobilo et al. 2014). AICAR blood–brain barrier permeability is very low (Marangos et al. 1990); therefore, its effects on cognition or on the brain in general are likely to be indirect. The beneficial effects of AICAR reported in the Kobilo et al.’s study were demonstrated to be mediated by muscle AMPK activation since mice overexpressing a muscle-specific dominant negative of AMPK α2 did not show any improvements following AICAR administration. These behavioral improvements were suggested to result from enhanced dentate gyrus neurogenesis in AICAR-treated animals (Kobilo et al. 2011). On the contrary, direct administration of AICAR in the brain by means of intracerebral infusions was found to impair memory functions (Dash et al. 2006) and lead to excitotoxicity in an HD mouse model (Ju et al. 2011). As a consequence, it is very important to take into account the drug used to activate or inhibit AMPK and its administration route to determine the impact of peripheral AMPK activation in addition to its central regulation before drawing conclusions.
It is very likely that AMPK could act both as a friend and as a foe during the course of these neurodegenerative diseases’ progression. Indeed, AMPK might be activated in the early stages of these diseases to help maintain or restore neuronal energy metabolism. However, chronic AMPK activation would eventually become detrimental to brain functions by repressing pathways that consume energy. Overall, several common mechanisms regulated by AMPK can be identified and are summarized in Fig. 7.5. For instance, the beneficial effects of AMPK often involve an increase of the autophagy pathway that might be involved in the clearance of misfolded proteins, protein aggregates, or defective mitochondria. It was also reported that AMPK might activate PP2A, thereby reducing the phosphorylation status of tau and α-synuclein. On the opposite, the deleterious impact of AMPK implies the phosphorylation of proteins which aggregates represent the common hallmarks of these diseases, including tau, Aβ, and α-synuclein. Additionally, AMPK chronic activation by repressing protein synthesis could, on the long term, impair synaptic integrity and plasticity and eventually lead to cell death.

AMPK in neurodegenerative diseases, friend or foe? At the onset of neurodegenerative diseases, activation of AMPK might be beneficial since it allows the restoration of energetic homeostasis and the elimination of protein aggregates which are often reported to be toxic for neurons. Indeed, AMPK promotes the formation of autophagosomes in order to induce protein aggregates and impaired mitochondria degradation through the autophagy/lysosomal pathway. On the other hand, in the late stages of these diseases, chronic AMPK activation becomes disadvantageous for neurons. This overactivation of AMPK could lead to neurodegeneration through several signaling pathways. Decreasing protein synthesis could drive synaptic loss and impair synaptic plasticity subsequently leading to neurodegeneration. Decreasing antiapoptotic factors could lead to induction of apoptosis and neurodegeneration. Finally, the production of autophagosomes combined with an alteration of lysosomal clearance (which is often reported to occur in these disorders), in the end, leads to the accumulation of autophagosomes and contributes to upsurge the levels of toxic protein aggregates and defective mitochondria
4 Conclusion
While the clinical manifestations, neuronal populations affected and proteins involved differ widely between these diseases, energy metabolism perturbations are often reported early in the course of these diseases' progression. These metabolic perturbations might result from the various environmental and genetic risk factors that drive these pathologies as it is already well acknowledged for mutations that affect directly mitochondrial functions. As a consequence, one can expect AMPK overactivation to be an additional early feature of these disorders. Hence, AMPK was suggested to participate in these diseases' progression by contributing in the establishment of the observed lesions mainly by regulating the clearance and posttranslational modifications of the proteins forming the respective aggregates. Additionally, AMPK chronic overactivation might participate in neurodegeneration by repressing energy-consuming pathways.
Given the demographic trend towards an aging population, the prevalence of these neurodegenerative diseases and thus their socioeconomic burden will continue to increase dramatically in the next decades. The current treatments are only symptomatic; there are no therapies available to cure these diseases. As a consequence, there is a need to better understand the underlying disease mechanisms in order to underpin the development of new diagnostic and therapeutic approaches. In this context, AMPK signaling pathways might be particularly interesting.
References
Abbud W, Habinowski S, Zhang JZ, Kendrew J, Elkairi FS, Kemp BE, Witters LA, Ismail-Beigi F (2000) Stimulation of AMP-activated protein kinase (AMPK) is associated with enhancement of Glut1-mediated glucose transport. Arch Biochem Biophys 380(2):347–352
Abnous K, Storey KB (2008) Skeletal muscle hexokinase: regulation in mammalian hibernation. Mol Cell Biochem 319(1–2):41–50
Bae BI, Hara MR, Cascio MB, Wellington CL, Hayden MR, Ross CA, Ha HC, Li XJ, Snyder SH, Sawa A (2006) Mutant huntingtin: nuclear translocation and cytotoxicity mediated by GAPDH. Proc Natl Acad Sci USA 103(9):3405–3409
Barberger-Gateau P, Lambert JC, Feart C, Peres K, Ritchie K, Dartigues JF, Alperovitch A (2013) From genetics to dietetics: the contribution of epidemiology to understanding Alzheimer’s disease. J Alzheimers Dis 33(Suppl 1):S457–S463
Beitz JM (2014) Parkinson’s disease: a review. Front Biosci (Schol Ed) 6:65–74
Bezprozvanny I, Mattson MP (2008) Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 31(9):454–463
Blokhuis AM, Groen EJ, Koppers M, van den Berg LH, Pasterkamp RJ (2013) Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol 125(6):777–794
Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, Bird ED, Beal MF (1997) Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol 41(5):646–653
Browne GJ, Finn SG, Proud CG (2004) Stimulation of the AMP-activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, serine 398. J Biol Chem 279(13):12220–12231
Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE (2005) Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol 57(5):695–703
Burke JR, Enghild JJ, Martin ME, Jou YS, Myers RM, Roses AD, Vance JM, Strittmatter WJ (1996) Huntingtin and DRPLA proteins selectively interact with the enzyme GAPDH. Nat Med 2(3):347–350
Cabezas-Opazo FA, Vergara-Pulgar K, Perez MJ, Jara C, Osorio-Fuentealba C, Quintanilla RA (2015) Mitochondrial dysfunction contributes to the pathogenesis of Alzheimer’s disease. Oxid Med Cell Longev 2015:509654
Chen Y, Zhou K, Wang R, Liu Y, Kwak YD, Ma T, Thompson RC, Zhao Y, Smith L, Gasparini L, Luo Z, Xu H, Liao FF (2009) Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription. Proc Natl Acad Sci USA 106(10):3907–3912
Chen Z, Shen X, Shen F, Zhong W, Wu H, Liu S, Lai J (2013) TAK1 activates AMPK-dependent cell death pathway in hydrogen peroxide-treated cardiomyocytes, inhibited by heat shock protein-70. Mol Cell Biochem 377(1–2):35–44
Choi JS, Park C, Jeong JW (2010) AMP-activated protein kinase is activated in Parkinson’s disease models mediated by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biochem Biophys Res Commun 391(1):147–151
Chou SY, Lee YC, Chen HM, Chiang MC, Lai HL, Chang HH, Wu YC, Sun CN, Chien CL, Lin YS, Wang SC, Tung YY, Chang C, Chern Y (2005) CGS21680 attenuates symptoms of Huntington’s disease in a transgenic mouse model. J Neurochem 93(2):310–320
Ciarmiello A, Giovacchini G, Orobello S, Bruselli L, Elifani F, Squitieri F (2012) 18F-FDG PET uptake in the pre-Huntington disease caudate affects the time-to-onset independently of CAG expansion size. Eur J Nucl Med Mol Imaging 39(6):1030–1036
Connolly NM, Dussmann H, Anilkumar U, Huber HJ, Prehn JH (2014) Single-cell imaging of bioenergetic responses to neuronal excitotoxicity and oxygen and glucose deprivation. J Neurosci 34(31):10192–10205
Coughlan KS, Mitchem MR, Hogg MC, Prehn JH (2015) “Preconditioning” with latrepirdine, an adenosine 5′-monophosphate-activated protein kinase activator, delays amyotrophic lateral sclerosis progression in SOD1(G93A) mice. Neurobiol Aging 36(2):1140–1150
Dash PK, Orsi SA, Moore AN (2006) Spatial memory formation and memory-enhancing effect of glucose involves activation of the tuberous sclerosis complex-Mammalian target of rapamycin pathway. J Neurosci 26(31):8048–8056
Davis JD, Lin SY (2011) DNA damage and breast cancer. World J Clin Oncol 2(9):329–338
Djouder N, Tuerk RD, Suter M, Salvioni P, Thali RF, Scholz R, Vaahtomeri K, Auchli Y, Rechsteiner H, Brunisholz RA, Viollet B, Makela TP, Wallimann T, Neumann D, Krek W (2010) PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. EMBO J 29(2):469–481
Domise M, Didier S, Marinangeli C, Zhao H, Chandakkar P, Buée L, Viollet B, Davies P, Marambaud P, Vingtdeux V (2016) AMP-activated protein kinase modulates tau phosphorylation and tau pathology in vivo. Sci Rep 6:26758
DuBoff B, Feany M, Gotz J (2013) Why size matters—balancing mitochondrial dynamics in Alzheimer’s disease. Trends Neurosci 36(6):325–335
Dulovic M, Jovanovic M, Xilouri M, Stefanis L, Harhaji-Trajkovic L, Kravic-Stevovic T, Paunovic V, Ardah MT, El-Agnaf OM, Kostic V, Markovic I, Trajkovic V (2014) The protective role of AMP-activated protein kinase in alpha-synuclein neurotoxicity in vitro. Neurobiol Dis 63:1–11
Dupuis L, Pradat PF, Ludolph AC, Loeffler JP (2011) Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol 10(1):75–82
Eckert T, Barnes A, Dhawan V, Frucht S, Gordon MF, Feigin AS, Eidelberg D (2005) FDG PET in the differential diagnosis of parkinsonian disorders. Neuroimage 26(3):912–21
Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ (2011) Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331(6016):456–461
Ferreira IL, Resende R, Ferreiro E, Rego AC, Pereira CF (2010) Multiple defects in energy metabolism in Alzheimer’s disease. Curr Drug Targets 11(10):1193–1206
Ferretta A, Gaballo A, Tanzarella P, Piccoli C, Capitanio N, Nico B, Annese T, Di Paola M, Dell’aquila C, De Mari M, Ferranini E, Bonifati V, Pacelli C, Cocco T (2014) Effect of resveratrol on mitochondrial function: implications in parkin-associated familiar Parkinson’s disease. Biochim Biophys Acta 1842(7):902–915
Fischer D, Mukrasch MD, Biernat J, Bibow S, Blackledge M, Griesinger C, Mandelkow E, Zweckstetter M (2009) Conformational changes specific for pseudophosphorylation at serine 262 selectively impair binding of tau to microtubules. Biochemistry 48(42):10047–10055
Fu J, Jin J, Cichewicz RH, Hageman SA, Ellis TK, Xiang L, Peng Q, Jiang M, Arbez N, Hotaling K, Ross CA, Duan W (2012) trans-(-)-epsilon-Viniferin increases mitochondrial sirtuin 3 (SIRT3), activates AMP-activated protein kinase (AMPK), and protects cells in models of Huntington Disease. J Biol Chem 287(29):24460–24472
Garcia-Escudero V, Martin-Maestro P, Perry G, Avila J (2013) Deconstructing mitochondrial dysfunction in Alzheimer disease. Oxid Med Cell Longev 2013:162152
Gouarne C, Tardif G, Tracz J, Latyszenok V, Michaud M, Clemens LE, Yu-Taeger L, Nguyen HP, Bordet T, Pruss RM (2013) Early deficits in glycolysis are specific to striatal neurons from a rat model of huntington disease. PLoS One 8(11):e81528
Gowans GJ, Hawley SA, Ross FA, Hardie DG (2013) AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab 18(4):556–566
Greco SJ, Sarkar S, Johnston JM, Tezapsidis N (2009) Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem Biophys Res Commun 380(1):98–104
Greco SJ, Hamzelou A, Johnston JM, Smith MA, Ashford JW, Tezapsidis N (2011) Leptin boosts cellular metabolism by activating AMPK and the sirtuins to reduce tau phosphorylation and beta-amyloid in neurons. Biochem Biophys Res Commun 414(1):170–174
Green KN, LaFerla FM (2008) Linking calcium to Abeta and Alzheimer’s disease. Neuron 59(2):190–194
Grimm A, Schmitt K, Eckert A (2016) Advanced mitochondrial respiration assay for evaluation of mitochondrial dysfunction in Alzheimer’s disease. Methods Mol Biol 1303:171–183
Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH (1996) Mitochondrial defect in Huntington’s disease caudate nucleus. Ann Neurol 39(3):385–389
Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30(2):214–226
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, Heun R, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O’Donovan M, Owen MJ, Williams J (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 41(10):1088–1093
Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG (2003) Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol 2(4):28
Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG (2005) Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2(1):9–19
Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA (2001) Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci 21(9):3017–3023
Horman S, Browne G, Krause U, Patel J, Vertommen D, Bertrand L, Lavoinne A, Hue L, Proud C, Rider M (2002) Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr Biol 12(16):1419–1423
Horman S, Vertommen D, Heath R, Neumann D, Mouton V, Woods A, Schlattner U, Wallimann T, Carling D, Hue L, Rider MH (2006) Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem 281(9):5335–5340
Hou YS, Guan JJ, Xu HD, Wu F, Sheng R, Qin ZH (2015) Sestrin2 protects dopaminergic cells against rotenone toxicity through AMPK-dependent autophagy activation. Mol Cell Biol 35(16):2740–2751
Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E (2000) alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol 157(2):401–410
Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA (2005) The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem 280(32):29060–29066
Hwang S, Disatnik MH, Mochly-Rosen D (2015) Impaired GAPDH-induced mitophagy contributes to the pathology of Huntington’s disease. EMBO Mol Med 7(10):1307–1326
Ingre C, Roos PM, Piehl F, Kamel F, Fang F (2015) Risk factors for amyotrophic lateral sclerosis. Clin Epidemiol 7:181–193
Inoki K, Zhu T, Guan KL (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115(5):577–590
Jager S, Handschin C, St-Pierre J, Spiegelman BM (2007) AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA 104(29):12017–12022
Jiang P, Gan M, Ebrahim AS, Castanedes-Casey M, Dickson DW, Yen SH (2013) Adenosine monophosphate-activated protein kinase overactivation leads to accumulation of alpha-synuclein oligomers and decrease of neurites. Neurobiol Aging 34(5):1504–1515
Ju TC, Chen HM, Lin JT, Chang CP, Chang WC, Kang JJ, Sun CP, Tao MH, Tu PH, Chang C, Dickson DW, Chern Y (2011) Nuclear translocation of AMPK-alpha1 potentiates striatal neurodegeneration in Huntington’s disease. J Cell Biol 194(2):209–227
Ju TC, Chen HM, Chen YC, Chang CP, Chang C, Chern Y (2014) AMPK-alpha1 functions downstream of oxidative stress to mediate neuronal atrophy in Huntington’s disease. Biochim Biophys Acta 1842(9):1668–1680
Keeney PM, Xie J, Capaldi RA, Bennett JP Jr (2006) Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci 26(19):5256–5264
Kickstein E, Krauss S, Thornhill P, Rutschow D, Zeller R, Sharkey J, Williamson R, Fuchs M, Kohler A, Glossmann H, Schneider R, Sutherland C, Schweiger S (2010) Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci USA 107(50):21830–21835
Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13(2):132–141
Kim TW, Cho HM, Choi SY, Suguira Y, Hayasaka T, Setou M, Koh HC, Hwang EM, Park JY, Kang SJ, Kim HS, Kim H, Sun W (2013) (ADP-ribose) polymerase 1 and AMP-activated protein kinase mediate progressive dopaminergic neuronal degeneration in a mouse model of Parkinson’s disease. Cell Death Dis 4:e919
Kim B, Figueroa-Romero C, Pacut C, Backus C, Feldman EL (2015) Insulin resistance prevents AMPK-induced tau dephosphorylation through Akt-mediated increase in AMPKSer-485 phosphorylation. J Biol Chem 290(31):19146–19157
Kish SJ, Lopes-Cendes I, Guttman M, Furukawa Y, Pandolfo M, Rouleau GA, Ross BM, Nance M, Schut L, Ang L, DiStefano L (1998) Brain glyceraldehyde-3-phosphate dehydrogenase activity in human trinucleotide repeat disorders. Arch Neurol 55(10):1299–1304
Kobilo T, Yuan C, van Praag H (2011) Endurance factors improve hippocampal neurogenesis and spatial memory in mice. Learn Mem 18(2):103–107
Kobilo T, Guerrieri D, Zhang Y, Collica SC, Becker KG, van Praag H (2014) AMPK agonist AICAR improves cognition and motor coordination in young and aged mice. Learn Mem 21(2):119–126
Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fievet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O, European Alzheimer’s Disease Initiative Investigators, de Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossu P, Piccardi P, Annoni G, Seripa D, Galimberti D, Hannequin D, Licastro F, Soininen H, Ritchie K, Blanche H, Dartigues JF, Tzourio C, Gut I, Van Broeckhoven C, Alperovitch A, Lathrop M, Amouyel P (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 41(10):1094–1099
Leuner K, Schutt T, Kurz C, Eckert SH, Schiller C, Occhipinti A, Mai S, Jendrach M, Eckert GP, Kruse SE, Palmiter RD, Brandt U, Drose S, Wittig I, Willem M, Haass C, Reichert AS, Muller WE (2012) Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid Redox Signal 16(12):1421–1433
Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, Gao B, Wierzbicki M, Verbeuren TJ, Shaw RJ, Cohen RA, Zang M (2011) AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab 13(4):376–388
Lim MA, Selak MA, Xiang Z, Krainc D, Neve RL, Kraemer BC, Watts JL, Kalb RG (2012) Reduced activity of AMP-activated protein kinase protects against genetic models of motor neuron disease. J Neurosci 32(3):1123–1141
Lin YT, Cheng JT, Liang LC, Ko CY, Lo YK, Lu PJ (2007) The binding and phosphorylation of Thr231 is critical for Tau’s hyperphosphorylation and functional regulation by glycogen synthase kinase 3beta. J Neurochem 103(2):802–813
Liu YJ, Ju TC, Chen HM, Jang YS, Lee LM, Lai HL, Tai HC, Fang JM, Lin YL, Tu PH, Chern Y (2015a) Activation of AMP-activated protein kinase alpha1 mediates mislocalization of TDP-43 in amyotrophic lateral sclerosis. Hum Mol Genet 24(3):787–801
Liu YJ, Lee LM, Lai HL, Chern Y (2015b) Aberrant activation of AMP-activated protein kinase contributes to the abnormal distribution of HuR in amyotrophic lateral sclerosis. FEBS Lett 589(4):432–439
Lu L, Zheng L, Si Y, Luo W, Dujardin G, Kwan T, Potochick NR, Thompson SR, Schneider DA, King PH (2014) Hu antigen R (HuR) is a positive regulator of the RNA-binding proteins TDP-43 and FUS/TLS: implications for amyotrophic lateral sclerosis. J Biol Chem 289(46):31792–31804
Ma TC, Buescher JL, Oatis B, Funk JA, Nash AJ, Carrier RL, Hoyt KR (2007) Metformin therapy in a transgenic mouse model of Huntington’s disease. Neurosci Lett 411(2):98–103
Ma T, Chen Y, Vingtdeux V, Zhao H, Viollet B, Marambaud P, Klann E (2014) Inhibition of AMP-activated protein kinase signaling alleviates impairments in hippocampal synaptic plasticity induced by amyloid beta. J Neurosci 34(36):12230–12238
Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F (2013) The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Abeta oligomers through Tau phosphorylation. Neuron 78(1):94–108
Mancuso R, del Valle J, Modol L, Martinez A, Granado-Serrano AB, Ramirez-Nunez O, Pallas M, Portero-Otin M, Osta R, Navarro X (2014) Resveratrol improves motoneuron function and extends survival in SOD1(G93A) ALS mice. Neurotherapeutics 11(2):419–432
Manwani B, McCullough LD (2013) Function of the master energy regulator adenosine monophosphate-activated protein kinase in stroke. J Neurosci Res 91(8):1018–1029
Marangos PJ, Loftus T, Wiesner J, Lowe T, Rossi E, Browne CE, Gruber HE (1990) Adenosinergic modulation of homocysteine-induced seizures in mice. Epilepsia 31(3):239–246
Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, Van den Berghe G, Carling D, Hue L (2000) Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol 10(20):1247–1255
Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK (2006) Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci 26(1):41–50
Mattson MP (2007) Calcium and neurodegeneration. Aging Cell 6(3):337–350
Mochel F, Durant B, Meng X, O’Callaghan J, Yu H, Brouillet E, Wheeler VC, Humbert S, Schiffmann R, Durr A (2012) Early alterations of brain cellular energy homeostasis in Huntington disease models. J Biol Chem 287(2):1361–1370
Momcilovic M, Hong SP, Carlson M (2006) Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem 281(35):25336–25343
Mosconi L (2005) Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. Eur J Nucl Med Mol Imaging 32(4):486–510
Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183(5):795–803
Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8(1):e1000298
Ng CH, Guan MS, Koh C, Ouyang X, Yu F, Tan EK, O’Neill SP, Zhang X, Chung J, Lim KL (2012) AMP kinase activation mitigates dopaminergic dysfunction and mitochondrial abnormalities in Drosophila models of Parkinson’s disease. J Neurosci 32(41):14311–14317
Nixon RA, Yang DS (2011) Autophagy failure in Alzheimer’s disease—locating the primary defect. Neurobiol Dis 43(1):38–45
Olah J, Klivenyi P, Gardian G, Vecsei L, Orosz F, Kovacs GG, Westerhoff HV, Ovadi J (2008) Increased glucose metabolism and ATP level in brain tissue of Huntington’s disease transgenic mice. FEBS J 275(19):4740–4755
Olanow CW, Tatton WG (1999) Etiology and pathogenesis of Parkinson’s disease. Annu Rev Neurosci 22:123–144
Perera ND, Sheean RK, Scott JW, Kemp BE, Horne MK, Turner BJ (2014) Mutant TDP-43 deregulates AMPK activation by PP2A in ALS models. PLoS One 9(4):e95549
Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T (2008) The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 118(6):2190–2199
Potter WB, O’Riordan KJ, Barnett D, Osting SM, Wagoner M, Burger C, Roopra A (2010) Metabolic regulation of neuronal plasticity by the energy sensor AMPK. PLoS One 5(2):e8996
Powers WJ, Videen TO, Markham J, McGee-Minnich L, Antenor-Dorsey JV, Hershey T, Perlmutter JS (2007) Selective defect of in vivo glycolysis in early Huntington’s disease striatum. Proc Natl Acad Sci USA 104(8):2945–2949
Reddy PH (2011) Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Res 1415:136–148
Renton AE, Chio A, Traynor BJ (2014) State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17(1):17–23
Rowland LP, Shneider NA (2001) Amyotrophic lateral sclerosis. N Engl J Med 344(22):1688–1700
Russell RR 3rd, Bergeron R, Shulman GI, Young LH (1999) Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am J Physiol 277(2 Pt 2):H643–H649
Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D (2007) Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J 403(1):139–148
Sato-Harada R, Okabe S, Umeyama T, Kanai Y, Hirokawa N (1996) Microtubule-associated proteins regulate microtubule function as the track for intracellular membrane organelle transports. Cell Struct Funct 21(5):283–295
Sayre LM, Perry G, Smith MA (2008) Oxidative stress and neurotoxicity. Chem Res Toxicol 21(1):172–188
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem 54(3):823–827
Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, Bis JC, Smith AV, Carassquillo MM, Lambert JC, Harold D, Schrijvers EM, Ramirez-Lorca R, Debette S, Longstreth WT Jr, Janssens AC, Pankratz VS, Dartigues JF, Hollingworth P, Aspelund T, Hernandez I, Beiser A, Kuller LH, Koudstaal PJ, Dickson DW, Tzourio C, Abraham R, Antunez C, Du Y, Rotter JI, Aulchenko YS, Harris TB, Petersen RC, Berr C, Owen MJ, Lopez-Arrieta J, Varadarajan BN, Becker JT, Rivadeneira F, Nalls MA, Graff-Radford NR, Campion D, Auerbach S, Rice K, Hofman A, Jonsson PV, Schmidt H, Lathrop M, Mosley TH, Au R, Psaty BM, Uitterlinden AG, Farrer LA, Lumley T, Ruiz A, Williams J, Amouyel P, Younkin SG, Wolf PA, Launer LJ, Lopez OL, van Duijn CM, Breteler MM, CHARGE Consortium, GERAD1 Consortium, EADI1 Consortium (2010) Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303(18):1832–1840
Shaw RJ (2009) LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 196(1):65–80
Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC (2004) The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA 101(10):3329–3335
Sheng ZH (2014) Mitochondrial trafficking and anchoring in neurons: new insight and implications. J Cell Biol 204(7):1087–1098
Sherer TB, Betarbet R, Greenamyre JT (2002) Environment, mitochondria, and Parkinson’s disease. Neuroscientist 8(3):192–197
Shin H, Kim MH, Lee SJ, Lee KH, Kim MJ, Kim JS, Cho JW (2013) Decreased metabolism in the cerebral cortex in early-stage Huntington’s disease: a possible biomarker of disease progression? J Clin Neurol 9(1):21–25
Shirendeb U, Reddy AP, Manczak M, Calkins MJ, Mao P, Tagle DA, Reddy PH (2011) Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum Mol Genet 20(7):1438–1455
Smith R, Bacos K, Fedele V, Soulet D, Walz HA, Obermuller S, Lindqvist A, Bjorkqvist M, Klein P, Onnerfjord P, Brundin P, Mulder H, Li JY (2009) Mutant huntingtin interacts with {beta}-tubulin and disrupts vesicular transport and insulin secretion. Hum Mol Genet 18(20):3942–3954
Son SM, Jung ES, Shin HJ, Byun J, Mook-Jung I (2012) Abeta-induced formation of autophagosomes is mediated by RAGE-CaMKKbeta-AMPK signaling. Neurobiol Aging 33(5):1006e11–1006e23
Song W, Chen J, Petrilli A, Liot G, Klinglmayr E, Zhou Y, Poquiz P, Tjong J, Pouladi MA, Hayden MR, Masliah E, Ellisman M, Rouiller I, Schwarzenbacher R, Bossy B, Perkins G, Bossy-Wetzel E (2011) Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat Med 17(3):377–382
Song L, Chen L, Zhang X, Li J, Le W (2014) Resveratrol ameliorates motor neuron degeneration and improves survival in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Biomed Res Int 2014:483501
Spasic MR, Callaerts P, Norga KK (2008) Drosophila alicorn is a neuronal maintenance factor protecting against activity-induced retinal degeneration. J Neurosci 28(25):6419–6429
Sui Y, Zhao Z, Liu R, Cai B, Fan D (2014) Adenosine monophosphate-activated protein kinase activation enhances embryonic neural stem cell apoptosis in a mouse model of amyotrophic lateral sclerosis. Neural Regen Res 9(19):1770–1778
Tabrizi SJ, Cleeter MW, Xuereb J, Taanman JW, Cooper JM, Schapira AH (1999) Biochemical abnormalities and excitotoxicity in Huntington’s disease brain. Ann Neurol 45(1):25–32
Tenreiro S, Reimao-Pinto MM, Antas P, Rino J, Wawrzycka D, Macedo D, Rosado-Ramos R, Amen T, Waiss M, Magalhaes F, Gomes A, Santos CN, Kaganovich D, Outeiro TF (2014) Phosphorylation modulates clearance of alpha-synuclein inclusions in a yeast model of Parkinson’s disease. PLoS Genet 10(5):e1004302
The Huntington’s Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72(6):971–983
Thornton C, Bright NJ, Sastre M, Muckett PJ, Carling D (2011) AMP-activated protein kinase (AMPK) is a tau kinase, activated in response to amyloid beta-peptide exposure. Biochem J 434(3):503–512
Tian J, Yan YP, Zhou R, Lou HF, Rong Y, Zhang BR (2014) Soluble N-terminal fragment of mutant Huntingtin protein impairs mitochondrial axonal transport in cultured hippocampal neurons. Neurosci Bull 30(1):74–80
Tschape JA, Hammerschmied C, Muhlig-Versen M, Athenstaedt K, Daum G, Kretzschmar D (2002) The neurodegeneration mutant lochrig interferes with cholesterol homeostasis and Appl processing. EMBO J 21(23):6367–6376
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304(5674):1158–1160
Vergouts M, Marinangeli C, Ingelbrecht C, Genard G, Schakman O, Sternotte A, Calas AG, Hermans E (2015) Early ALS-type gait abnormalities in AMP-dependent protein kinase-deficient mice suggest a role for this metabolic sensor in early stages of the disease. Metab Brain Dis 30(6):1369–1377
Verstraeten A, Theuns J, Van Broeckhoven C (2015) Progress in unraveling the genetic etiology of Parkinson disease in a genomic era. Trends Genet 31(3):140–149
Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, Janle EM, Lobo J, Ferruzzi MG, Davies P, Marambaud P (2010) AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem 285(12):9100–9113
Vingtdeux V, Chandakkar P, Zhao H, d’Abramo C, Davies P, Marambaud P (2011a) Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-beta peptide degradation. FASEB J 25(1):219–231
Vingtdeux V, Davies P, Dickson DW, Marambaud P (2011b) AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol 121(3):337–349
Walker FO (2007) Huntington’s disease. Lancet 369(9557):218–228
Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E (2010) Synergy and antagonism of macroautophagy and chaperone-mediated autophagy in a cell model of pathological tau aggregation. Autophagy 6(1):182–183
Wang ZX, Tan L, Yu JT (2015) Axonal transport defects in Alzheimer’s disease. Mol Neurobiol 51(3):1309–1321
Weisova P, Concannon CG, Devocelle M, Prehn JH, Ward MW (2009) Regulation of glucose transporter 3 surface expression by the AMP-activated protein kinase mediates tolerance to glutamate excitation in neurons. J Neurosci 29(9):2997–3008
Weisova P, Davila D, Tuffy LP, Ward MW, Concannon CG, Prehn JH (2011) Role of 5′-adenosine monophosphate-activated protein kinase in cell survival and death responses in neurons. Antioxid Redox Signal 14(10):1863–1876
Weisova P, Alvarez SP, Kilbride SM, Anilkumar U, Baumann B, Jordan J, Bernas T, Huber HJ, Dussmann H, Prehn JH (2013) Latrepirdine is a potent activator of AMP-activated protein kinase and reduces neuronal excitability. Transl Psychiatry 3:e317
Won JS, Im YB, Kim J, Singh AK, Singh I (2010) Involvement of AMP-activated-protein-kinase (AMPK) in neuronal amyloidogenesis. Biochem Biophys Res Commun 399(4):487–491
Wong YC, Holzbaur EL (2014) The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci 34(4):1293–1305
Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D (2003) LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 13(22):2004–2008
Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D (2005) Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab 2(1):21–33
Wu Y, Li X, Zhu JX, Xie W, Le W, Fan Z, Jankovic J, Pan T (2011) Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neurosignals 19(3):163–174
Wu CA, Chao Y, Shiah SG, Lin WW (2013) Nutrient deprivation induces the Warburg effect through ROS/AMPK-dependent activation of pyruvate dehydrogenase kinase. Biochim Biophys Acta 1833(5):1147–1156
Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, Saiu P, Howell SA, Aasland R, Martin SR, Carling D, Gamblin SJ (2011) Structure of mammalian AMPK and its regulation by ADP. Nature 472(7342):230–233
Xu Y, Liu C, Chen S, Ye Y, Guo M, Ren Q, Liu L, Zhang H, Xu C, Zhou Q, Huang S, Chen L (2014) Activation of AMPK and inactivation of Akt result in suppression of mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death in in vitro models of Parkinson’s disease. Cell Signal 26(8):1680–1689
Yang TT, Shih YS, Chen YW, Kuo YM, Lee CW (2015) Glucose regulates amyloid beta production via AMPK. J Neural Transm 122(10):1381–1390
Yoon SO, Park DJ, Ryu JC, Ozer HG, Tep C, Shin YJ, Lim TH, Pastorino L, Kunwar AJ, Walton JC, Nagahara AH, Lu KP, Nelson RJ, Tuszynski MH, Huang K (2012) JNK3 perpetuates metabolic stress induced by Abeta peptides. Neuron 75(5):824–837
Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12(1):9–14
Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Naslund J, Mathews PM, Cataldo AM, Nixon RA (2005) Macroautophagy—a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol 171(1):87–98
Zala D, Hinckelmann MV, Yu H, Lyra da Cunha MM, Liot G, Cordelieres FP, Marco S, Saudou F (2013) Vesicular glycolysis provides on-board energy for fast axonal transport. Cell 152(3):479–491
Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, Pagani W, Lodin D, Orozco G, Chinea A (2015) A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int 6:171
Zhao Z, Sui Y, Gao W, Cai B, Fan D (2015) Effects of diet on adenosine monophosphate-activated protein kinase activity and disease progression in an amyotrophic lateral sclerosis model. J Int Med Res 43(1):67–79
Zheng D, MacLean PS, Pohnert SC, Knight JB, Olson AL, Winder WW, Dohm GL (2001) Regulation of muscle GLUT-4 transcription by AMP-activated protein kinase. J Appl Physiol (1985) 91(3):1073–1083
Zuccato C, Valenza M, Cattaneo E (2010) Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev 90(3):905–981
Acknowledgments
This work was supported by the French Fondation pour la cooperation Scientifique—Plan Alzheimer 2008–2012 (Senior Innovative Grant 2013) and in part through the Labex DISTALZ (Development of Innovative Strategies for a Transdisciplinary Approach to Alzheimer’s Disease). MD holds a doctoral fellowship from Lille 2 University.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Domise, M., Vingtdeux, V. (2016). AMPK in Neurodegenerative Diseases. In: Cordero, M., Viollet, B. (eds) AMP-activated Protein Kinase. Experientia Supplementum, vol 107. Springer, Cham. https://doi.org/10.1007/978-3-319-43589-3_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-43589-3_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-43587-9
Online ISBN: 978-3-319-43589-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)