
Abstract
Traumatic brain injury (TBI) may be due to a bump, blow, or jolt to the head or a penetrating head injury that disrupts normal brain function; it presents an ever-growing, serious public health problem that causes a considerable number of fatalities and cases of permanent disability annually. Physical exercise restores the healthy homeostatic regulation of stress, affect and the regulation of hypothalamic–pituitary–adrenal axis. Physical activity attenuates or reverses the performance deficits observed in neurocognitive tasks. It induces anti-apoptotic effects and buttresses blood–brain barrier intactness. Exercise offers a unique non-pharmacologic, non-invasive intervention that incorporates different regimes, whether dynamic or static, endurance, or resistance. Exercise intervention protects against vascular risk factors that include hypertension, diabetes, cellular inflammation, and aortic rigidity. It induces direct changes in cerebrovasculature that produce beneficial changes in cerebral blood flow, angiogenesis and vascular disease improvement. The improvements induced by physical exercise regimes in brain plasticity and neurocognitive performance are evident both in healthy individuals and in those afflicted by TBI. The overlap and inter-relations between TBI effects on brain and cognition as related to physical exercise and cognition may provide lasting therapeutic benefits for recovery from TBI. It seems likely that some modification of the notion of scaffolding would postulate that physical exercise reinforces the adaptive processes of the brain that has undergone TBI thereby facilitating the development of existing networks, albeit possibly less efficient, that compensate for those lost through damage.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
A common cause of traumatic brain injury (TBI) is the impact of a mechanical insult, an external force, to the brain that results in tissue damage, cerebral inflammation and neurodegeneration in the central nervous system (cf., Rice et al. 2003). After mechanical impact, the activation of secondary systems contributes to ischemic damage due to circulatory disturbances, compromisation of the blood–brain barrier (BBB), and excitotoxic loss of neurons (Xiong et al. 1997). It has been estimated that about 1.7 million citizens of the USA sustain TBI annually (Faul et al. 2010); the incidence in Sweden has varied from one study reporting 540 individuals per 100,000 in Western Sweden (Andersson et al. 2003), to another, 20-year-old study, reporting 249 per 100,000 in Northern Sweden (Johansson et al. 1991), while a recent National registry-based study reported about 250 per 100,000 individuals annually (Borg et al. 2011). Since psychiatric disorders are common following TBI, the timing of onset may differ according to pre-injury history with different trajectories for anxiety and depressive disorders, thereby posing implications for identifying the time individuals are most at risk for psychiatric disorders post-injury (Bryant 2011; Gould et al. 2011). TBI diagnosis covers a wide range of short- and long-term impairments in physical, cognitive, behavioral, and emotional domains, depending upon injury extent, severity, and location (Albensi 2001). In mild sport-related TBI, memory problems, not least spatial navigation memory deficits and alterations in brain networks suggest widespread behavioral changes (Slobounov et al. 2010). For example regarding affective status, TBI patients report significant changes in self-concept with the post-injury self-image experienced negatively in comparison with pre-injury self-image. Their perceived change in self-identity was positively associated with depression and grief but negatively associated with self-esteem and awareness. Awareness in this context was negatively associated with self-esteem and positively associated with depression (cf. Carroll and Coetzer 2011). Anxiety following childhood TBI appears to be part of a broader problem of affective dysregulation related to a damaged dorsal frontal lobe and frontal white matter systems (Max et al. 2011). Not least, quality-of-life issues are critical to considerations of post-injury recovery (cf. Hawthorne et al. 2011).
The tragic and wasteful consequences of TBI that are expressed even under a cursory examination of the pathophysiology of TBI suggest that it is both important and of necessity that the particular benefits of physical exercise as an intervention ought to be addressed. The variable nature of TBI pathology is characterized by the deficits observed in the capacity of neurons to metabolize energy, sustain synaptic function, often with consequent cognitive–emotional and debilitating disorders that disrupt homeostatic mechanisms (Wu et al. 2011). Exercise influences the homeostatic imbalance that arises under conditions of prevailing HPA dysregulation. The anti-neurodegenerative effects of exercise emerge beneficially through expressions of neurocognitive functioning and neuroplasticity, whereas the destructive and wasteful consequences of TBI for cellular inflammation and vascular pathophysiology appear to be assuaged by carefully selected exercise/activity regimes. The notion of Scaffolding in injured/aging brains may provide a conceptual ‘hub’ to elucidate the unique utility and incentive of exercise in promoting restorative gains against greater or lesser damage to structures at various levels and their related functions.
Pathophysiological Consequences of TBI
The main pathophysiological outcomes of TBI, taking into account several individual difference moderating factors such as general health and conditions, extent of injury and “age-at-injury”, include inflammation, ischemia, oxidative stress (which may be integrated with mitochondrial function), alterations in wide-scale networks, e.g., the default mode network, impaired mitochondrial function, altered BBB permeability, and lasting changes in white matter integrity. These outcomes, sited within the brain and CNS, precede, to greater or lesser extent, the plethora of alterations in behavioral performance within cognitive, emotional, sensory and motor domains. The consequences of TBI ought to be viewed at (i) molecular and cellular levels, (ii) at macro levels, including circuitry and regional levels, for instance as exemplified by neuroimaging techniques and functional magnetic resonance imaging (fMRI) networks, and (iii) at the levels of neurobehavioral outcomes and expressions that may offer the initial and preliminary determinants of health status and function. Ancilliary to the notion of TBI levels the moderation by individual difference factors upon outcome implies that exercise may not necessarily present the primary moderator but rather influence and/or be influenced by individual characteristics and predispositions that precede injury. The convergence of adverse factors could result severe clinical outcome. A meta-analysis by Molloy et al. (2011) supports the contention of an increased risk for schizophrenia spectrum disorder following TBI; the authors observed a larger effect in those individuals with a genetic predisposition to psychosis. Thus, the pathological outcomes of TBI take on greater consequence with closer examinations of the affected domains.
TBI is a major cause of death and disability worldwide, according to some sources especially in children and young adults. The causes of TBI include falls, vehicle accidents (highly prevalent), sports injuries, and physical violence. Cerebral inflammation following TBI plays a critical role in the pathophysiology of brain diseases of high prevalence and economic impact, such as major depression, schizophrenia, post-traumatic stress disorder, and Parkinson’s and Alzheimer’s diseases. The assessment and diagnosis of TBI must take into account a multitude of external and internal (individual) factors that interact at the point-of-confrontation starting from the mechanical forces that induce shearing and compression of neuronal and vascular tissue at the time of impact (Moppett 2007). This is followed by a series of pathological events may then ensue, thereby leading to further brain injury. The presentations of secondary injury may be amenable to intervention but cause deterioration through secondary physiological insults. The presence of various risk factors that lead to poor outcome after TBI may lead to further impairments in tissue integrity. Most of these risk factors are fixed at the time of injury such as age, gender, mechanism of injury, and presenting signs (Glasgow Coma Scale and pupillary signs), but some such as hypotension and hypoxia are potential areas for medical intervention. The diagnosis of TBI and its severity ought to describe cognitive, physical, and psychological domains, with physical deficits include ambulation, balance, coordination, fine motor skills, strength, and endurance. The cognitive deficits ought to include expressions language and communication, information processing, memory, affective functions, coping behavior, and perceptual skills. Psychological status may be altered all-too-often with the debut of maladaptive behaviors such as substance abuse and problems of impulse control. Not least, adjustment to disability issues and confrontational episodes are frequently encountered by individuals presenting TBI.
At the level of outcome, Kim (2011) has presented a systematic review of patient outcomes in TBI with the following factors associated with unfavorable outcomes: (i) sociodemographic factors, including older age, male gender, lower level of education; (ii) clinical factors, including lower Glasgow Coma Scale score, motor vehicle crash injury, hypotension, hypoxia, increased intracranial pressure, no pupil reaction, hypo- or hyperglycaemia, anemia, coagulophy (clotting and bleeding disorders), hypo- or hyperthermia, abnormal levels of electrolytes, duration of coma; (iii) higher level of computerized tomography classification by Marshall score category [a classification scale of intracranial pathology on head computed axial tomography (CT)]; and (iv) type of intracerebral lesion. Alcohol, another complicating factor, potentiates severity of TBI (Cunningham et al. 2002). Ischemia, the inadequate supply of blood to organ tissues due to blockade, induces poor oxygen supply or cerebral hypoxia thereby leading to the death of brain tissue or cerebral infarction/ischemic stroke and constitutes a particular type of TBI. In cerebral ischemia, excitotoxic cell death results from glutamate release by injured neurons resulting in hyperactive glutamate receptors inducing excessive intracellular Ca2+ influx (Zhang et al. 2002). This increase in intracellular Ca2+ leads to activation of caspase-3 with eventual apoptotic and necrotic cell death (Raghupathi 2004; Robertson 2004). Free radical production, facilitated by the arachidonic acid cascade in experimentally induced focal ischemia in the rat brain, induces lipid peroxidation in neuronal and glial cell membranes as well as DNA damage in neuronal cells (Djebaili et al. 2005; Lee et al. 2005). Clausen et al. (2011) have shown a close relationship between oxidative stress and excitotoxicity following TBI in humans. fMRI has shown that alterations in the brain resting state default mode network, in the subacute phase of injury, are important in assessing TBI severity and pathophysiology of the disorder (Johnson et al. 2011).
At molecular and cellular levels, TBI induces impaired mitochondrial function, oxidative stress and altered antioxidant activity in the brain and spinal cord (Azbill et al. 1997; Niizuma et al. 2009). Neurons and glial cells in proximity to the damaged region induce apoptotic cell death at the early post-TBI stage (Itoh et al. 2009, 2010), with accompanying cerebral dysfunction (Dressler and Vemuganti 2009). Experimental TBI reliably models the functional deficits, within cognitive, emotional, sensory, and motor domains that are observed in TBI patients (Ekmark-Lewén et al. 2010; Sigurdardottir et al. 2010) thereby providing analyses at all levels of TBI pathophysiology. The notion of ‘age-at-injury’ poses an important aspect of both clinical and experimental TBI since the immature brain may be particularly vulnerable to injury during critical periods of development (Serra-Grabulosa et al. 2005), whereas considerable evidence indicates that outcomes from TBI are worse in elderly individuals (Marklund et al. 2009; Onyszchuk et al. 2008). Applying a diffuse TBI model in rat pups to exemplify the level of behavioral outcomes, Cernak et al. (2010) showed motor deficits that persisted even after the pups had reached adulthood, as well as reduced cognitive performance 2 weeks after injury. In addition, it was observed that the model induced prominent edema, particularly evident in 7- and 14-day-old animals, as measured by both the wet weight/dry weight method and diffusion-weighted MRI, often seen in pediatric TBI. BBB permeability, as measured by the Evans blue dye technique, peaked at 20 min after trauma in all age groups, with a second peak found only in adult animals at 24 h after injury. The BBB level of cellular pathology presents both global and regional extents of damage with the likelihood that the intervention may take on the role of damage control. BBB disruption in epileptic patients following even mild TBI is not an infrequent observation (Tomkins et al. 2011). Mild TBI induces neuropathological insult ranging from white matter damage to long-lasting neurocognitive deficits (Bazarian et al. 2006; McAllister et al. 2006; Niogi et al. 2008), with or without affective disorder symptoms (Chan et al. 2008; Lanctôt et al. 2010; Rapoport et al. 2008). Zohar et al. (2011), in a mouse model of mild TBI, have observed profound and long-lasting, irreversible cognitive impairments as well as permanent depressive-like behavior. Thus, any understanding of putative interventional efforts ought to present evidence for positive gains at the various levels and loci of injury.
Physical Exercise as Intervention
The common features of activities that may contribute to physical exercise ought to be presented and examined for purposes of determining eventual appropriateness for interventional status. Physical exercise has been described as any and all activity that generates force through muscular activity that disrupts a homeostatic state (McArdle et al. 1978). Although daily physical activity holds benefits for general measures of function, quality-of-life and physical strength, as well as increasing endurance (Dechamps et al. 2010; Marks et al. 2009), much evidence presents the manifest advantages for cerebral integrity and neurocognition (Kramer et al. 1999; Lustig et al. 2009; Marks et al. 2010). Any bodily activity that enhances or maintains physical fitness implies the involvement of regular and frequent exercise. Morris and Schoo (2004) have defined exercise as a planned, structured physical activity with the purpose of improving one or more aspects physical fitness and functional capacity. It has been characterized on the basis of type, intensity, frequency, and duration, with either endurance or resistance as the training end-point (Mougios 2010). Endurance exercise develops the capacity to exert oneself over long periods whereas resistance exercise implies the resistance to the force of muscular contraction and elastic or hydraulic resistance, a specific type of strength training that utilizes elastic or hydraulic tension to provide this resistance (Ormsbee et al. 2009). Several molecular agents feature in different aspects within the context of physical exercise and increasingly there is much consideration of how their actions converge to impact the structure and function (e.g., mobility, learning and memory domains) of the brain and CNS (Ang and Gomez-Pinilla 2007). For example, serum brain-derived neurotrophic factor (BDNF) is known to increase with exercise and this increase is believed to originate from the brain and it is suggested that monoamines are involved in BDNF regulation (Maisonpierre et al. 1991).
The nature of physical exercise may vary enormously must needs be tailored to the special requirements of each individual even before proceeding with considerations of cellular, macro-level, outcome-level, and the domains of neurologic/psychiatric condition. The type, intensity, frequency, and duration of exercise determines the extent to which muscle contractions induce generation of reactive oxygen- and nitrogen species (RONS) with strenuous exercise causing oxidation of proteins, lipids, DNA, release of cytosolic enzymes, and other markers of cell damage (Cubrilo et al. 2011; Powers et al. 2011), but only exhaustive (very strenuous) exercise may be detrimental (Gomez-Cabrera et al. 2009). As a general paradigm, running exercise, whether wheel running, treadmill running, running of various other types or fast and strenuous (quadropedal) ‘Nordic’ walking, is associated with cardiovascular, proprioceptive, metabolic, motor, motivational, and/or general arousal system mobilization with multiple benefits for individuals (Deslandes et al. 2009; Qiu et al. 2010; Zhang et al. 2011). Mitochondrial biogenesis is critical to normal cellular functioning; exercise, particularly aerobic, activates mitochondriogenesis (Eynon et al. 2011). Low mitochondriogenesis is implicated in loss of muscle function in aging and in the development of frailty. Thus, the positive effects of exercise on mitochondriogenesis are limited (Viňa et al. 2009), possibly due to depletion of peroxisome proliferator-activated receptor-γ coactivator-1α (Derbré et al. 2011). Physical exercise has been manifested in marked improvements both in function and biomarker integrity (Archer 2011; Archer and Fredriksson 2010; Archer and Kostrzewa 2011; Archer et al. 2010, 2011a, b, Fredriksson et al. 2011). Ang et al. (2003) have indicated that neuroprotection after physical exercise may be the consequence of elevated levels of an endogenous neurotrophic factor/nerve growth factor and the proliferation of its receptive cholinergic neurons.
It is essential to remain aware of the stress-provoking aspect of exercise at both behavioral and biomarker expressions continually. Strenuous, or markedly strenuous, levels of physical exertion, such as during marathons/ultramarathons/triathalons, induce large elevations in plasma cytokine levels (Nieman et al. 2001, 2005; Smetanka et al. 1999). Exercise-induced cytokine release under these conditions may be due to elevation in catecholamines and cortisol, high core body temperature, endotoxemia, etc. (Bosenberg et al. 1988; Camus et al. 1997; Jeukendrup et al. 2000). Notwithstanding these effects of strenuous exercise, physical activity exerts anti-inflammatory effects (Pedersen 2006), and it may be applied as a means to control low-grade systemic inflammation (Mathur and Pedersen 2011). As indicated above and below, it is quite evident that physical exercise presents a form of stress with the exercise–stress–inflammation relationship a model of neuroendocrine interaction. It stimulates the innate immune responses with effects upon inflammatory responses mediated by the sympathetic nervous system and the hypothalamic–pituitary–adrenal axis (HPA), yet subject to the dictates of moderate and long-term necessity (Ortega 2003). Martin-Cordero et al. (2011), using an experimental model of metabolic syndrome, have shown that a physical exercise training schedule on a treadmill (from 25 cm/s for 10 min in the 1st weeks to 35 cm/s in the last month 5 days/week over 14 weeks) produced a reinstatement of the interleukin-6 (Il-6)-noradrenaline (NA) feedback mechanism in response to acute exercise (5 min at 17 cm/s followed by 25-35 min at 35 cm/s with no slope).
The notion of scaffolding provokes the concept of transient measures, external to the buildings, that provides for the construction and maintenance of buildings but not the building itself. In this regard, physical exercise regimes, representing environmental mediatory factors or the transient external-locus scaffolding, have provided measureable improvements at both behavioral and biomarker levels under several brain deficit conditions. The notion is analogous to that of parental scaffolding of executive functioning in preschool children (Hammond et al. 2011), presupposing the necessity for some manner of construction, development, maintenance and neuronal repair. Although the notion of “Scaffolding” concerns loss of cognition during aging in the context of a dynamically adaptive brain and central nervous system, it lends itself plausibly the interventional role of physical exercise in brain disorders. According to Park and Reuter-Lorentz (2009), scaffolding presents a normal process that continues across the lifespan involving the application and development of complementary, alternative neural circuits to achieve a particular cognitive goal; it is protective of cognition in the aging (or disabled) brain and is reinforced by physical exercise and cognitive engagement (which is harnessed during exercise). With regard to the brain afflicted by TBI, the notion of scaffolding offers the viewpoint that exercise buttresses the more or less, dependent upon injury extent, surviving adaptive and compensatory neuroreparative processes. Voss et al. (2010) have shown the scaffolding effect of a 1-year walking exercise intervention that increased the resting functional connectivity and efficiency in higher level cognitive networks that included frontal, temporal, and posterior cortices in the Default Mode Network and Frontal Executive Network. Within the context of TBI situations where long-lasting rehabilitational advantages are sought, physical activity/exercise was shown to provide beneficial outcomes for both stroke and TBI patients: Dejong et al. (2011) showed that two types of physical therapy activities (gait training and community mobility) were both positively associated with discharge motor Functional Independence Measure outcomes across for these patients groups.
Exercise Influence Upon HPA Dysregulation: Stress and Affect Following TBI
Neuroendocrine disturbances are common after TBI, and the detrimental effects of TBI on brain integrity and functioning are markedly potentiated by the effects of stress biomarkers (Griesbach et al. 2011). This situation is hardly surprising since strong correlations were obtained between affective distress and self-discrepancy (Cantor et al. 2005). The initial, protective response to TBI involves acute activation of the HPA axis through promotion of intravascular fluid retention and elevated cortical levels modulating immune/inflammatory responding and elevated metabolic substrate availability (Johnston 2006). Endocrine failure may induce clinically critical consequences during acute and convalescent care after TBI; this condition may be caused by direct injury to the HPA axis, neuroendocrinological effects from catecholamines and cytokines, or from systemic infection/inflammation that produces primary gland failure (Powner et al. 2006). Both acute and chronic dysfunction of the HPA axis expressed through GH, quality-of-life and neurobehavioral deficits, as well as adrenal insufficiency, are observed after mild, moderate and severe TBI (Cohan et al. 2005; Dusick et al. 2008; Kelly et al. 2006). Given the cerebrovascular and anatomical vulnerability exacerbated by the diffuse and variable nature of TBI, HPA complex regulation may be defective to greater or lesser extent, e.g., following head wounds, pituitary damage is not uncommon (Benvenga 2005). The type and duration of pituitary damage varies with TBI characteristics and patient heterogeneity (Tanriverdi et al. 2007, 2010a, b, c). TBI-induced HPA dysregulation due to adrenocorticotropic hormone (ACTH) and/or adrenal insufficiency has been observed in patients with mild to severe TBI (Tanriverdi et al. 2006, 2008a, b). Both among mild TBI patients and in an animal model of TBI, controlled cortical impact injury, severity-dependent disruptions of HPA axis are described (Taylor et al. 2008, 2010). The response of the HPA axis to psychosocial stress and acute exercise is similar although the latter induces a stronger response (Negrao et al. 2000).
There is much evidence that regular physical exercise counteracts some of the effects of stress, although several studies have suggested that prior exercise does not alter the acute HPA axis responses to stress (e.g., Fatouros et al. 2010). Nevertheless, Campeau et al. (2010) have presented results showing that 6 weeks of daily or intermittent, voluntary wheel-running exercise constrains the HPA axis response to mild, but not more intense stressors, and that this regulation may be mediated at a central level beyond the primary sensory input. Regular physical exercise alleviates most of the symptoms associated with stress, affective syndromes from various causes and associated health problems often linked to TBI, such as anxiety. The sympatho-adrenal system with its stress-induced activation and increased release of catecholamines presents a major system involved in the response to stressful events. Exercise training offers as an important modulator of sympatho-adrenal system, adrenal medulla, and stellate ganglia being two components of this system. Gavrilovic et al. (2011) investigated physical exercise-related changes in gene expression of catecholamine biosynthetic enzymes tyrosine hydroxylase, dopamine-β-hydroxylase, and phenylethanolamine N-methyltransferase in the adrenal medulla and stellate ganglia of chronically psychosocially stressed adult rats exposed daily to 20-min treadmill exercise for 12 weeks. They observed that treadmill exercise induced decreased gene transcription of catecholamine biosynthetic enzymes in stellate ganglia and an attenuation of cardiac NA production during stressful situations. The reduction of catecholamine synthesis in stellate ganglia may have been linked to the beneficial effects of treadmill exercise on cardiovascular system in stressed animals.
Several studies suggest that physical exercise may alleviate depressive symptoms in clinical/nonclinical populations to greater or lesser extent (Krogh et al. 2010; Lawlor and Hopker 2001; Rethorst et al. 2009). Takada et al. (2009) examined the links between lifestyle, working environment, depressive symptoms and suicide ideation in 4,118 Japanese business employees (2,834 male and 1,284 female). They found that the factors associated with depressive symptoms were: high levels of job stress, problem drinking, insufficient sleep, lack of social support and absence of stress reduction techniques over both genders, such as lack of physical exercise and sedentary lifestyle. A wide range of studies have shown that regular physical exercise reduces stress symptoms, mood disorder, anxiety and depressiveness (Broman-Fulks and Storey 2008; Janisse et al. 2004; Smith et al. 2007; Tsang et al. 2008; Wang et al. 2010). Acute resistance exercise induced catecholaminergic rather than HPA axis stimulation (Fatouros et al. 2010). Exercise stimulates GH, prolactin and cortisol release (Karkoulias et al. 2008; Weltman et al. 2003) with exercise training, in humans, modulating the neuroendocrine response to challenge (Brooks et al. 2001, 2003), and exerting an antidepressant effect (Mead et al. 2008). Campbell et al. (2009) demonstrated that voluntary wheel running exercise initially caused hyperactivation of the HPA axis, due to enhanced adrenal sensitivity to ACTH, and consequently these alterations in HPA activity were eliminated to give restored normal levels completely by 8 weeks of exercise training.
High rates of depression have been reported in individuals with TBI with estimates ranging from 6 to 77% (Jorge et al. 2004; Kreutzer et al. 2001) and later life-long risk (Dikman et al. 2004). Both cognitive and psychosocial behavior impairments may be observed in the depression following TBI (Chamelian and Feinstein 2006; Fann et al. 1995; Hibbard et al. 2004). Neuropathologically, it appears that an imbalance of left versus right frontal and parietal viable brain volumes is related to the development of depression (Schönberger et al. 2011). Hudak et al. (2011) studied the post-injury atrophy of brain regions of interest in TBI patients with depressive symptoms assessed by the Beck Depression Inventory-II. They found that three regions of interest, left rostral anterior cingulate and bilateral orbitofrontal cortex also relevant to spontaneous depression, were found to be significantly correlated with depressive symptoms. The efficacy of exercise in the treatment of depression has been documented (e.g. Blumenthal et al. 2007; Dunn et al. 2005; Greer and Trivedi 2009). Among TBI survivors presenting depression, physical exercise intervention was highly preferred (Fann et al. 2009), improving several aspects of depressive symptoms including sleep problems, anxiety, fatigue and quality-of-life (Gordon et al. 1998; Lai et al. 2006; Ströhle 2009), as well as cognition (Grealy et al. 1999; Kleim et al. 2003). Schwandt et al. (2011) tested the effect of an aerobic exercise intervention on symptoms of depression among individuals with TBI (more than 11 months previously) using the Hamilton Rating Scale for Depression, aerobic capacity (cycle ergometer, heart rate at reference resistance, perceived exertion), the Rosenberg Self-Esteem Scale and the program perception survey. They have reported that with post-exercise intervention all the participants presented fewer symptoms of depression, improved aerobic capacity and higher self-esteem following the intervention. High levels of satisfaction with the program, improved mood and cardiovascular were reported without any adverse effects. Finally, Hoffman et al. (2010) have reported that TBI patients with higher levels of exercise expressed improved levels of sleep, community participation and overall quality-of-life.
Exercise and Neurocognition Following TBI
With an estimated 10 million individuals affected worldwide annually, the World Health Organization estimates that TBI will surpass other diseases as the major cause of death and disability by the year 2020. The burden of mortality and morbidity that this condition imposes upon individuals, caregivers and society, renders TBI a pressing public health and medical problem. It appears that 40-50% of the afflicted patients express cognitive deficits (Greve et al. 2003). More recently, 33% of adults with TBI were reported to present impaired cognitive function on discharge from hospital, although limited recovery was observed during the 1st year post-injury (Lin et al. 2010). Cognitive impairments associated with TBI, if not permanent, may last decades, including memory deficits, reduced abstract thinking, retarded information processing, poor concentration, information processing and sequencing deficits, slowed reaction time, dysarthia (problems articulating speech), anomia (problems of word/name recollection), impaired auditory comprehension, loss of verbal fluency, deficits in general intelligence and problems in planning and organization (Rimel et al. 1982), which lead to impulse control deficits. Even mTBI causes retarded information processing, poor concentration and memory deficits (Rimel et al. 1981). Adolescents with more severe TBI may underestimate their own degree of executive dysfunction in daily life, particularly aspects of metacognitive abilities (Wilson et al. 2011). Diffusion tensor imaging (DTI) analyses have shown that greater white matter pathology predicted greater cognitive deficits (Kraus et al. 2007). Wilde et al. (2011) assessed the neural correlates with fMRI and DTI of working memory, using the Sternberg Item Recognition Task in 40 children with moderate-to-severe TBI compared to 41 demographically comparable children with orthopedic injury. They observed that diminished white matter integrity of the frontal lobes and cingulum bundle, as measured by DTI, was associated with longer reaction times on the Sternberg Item Recognition Task. Across modalities, the cingulate emerged as a common structure related to performance after TBI. In view of the marked cognitive deficits and associated neuropathology, the application of physical exercise as an interventional parameter in TBI ought to be of critical importance.
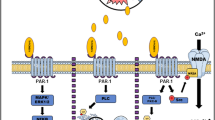
In both human and laboratory animal studies with forced or voluntary exercise, improvements in the cognitive functioning domain (including learning ability and memory capacity) were obtained under conditions of stroke, aging and life-span (McDonnell et al. 2011a; Tseng et al. 2011; Voss et al. 2011). These observations suggest that voluntary exercise programs may be efficacious against cognitive deficits in TBI (e.g., Grealy et al. 1999). For example, older healthy women can improve episodic and working memory through spending time on a challenging physical or mental activity (Evers et al. 2011). Little et al. (2010) have shown that TBI causing diffuse axonal injury that results in damage to the thalamic projection fibers, and physical exercise, rota-rod and treadmill training, contributed to increased expression of synaptophysin in subcortical regions of the ischemic hemisphere including the hippocampus, dentate gyrus, and thalamus (Seo et al. 2010). Synaptophysin is an abundant synaptic vesicle protein without a definite synaptic function. McMorris et al. (2011) concluded that acute, intermediate intensity exercise induced a strong beneficial effect on speed of response in working memory tasks but a low to moderate, detrimental one on accuracy, suggesting that exercise-induced increases in brain concentrations of catecholamines resulted in faster processing whereas increased neural noise may negatively affect accuracy (see also Pontifex et al. 2009). Gosselin et al. (2011) used fMRI and event-related potentials (ERP) to study cerebral dysfunction in 14 mTBI symptomatic patients (5.7 ± 2.9 months post-injury) compared to 23 healthy controls, during the post-acute phase, using a visual externally ordered working memory task. They observed attenuated blood oxygen level dependent (BOLD) signal changes in the left and right mid-dorsolateral prefrontal cortex (mid-DLPFC), the putamen, the body of the caudate nucleus, and the right thalamus in the mTBI group compared with the controls. Furthermore, symptom severity and BOLD signal changes were correlated: patients with more severe symptoms had lower BOLD signal changes in the right mid-DLPFC. For ERP, a group × task interaction was observed for N350 amplitude. A larger amplitude for the working memory task than for the control task was found in the controls, but not in mTBI patients, who presented weak amplitudes on both tasks. Figure 1 presents selected neurocognitive deficits with related brain pathology following TBI that were alleviated through exercise intervention together with inter-relationships between TBI effects on the brain and cognitive performance, exercise effects on brain and cognition, and the overlap that offers putative treatment possibilities that promote the notion of scaffolding.

Selected neurocognitive deficits with related brain pathology following TBI that were alleviated through exercise intervention (see text for descriptions and references). Inter-relationships between TBI effects on the brain and cognitive performance, exercise effects on brain and cognition, and the overlap that offers putative treatment possibilities that promote the notion of scaffolding. *↑, Increased expression of synaptophysin; ▼↓/↑, decreased/increased blood oxygen level dependent (BOLD) signals in right and left mid-dorsolateral prefrontal cortex
Both correlational and regressional studies have demonstrated that healthy (non-clinically debilitated) adults individuals who are generally more active physically are more likely than individuals who are generally inactive to show higher levels of performance on several different cognitive tasks, including executive function and working memory (Ratey and Loehr 2011). Higher levels of self-reported physical activity were associated also with better performance on tasks of selective attention (Hillman et al. 2006), task-switching performance (Kamijo and Takeda 2010), and superior processing speed, memory, mental flexibility and overall cognition (Angevaren et al. 2007). Exercise even improved cognition in older adults who did not present cognitive impairment (Angevaren et al. 2008). The randomized control trial study carried out by McMillan et al. (2002) comparing attentional control training, physical exercise and a control condition in 130 adults presenting TBI, 3 months and 1-year post-TBI and living at home with supervised exercise sessions, is illustrative. Tests of outcome included test of everyday attention, the Adult Memory of Information Processing Battery, Paced Auditory Serial Addition test, Trail Making test, Sunderland Memory questionnaire, and Cognitive Failures Questionnaire, incorporated aspects of both executive function and working memory with recording sessions at baseline, 6 months, and 12 months, with an addition questionnaire at 12 months. Their evidence suggested the aerobic exercise improved performance in speed of information processing, global cognition, attention and cognitive flexibility. Nevertheless, the prevailing constraint being the complexity and inherent inconsistencies of cognition and its expression remains a continual straight-jack to undue optimism (McDonnell et al. 2011b).
Many of the benefits from physical activity augmenting neurocognition appear to be mediated by growth factors, neurotrophins and other biomarkers for the development of greater brain tissue integrity (Berchtold et al. 2010). The benefits of physical exercise for a variety of factors that influence cognitive performance, as well as measures of cognition are well-documented, including neuronal protection (Stummer et al. 1994), enhanced neurogenesis (Kannangara et al. 2010; Van Praag et al. 1999), growth factors, besides BDNF, like IGF1 and VEGF, acting synergistically to benefit neural integrity and function (Carro et al. 2001; Cotman et al. 2007) and cognitive ability (Fordyce and Wehner 1992; Samorajski et al. 1985). BDNF, itself, mediates multiple morphologic alterations at neuronal levels, such as dendritic arborization (Imamura and Greer 2009; Zhou et al. 2008), axonal and dendritic remodeling (Yacobian and Lo 2000), synaptogenesis (Lu et al. 2009; Menna et al. 2009), synaptic efficacy (Boulanger and Poo 1999; Sallert et al. 2009) and neural stem cell efficacy (Xuan et al. 2008). Griesbach et al. (2009) have shown that physical exercise counteracted the cognitive deficits associated with the head injury. Western blot analyses demonstrated that exercise elevated the mature form of BDNF, synapsin I and cyclic-AMP response element-binding protein (CREB) in the vehicle treated Sham (intact)-RW (exercised) group whereas only the mature form of BDNF and CREB were increased in the vehicle treated FPI (TBI)-RW group. The blockade of BDNF (through the pre-administration of TrkB-IgG, an immunoadhesin chimera that inactivates BDNF) greatly reduced the molecular effects of exercise in that exercise-induced increases of BDNF, synapsin I and CREB were not observed.
Exercise and Neuroplasticity Following TBI
Neuroplasticity refers to capacity of the brain and central nervous system to remodel itself at several different levels (Hallett 2005): (i) individual neurons and events responsible for remodeling that occur within the cell, (ii) groups of neurons and their functions that can evolve to alter function and daily behavior, (iii) multiple processes that may occur both in parallel and serially. Some of these processes are fast yet transient, other longer lasting but more permanent; it appears that the more persistent the occurrence of early change, the greater its likelihood of its permanence. Exercise impacts brain plasticity by involving the actions of BDNF via an influence upon the expression of select molecular systems related to the effects of BDNF on synaptic plasticity (Ding et al. 2011), but also upon the growth, differentiation and maintenance of neuronal systems (Satomura et al. 2011); in this regard, BDNF, whether induced through exercise or otherwise presents a likely substrate for expressions of scaffolding.
The fastest type of change is a simple effect of BDNF, an essential neurotrophin intimately connected with brain metabolism and homeostasis; the neurotrophic factor induces a cascade of molecular and cellular processes. Kaplan et al. (2010) have suggested treatment approaches that enhance BDNF-related signaling and have the potential to restore neural connectivity. Such treatment approaches could facilitate neuroplastic changes that lead to adaptive neural repair and reverse cognitive and emotional deficits in both TBI and PTSD. In juvenile TBI, molecular responses related to growth, development and metabolism may play a particularly important role in the injury response and the recovery trajectory following developmental TBI (Babikian et al. 2010). Lombardi (2008) has discussed the relative therapeutic effects of psychostimulant drugs co-administered with sensory–motor exercise inventions that have been shown to induce a steady acceleration of motor recovery in TBI laboratory animals; this improvement in turn is considered to exert a facilitation of the neurological recovery process. Griesbach et al. (2007) have demonstrated that an exercise-induced increase in hippocampal BDNF is dependent upon when the exercise schedule is initiated after TBI. They had observed that the introduction of voluntary exercise 2 weeks after TBI-induction with a mild fluid-percussion injury produced an increase in BDNF and an improvement in the behavioral outcome. Adult rats were allowed running wheel access either 0–6, 14–20, or 30–36 days post-injury day showing significant increases in BDNF, synapsin I and CREB. Synapsin I is a neuron-specific phosphoprotein, a substrate for cAMP-dependent and Ca/calmodulin-dependent protein kinases, and is implicated in synaptogenesis and the modulation of neurotransmitter release. CREB (cAMP response element-binding) is a cellular transcription factor with an essential role in neuronal plasticity and long-term memory formation.
According to the analysis of exercise-induced effects of BDNF release in humans (Knaepen et al. 2010), 69% of studies in healthy volunteers and in patients presenting chronic disease/damage displayed only transient increases in serum or plasma BDNF levels after an acute aerobic exercise, with endurance (aerobic) exercise more effective than resistance. It is likely that physical exercise/training elevates basal BDNF only temporarily concurrent with an up-regulation of the cellular processing of BDNF (i.e., synthesis, release, absorption, and degradation). Prolonged physical exercise, rather than limited or discrete periods, ought then to produce higher and more continuous release of the neurotrophin into blood circulation, and in turn through more efficient absorption to peripheral and central tissue, contribute to a cascade of neuroplastic and neuroprotective effects. The particular timing of exercise intervention seems of vital importance since the up-regulation of plasticity-related proteins after TBI (administered as FPI) was compromised by premature voluntary wheel-running exercise (Griesbach et al. 2004a). Contrastingly, it was seen that voluntary exercise endogenously up-regulated BDNF and enhance recovery when it was delayed after TBI (Griesbach et al. 2004b). Finally, Goldshtrom et al. (2010) have reported the case of a 24-year-old female patient, 9-year post hemispherectomy following TBI that caused right hemiparesis. The patient was trained to perform Rhythmic Exercises with Auditory Cues (REAC) with gait pattern, functional assessment, cognitive, and psychological pre- and post-intervention. It was shown that gait pattern improved with reduced ‘hip-hiking’ and increased cadence, decreased spasticity in right arm and leg together with regained sensation, and improved cognitive performance.
Anti-Inflammatory and Angiogenic Effects of Exercise: TBI
Physical exercise regimes diminish inflammation (Beavers et al. 2010), and elevate the release of adrenaline, cortisol, growth hormones and neurotrophins, prolactin and other agents equipped with immunomodulatory functions (Handschin and Spiegelman 2008). Exercise and in particular strenuous exercise increases the circulating levels of several cytikines/chemokines (Petersen and Pedersen 2005). Physical exercise induces a rapid increase in peripheral blood lymphocytes (Gleeson 2007; Kruger and Mooren 2007) accompanied by monoaminergic involvement, e.g., adrenergic influence (Kruger et al. 2008). There appears to be a consensus that lymphocytosis is observed during and after exercise, proportional to intensity and duration. Both acute and chronic exercises alter the number and function of circulating cells of the innate immune system. Acute bouts of moderate exercise cause little change in mucosal immunity, but prolonged exercise and intensified training may alter both immune functioning and efficacy of the HPA axis. The prophylactic effects of exercise may be linked to anti-inflammatory actions depending upon which aspect of exercise characteristics present the most efficacious levels of protection (Walsh et al. 2011). Exercise training decreased chronic low-level systemic inflammation associated with obesity and a sedentary lifestyle (Kizaki et al. 2011). Given the post-traumatic cerebral inflammation following severe head injury, any eventual anti-inflammatory effects linked to exercise, post-TBI, ought to offer prospects for restorative scaffolding. Infarct size following forebrain ischemia was shown to present altered inflammatory status by profile performing exercise before brain trauma with prophylactic effects on brain damage (Endres et al. 2003; see also Ding et al. 2006). Pre-TBI physical training was found to induce interleukin-10 increase per se and protected against cerebral interleukin-1β and tumor necrosis factor-α increases and fluid perfusion induced brain damage decrements in interleukin-10 (Mota et al. 2011).
Given the sharp increase in TBI since 2001 following US involvement in Afghanistan and Iraq (Martin et al. 2008; Warden 2006) with ca., 5% of a huge military personnel (1.5 million) suffering head injury accompanied by loss of consciousness (Hoge et al. 2008), the utilty of non-invasive interventions is undeniable. Several studies indicate that while stress exacerbates neuropathological changes associated with brain disorder, exercise reduces these changes (Dishman et al. 1998). In this regard, Nation et al. (2011) have presented a neurovascular pathway of neurodegenerative disorder that underlines exercise as an intervention against vascular risk factors that include hypertension, diabetes, and aortic rigidity as well as direct changes in cerebrovasculature that involve changes in cerebral blood flow, angiogenesis and vascular disease improvement. Mota et al. (2011) have shown that exercise preconditioning reduced cerebral inflammation and protected against TBI-induced toxicity; cardiovascular adaptation to physical exercise (Scheuer and Tipton 1977) offers a necessary adjunct to cerebrovascular integrity. Aerobic exercise reduces mortality in cardiovascular disorder (Taylor et al. 2004), benefits mood (Blumenthal et al. 1999), decreased impulsiveness in ADHD children (Medina et al. 2010), diminishes anxiety and depression (Lewin et al. 1992), and improves cognition (Colcombe and Kramer 2003; Gunstad et al. 2005). In older individuals, physical exercise induced a decrease in cerebral vascular conductance during moderate intensity cycling, compared to younger subjects (Fisher et al. 2008), whereas mean arterial pressure was increased in older subjects (Ogoh et al. 2011). Exercise increased also critical closing pressure (CCP) in young healthy subjects (Ogoh et al. 2010). In the context of TBI, Body Weight-Supported Treadmill Training (BWSTT) on a treadmill with rhythmic passive/active activation over 2-weeks (60 h) total training (de Bode et al. 2007) has been shown beneficial, not least as confirmed by neuroimaging techniques (Horenstein et al. 2009; Luft et al. 2004; Richards et al. 2008; Stewart et al. 2006; Thaut et al. 2007).
The positive benefits of exercise for TBI-induced inflammation and vascular pathology offer a non-invasive form of intervention, tailored to individual requirements and propensities that focuses of general aspects of health and physiology that form the foundations of brain integrity and function.
Conclusion: Exercise as Scaffolding to Alleviate TBI
The localization and severity of mild, moderate, or severe TBI causes both structural and functional destruction that, in addition to presenting symptoms, includes BBB breakdown, apoptosis and excitotoxicity, cerebral vascular pathophysiology, edema, and cerebral inflammation. Other disruptive influences pertain to emotional, neurocognitive and behavioral domains whereby hyperreactivity to stressors, HPA dysregulations and affective conditions, cognitive dysfunctions and loss of neuroplasticity exacerbate further the gravity of the TBI situation. Physical exercise facilitates improvements in debilities due to stress, affect and HPA dysregulation following TBI, augments neuroplasticity, and either reverses or attenuates the cognitive performance impairments due to TBI. It ought to be noted that the complete scope of physical exercise applications in TBI remains apparent rather than real. Chen et al. (2011) have shown that there is an increased risk of stroke among individuals who have sustained a TBI suggesting the need for more intensive medical monitoring and wider health education programs following TBI, especially during the first few months and years. It is likely that exercise models of recovery from stroke may contribute to a further understanding of TBI-exercise interventions, not least with relevance for determining exact parameters regard treatment introduction. As argued by Greisbach (2011), the premature application of post-concussive exercise may potentiate, rather than ameliorate, deficits by exacerbating post-concussive symptomatology and disrupting restorative processes. Nevertheless, it may be concluded that physical exercise/activity offers the critical factor mediating the increased BDNF levels and brain regional and cellular gains (Kobilo et al. 2011).
According to the modified notion of scaffolding (above), physical exercise reinforces the adaptive processes of the TBI brain in facilitating development of networks, albeit less efficient than pre-TBI, that compensate for those lost through cerebral damage. The essential aspect is that of an adaptive brain that through the scaffolding process is ensured optimal aging over an individual’s lifespan. It is important to distinguish between what constitutes scaffolding and what constitutes the building itself (Petrik et al. 2012), whereas exercise regimes have been shown to induce neurogenesis, neurorepair, and damage control that buttress the brain against neurodegenerative processes it is the molecular, cellular, circuitry and regional levels, the ‘steel and concrete’ that form the brain construction within the affected domains. Whether this metaphor facilitates the notion of scaffolding to elucidate the role of exercise in TBI-afflicted individuals depends on the extent to the restorative actions of physical are investigated with this end in view; it ought to be noted that scaffolding has been applied from a perspective affective and anxiety disorders (Petrik et al. 2012). Nevertheless, it is essential that the major differences in brain responses to chronic neurodegenerative aging and the eventual recovery from sudden impact damage not be neglected. Whether or not the aging brain undergoes a qualitative shift of activation to induce complementary or alternative brain networks remains an issue (Gratton et al. 2009; Peltz et al. 2011; Schneider-Garces et al. 2010). According to Voss et al. (2010), the facilitation of the shift of brain networks in the direction of young adults instead of toward alternative network formation among older adults was observed with the implementation of aerobic exercise regimes (see also Voss et al. 2012). Scaffolding hypothesis posits that qualitatively different networks emerge during aging to support/compensate for age-related brain dysfunction. Contrastingly, from the context of TBI given that impacted brain injury may actually impair function completely in certain regions, it is feasible that qualitatively different patterns of activation and/or networks may emerge, under the ‘scaffolding’ of exercise regimes that compensate for specific loss in specific cases. Under the conditions of disease/disorder following TBI, deficits in cognitive, motor, emotional or neuroimmune functioning that express the critical disturbance in adaptive capacity may be buttressed by the scaffolding that is provided by explicit physical exercise and activity. Possibly, the scaffolding process will be less effectively generated in the aged brain and in the very young. Despite this, much evidence indicates the physical exercise induces functional plasticity in large-scale brain systems not only in the TBI brain but brains affected by the ravages of time.
If indeed the notion of scaffolding following physical exercise presents a useful conceptualization for the observed evidence of restored structure and function, the question of which biological substrates (the steel and concrete) are available for consideration arises. Most likely, the widespread contributions of BDNF and other neurotrophins to neurogenesis, dendritic plasticity/arborization and neuronal repair present a primary target for further observation and further targeting of conditions influencing BDNF concentrations whether systemic or in regions of the brain. Thus, a reliable prospect for future application of the scaffolding notion to design cohesive studies appears readily available. Finally, the importance of general high level of physical condition in case of TBI affliction in order to enjoy the most advantageous prognosis has been shown to be an essential prerequisite from laboratory, clinical and retrospective studies (cf. Hassett et al. 2011). Suffice it to say that for this reason alone individuals ought to adhere routinely to physical exercise schedules to ensure their own health conditions.
References
Albensi BC (2001) Models of brain injury and alterations in synaptic plasticity. J Neurosci Res 65:279–283
Andersson EH, Björklund R, Emanuelson I, Stålhammar D (2003) Epidemiology of traumatic brain injury: a population based study. Acta Neurol Scand 107:256–259
Ang ET, Gomez-Pinilla F (2007) Potential therapeutic effects of exercise to the brain. Curr Med Chem 14:2564–2571
Ang ET, Wong PT, Moochhala S, Ng YK (2003) Neuroprotection associated with running: is it a result of increased endogenous neurotrophic factors? Neuroscience 118:335–345
Angevaren M, Aufdemkampe G, Verhaar HJ, Aleman A, Vanhees L (2008) Physical activity and enhanced fitness to improve cognitive function in older people without known cognitive impairment. Cochrane Database Syst Rev 3:CD005381
Angevaren M, Vanhees L, Wendel-Vos W et al (2007) Intensity, but not duration, of physical activities is related to cognitive function. Eur J Cardiovasc Prev Rehabil 14:825–830
Archer T (2011) Physical exercise alleviates debilities of normal aging and Alzheimer’s disease. Acta Neurol Scand 123:221–238
Archer T, Fredriksson A (2010) Physical exercise attenuates MPTP-induced deficits in mice. Neurotoxic Res 18:313–327
Archer T, Kostrzewa RM (2011) Physical exercise alleviates ADHD symptoms: regional deficits and developmental trajectory. Neurotox Res. doi:10.1007/s12640-011-9260-0
Archer T, Kostrzewa RM, Beninger RJ, Palomo T (2010) Staging perspectives in neurodevelopmental aspects of neuropsychiatry: agents, phases and ages at expression. Neurotoxicity Res 18:287–305. doi:10.1007/s12640-010-9162-6
Archer T, Fredriksson A, Johansson B (2011) Exercise alleviates Parkinsonism: clinical and laboratory evidence. Acta Neurol Scand. doi:10.1111/j.1600-0404.2010.01360.x, 2010
Archer T, Fredriksson A, Schütz E, Kostrzewa RM (2011b) Influence of physical exercise on neuroimmunological functioning and health: aging and stress. Neurotoxic Res 20:69–83
Azbill RD, Mu X, Bruce-Keller AJ, Mattson MP, Springer JE (1997) Impaired mitochondrial function, oxidative stress and altered antioxidant enzyme activities following traumatic spinal cord injury. Brain Res 765:283–290
Babikian T, Prins ML, Cai Y, Barkhoudarian G, Hartonian I, Hovda DA, Giza CC (2010) Molecular and physiological responses to juvenile traumatic brain injury: focus on growth and metabolism. Dev Neurosci 32:431–441
Bazarian JJ, Blyth B, Cimpello L (2006) Bench to bedside: evidence for brain injury after concussion—looking beyond the computed tomography scan. Acad Emerg Med 13:199–214
Beavers KM, Brinkley TE, Nicklas BJ (2010) Effect of exercise training on chronic inflammation. Clin Chim Acta 411:785–793
Benvenga S (2005) Brain injury and hypopituitarism: the historical background. Pituitary 8:193–195
Berchtold NC, Castello N, Cotman CW (2010) Exercise and time-dependent benefits to learning and memory. Neurosci 167:588–597
Blumenthal J, Babyak M, Moore KA, Craighead WE, Herman S (1999) Effects of exercise training on older patients with major depression. Arch Intern Med 159:2349–2356
Blumenthal J, Babyak M, Doraiswamy P et al (2007) Exercise and pharmacotherapy in the treatment of major depressive disorder. Psychosom Med 69:587–596
Borg J, Röe C, Nordenbo A, Andelic N, de Boussard C, Af Geijerstam JL (2011) Trends and challenges in the early rehabilitation of patients with traumatic brain injury: a Scandinavian perspective. Am J Phys Med Rehabil 90:65–73
Bosenberg AT, Brock-Utne JG, Gaffin SL et al (1988) Strenuous exercise causes systemic endotoxemia. J Appl Physiol 65:106–108
Boulanger LM, Poo MM (1999) Presynaptic depolarization facilitates neurotrophin-induced synaptic potentiation. Nat Neurosci 2:346–351
Broman-Fulks JJ, Storey KM (2008) Evaluation of a brief aerobic exercise intervention for high anxiety sensitivity. Anxiety Stress Coping 21:117–128
Brooks A, Meyer T, Gleiter C et al (2001) Effect of aerobic exercise on behavioral and neuroendocrine responses to meta-chlororphenylpiperazine and ipsapirone in untrained healthy subjects. Psychopharmacology 155:234–241
Brooks A, Meyer T, Opitz M et al (2003) 5HT1A responsivity in patients with panic disorder before and after treatment with aerobic exercise, clomipramine or placebo. Eur Neuropsychopharmaco l13:153–164
Bryant RA (2011) Mental disorders and traumatic injury. Depress Anxiety 28:99–102. doi:10.1002/da.20786
Campbell JE, Rakhshani N, Fediuc S, Bruni S, Riddell MC (2009) Voluntary wheel running initially increases adrenal sensitivity to adrenocorticotrophic hormone, which is attenuated with long-term training. J Appl Physiol 106:66–72
Campeau S, Nyhuis TJ, Sasse SK, Kryskow EM, Herlihy L, Masini CV, Babb JA, Greenwood BN, Fleshner M, Day HE (2010) Hypothalamic pituitary adrenal axis responses to low-intensity stressors are reduced after voluntary wheel running in rats. J Neuroendocrinol 22:872–888
Camus G, Poortmans J, Nys M et al (1997) Mild endotoxemia and the inflammatory response induced by a marathon race. Clin Sci 92:415–422
Cantor JB, Ashman TA, Schwartz ME, Gordon WA, Hibbard MR, Brown M, Spielman L, Charatz HJ, Cheng Z (2005) The role of self-discrepancy theory in understanding post-traumatic brain injury affective disorders: a pilot study. J Head Trauma Rehabil 20:527–543
Carro E, Trejo JL, Busiguina S, Torres-Aleman I (2001) Circulating insulin-like growth factor 1 mediates the protective effects of physical exercise against brain insults of different etiology and anatomy. J Neurosci 21:5678–5684
Carroll E, Coetzer R (2011) Identity, grief and self-awareness after traumatic brain injury. Neuropsychol Rehabil 21:289–305
Cernak I, Chang T, Ahmed FA, Cruz MI, Vink R, Stoica B, Faden AI (2010) Pathophysiological response to experimental diffuse brain trauma differs as a function of developmental age. Dev Neurosci 32:442–453
Chamelian L, Feinstein C (2006) The effect of major depression on subjective and objective cognitive deficits in moild to moderate traumatic brain injury. J Neuropsychiatr Clin Neurosci; 18:33–38
Chan F, Lanctôt KL, Feinstein A, Herrmann N, Strauss J, Sicard T, Kennedy JL, McCullagh S, Rapoport MJ (2008) The serotonin transporter polymorphisms and major depression following traumatic brain injury. Brain Inj 22:471–479
Chen YH, Kang JH, Lin HC (2011) Patients with traumatic brain injury: population-based study suggests increased risk of stroke. Stroke 42:2733–2739
Clausen F, Marklund N, Lewén A et al (2011) Interstitial F2-isoprostane 8-iso-PGF2α as a biomarker of oxidative stress following severe human traumatic brain injury. J Neurotrauma. doi:10.1089/neu.2011.1754
Cohan P, Wang C, McArthur DL, Cook SW, Dusick JR, Armin B, Swerdloff R, Vespa P, Muizelaar JP, Cryer HG, Christenson PD, Kelly DF (2005) Acute secondary adrenal insufficiency after traumatic brain injury: a prospective study. Crit Care Med 33:2358–2366
Colcombe S, Kramer AF (2003) Fitness effects on the cognitive function of older adults: a meta-analytic study. Psychol Sci 14:125–130
Cotman CW, Berchtold NC, Christie LA (2007) Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci 30:464–472
Cubrilo D, Djordjevic D, Zivkovic V, Djuric D, Blagojevic D, Spasic M, Jakovljevic V (2011) Oxidative stress and nitrite dynamics under maximal load in elite athletes: relation to sport type. Mol Cell Biochem 355:273–279
Cunningham RM, Maio RF, Hill EM, Zink BJ (2002) The effects of alcohol on head injury in the motor vehicle crash victim. Alcohol Alcohol 37:236–240
De Bode S, Mathern GW, Bookheimer S, Dobkin B (2007) Locomotor training remodels fMRI sensorimotor cortical activations in children after cerebral hemispherectomy. Neurorehabil Neural Repair 21:497–508
Dechamps A, Diolez P, Thiaudiere E et al (2010) Effects of exercise programs to prevent decline in health related quality of life in highly deconditioned institutionalized elderly persons: a randomized controlled trial. Arch Intern Med 170:162–169
Dejong G, Hsieh CH, Putman K, Smout RJ, Horn SD, Tian W (2011) Physical therapy activities in stroke, knee arthroplasty, and traumatic brain injury rehabilitation: their variation, similarities, and association with functional outcomes. Phys Ther 91:1826–1837
Derbré F, Gomez-Cabrera MC, Nascimento AL, Sanchis-Gomar F, Martinez-Bello VE, Tresguerres JA, Fuentes T, Gratas-Delamarche A, Monsalve M, Viña J (2011) Age associated low mitochondrial biogenesis may be explained by lack of response of PGC-1α to exercise training. Age. doi:10.1007/s11357-011-9264-y
Deslandes A, Moraes H, Ferreira C et al (2009) Exercise and mental health: many reasons to move. Neuropsychobiolog 59:191–198
Dikman S, Bombardier C, Machamer J et al (2004) Natural history of depression in traumatic brain injury. Arch Phys Med Rehabil 85:1457–1464
Ding Q, Vaynman S, Akhavan M, Ying Z, Gomez-Pinilla F (2006) Insulin-like growth factor I interfaces with brain-derived neurotrophic factor-mediated synaptic plasticity to modulate aspects of exercise-induced cognitive function. Neuroscience 140(3):823–833
Ding Q, Ying Z, Gomez-Pinilla F (2011) Exercise influences hippocampal plasticity by modulating brain-derived neurotrophic factor processing. Neuroscience 192:773–780
Dishman RK, Bunnell BN, Youngstedt SD, Yoo HS, Mougey EH, Meyerhoff JL (1998) Activity wheel running blunts increased plasma adrenocorticotrophin (ACTH) after footshock and cage-switch stress. Physiol Behav 63:911–917
Djebaili M, Guo Q, Pettus EH, Hoffman SW, Stein DG (2005) The neurosteroids progesterone and allopregnanolone reduce cell death, gliosis, and functional deficits after traumatic brain injury in rats. J Neurotrauma 22:106–118
Dressler J, Vemuganti R (2009) Apoptosis and gene expression after TBI. Leg Med (Tokyo) 11(Suppl 1):S54–S55
Dunn A, Trivedi M, Kampert J et al (2005) Exercise treatment for depression: efficacy and dose response. Am J Prev Med 28:1–8
Dusick JR, Wang C, Cohan P, Swerdloff R, Kelly DF (2008) Chapter 1: pathophysiology of hypopituitarism in the setting of brain injury. Pituitary. May 15. PMID: 18481181
Ekmark-Lewén S, Lewén A, Meyerson BJ, Hillered L (2010) The multivariate concentric square field test reveals behavioral profiles of risk taking, exploration, and cognitive impairment in mice subjected to traumatic brain injury. J Neurotrauma 27:1643–1655
Endres M, Gertz K, Lindauer U, Katchanov J, Schultze J, Schröck H, Nickenig G, Kuschinsky W, Dirnagl U, Laufs U (2003) Mechanisms of stroke protection by physical activity. Ann Neurol 54:582–590
Evers A, Klusmann V, Schwarzer R, Heuser I (2011) Improving cognition by adherence to physical or mental exercise: a moderated mediation analysis. Aging Ment Health 15:446–455
Eynon N, Moran M, Birk R, Lucia A. (2011) The champions’ mitochondria: is it genetically determined? A review on mitochondrial DNA and elite athletic performance. Physiol Genomics 43:789–798
Fann J, Katon W, Uomoto J, Esselman P (1995) Psychiatric disorders and functional disability in outpatients with traumatic brain injuries. Am J Psychiatry 152:1493–1499
Fann J, Jones A, Dikman S et al (2009) Depression treatment preferences after traumatic brain injury. J Head Trauma Rehabil 24:272–278
Fatouros I, Chatzinikolaou A, Paltoglou G, Petridou A, Avloniti A, Jamurtas A, Goussetis E, Mitrakou A, Mougios V, Lazaropoulou C, Margeli A, Papassotiriou I, Mastorakos G (2010) Acute resistance exercise results in catecholaminergic rather than hypothalamic-pituitary-adrenal axis stimulation during exercise in young men. Stress 13:461–468
Faul M, Xu L, Wald MM, Coronado VG (2010) Emergency department visits, hospitalizations and deaths 2002–2006. Centers for Disease Control and Prevention, National Center for injury Prevention and Control, Atlanta
Fisher JP, Ogoh S, Young CN, Raven PB, Fadel PJ (2008) Regulation of middle cerebral artery blood velocity during dynamic exercise in humans: influence of aging. J Appl Physiol 105:266–273
Fordyce DE, Wehner JM (1992) Physical activity enhances spatial learning performance with an associated alteration in hippocampal protein kinase C activity in C57Bl/6 and DBA/2 mice. Brain Res 619:111–119
Fredriksson A, Stigsdotter IM, Hurtig A, Ewalds-Kvist B, Archer T (2011) Running wheel activity restores MPTP-induced deficits. J Neural Transm 118:407–420
Gavrilovic L, Spasojevic N, Dronjak S (2011) Modulation of catecholamine-synthesizing enzymes in adrenal medulla and stellate ganglia by treadmill exercise of stressed rats. Eur J Appl Physiol. doi:10.1007/s00421-011-2046-5
Gleeson M (2007) Immune function in sport and exercise. J Appl Physiol 103:693–699
Goldshtrom Y, Knorr G, Goldshtrom I (2010) Rhythmic exercises in rehabilitation of TBI patients: a case report. J Bodyw Mov Ther 14:336–345
Gomez-Cabrera MC, Viňa J, Li LL (2009) Interplay of oxidants and antioxidants during exercise: implications for muscle health. Phys Sports Med 37:116–123
Gordon W, Sliwinski M, Echo J et al (1998) The benefits of exercise in individuals with traumatic brain injury: a retrospective study. J Head Trauma Rehabil 13:58–67
Gosselin N, Bottari C, Chen JK, Petrides M, Tinawi S, de Guise E, Ptito A (2011) Electrophysiology and functional MRI in post-acute mild traumatic brain injury. J Neurotrauma 28:329–341
Gould KR, Ponsford JL, Johnston L, Schönberger M (2011) The nature, frequency and course of psychiatric disorders in the first year after traumatic brain injury: a prospective study. Psychol Med 11:1–11
Gratton G, Wee E, Rykhlevskaia EI, Leaver EE, Fabiani M (2009) Does white matter matter? Spatio-temporal dynamics of task switching in aging. J Cogn Neurosci 21:1380–1395
Grealy M, Johnson D, Rushton S (1999) Improving cognitive function after brain injury: the use of exercise and virtual reality. Arch Phys Med Rehabil 80:661–667
Greer T, Trivedi M (2009) Exercise in the treatment of depression. Curr Psychiatr Rep 11:466–472
Greve KW, Bianchini KJ, Mathias CW, Houston RJ, Crouch JA (2003) Detecting malingered performance on the Wechsler Adult Intelligence Scale. Validation of Mittenberg’s approach in traumatic brain injury. Arch Clin Neuropsychol 18:245–260
Griesbach GS (2011) Exercise after traumatic brain injury: is it a double-edged sword? PM&R 3(6):S64–S72
Griesbach GS, Gomez-Pinilla F, Hovda DA (2004a) The upregulation of plasticity-related proteins following TBI is disrupted with acute voluntary exercise. Brain Res 1016:154–162
Griesbach GS, Hovda DA, Molteni R, Wu A, Gomez-Pinilla F (2004b) Voluntary exercise following traumatic brain injury: brain-derived neurotrophic factor upregulation and recovery of function. Neuroscience 125:129–139
Griesbach GS, Gómez-Pinilla F, Hovda DA (2007) Time window for voluntary exercise-induced increases in hippocampal neuroplasticity molecules after traumatic brain injury is severity dependent. J Neurotrauma 24:1161–1171
Griesbach GS, Hovda DA, Gomez-Pinilla F (2009) Exercise-induced improvement in cognitive performance after traumatic brain injury in rats is dependent on BDNF activation. Brain Res 1288:105–115
Griesbach GS, Hovda DA, Tio DL, Taylor AN (2011) Heightening of the stress response during the first weeks after a mild traumatic brain injury. Neuroscience 178:147–158
Gunstad J, MacGregor K, Paul R, Poppas A, Jefferson A, Todaro J, Cohen R (2005) Cardiac rehabilitation improves cognitive performance in older adults with cardiovascular disease. J Cardiopulm Rehabil 25:173–175
Hallett M (2005) Neuroplasticity and rehabilitation. J Rehabil Res Dev 42(4):xvii–xxii
Hammond SI, Müller U, Carpendale JI, Bibok MB, Liebermann-Finestone DP (2011) The effects of parental scaffolding on preschoolers’ executive function. Dev Psychol. doi:10.1037/a0025519
Handschin C, Spiegelman BM (2008) The role of exercise and PGC1alpha in inflammation and chronic disease. Nature 454:463–469
Hassett LM, Tate RL, Moseley AM, Gillett LE (2011) Injury severity, age and pre-injury exercise history predict adherence to a home-based exercise programme in adults with traumatic brain injury. Brain Inj 25:698–706
Hawthorne G, Kaye AH, Gruen R, Houseman D, Bauer I (2011) Traumatic brain injury and quality of life: initial Australian validation of the QOLIBRI. J Clin Neurosci 18:197–202
Hibbard M, Ashman T, Spielman L et al (2004) Relationship between depression and psychosocial functioning after traumatic brain injury. Arch Phys Med Rehabil 85(Suppl 2):S43–S53
Hillman CH, Motl RW, Pontifex MB et al (2006) Physical activity and cognitive function in a cross-section of younger and older community-dwelling individuals. Health Psychol 25:678–687
Hoffman JM, Bell KR, Powell JM, Behr J, Dunn EC, Dikman S, Bombardier CH (2010) A randomized controlled trial of exercise to improve mood after traumatic brain injury. PM&R 2:911–919
Hoge C, McGurk D, Thomas J, Cox A, Engel C, Castro C (2008) Mild traumatic brain injury in US soldiers returning from Iraq. New Eng J Med 358:453–463
Horenstein C, Lowe MJ, Koenig KA, Phillips MD (2009) Comparison of unilateral and bilateral complex finger tapping-related activation in premotor and primary motor cortex. Human Brain Mapp 30:1397–1412
Hudak A, Warner M, Marquez de la Plata C, Moore C, Harper C, Diaz-Arrastia R (2011) Brain morphometry changes and depressive symptoms after traumatic brain injury. Psychiatry Res 191:160–165
Imamura F, Greer CA (2009) Dendritic branching of olfactory bulb mitral and tufted cells: regulation by TrkB. PLoS ONE 4:e6729
Itoh T, Satou T, Nishida S, Tsubaki M, Hashimoto S, Ito H (2009) Improvement of cerebral function by anti-amyloid precursor protein antibody infusion after brain injury in rats. Mol Cell Biochem 324:191–199
Itoh T, Satou T, Nishida S, Tsubaki M, Imano M, Hashimoto S, Ito H (2010) Edaravone protects against apoptotic neuronal cell death and improves cerebral function after traumatic brain injury. Neurochem Res 35:348–355
Janisse HC, Nedd D, Escamille S, Nies MA (2004) Physical activity, social support, and family structure as determinants of mood among European-American and African-American women. Women Health 39:101–116
Jeukendrup AE, Vet-Joop K, Sturk A et al (2000) Relationship between gastro-intestinal complaints and endotoxemia, cytokine release and the acute-phase reaction during and after a long-distance triathalon in highly trained men. Clin Sci 98:47–55
Johansson E, Rönnkvist M, Fugl-Meyer AR (1991) Traumatic brain injury in northern Sweden. Incidence and prevalence of long-standing impairments and disabilities. Scand J Rehabil Med 23:179–185
Johnson B, Zhang K, Gay M, Horovitz S, Hallett M, Sebastianelli W, Slobounov S (2011) Alteration of brain default network in subacute phase of injury in concussed individuals: resting-state fMRI study. Neuroimage. doi:10.1016/j.neuroimage.2011.07.081
Johnston JA (2006) The hypothalamic-pituitary-adrenal axis in critical illness. AACN Clin Issues 17:39–49
Jorge R, Robinson R, Moser D et al (2004) Major depression following traumatic brain injury. Arch Gen Psychiatry 61:42–50
Kamijo K, Takeda Y (2010) Regular physical activity improves executive function during task switching in young adults. Int J Psychophysiol 75:304–311
Kannangara TS, Lucero MJ, Gil-Mohapel J et al (2010) Running reduces stress and enhances cell genesis in aged mice. Neurobiol Aging 32:2279–2286
Kaplan GB, Vasterling JJ, Vedak PC (2010) Brain-derived neurotrophic factor in traumatic brain injury, post-traumatic stress disorder, and their comorbid conditions: role in pathogenesis and treatment. Behav Pharmacol 21:427–437
Karkoulias K, Habeos I, Charokopos N et al (2008) Hormonal responses to marathon running in non-elite athletes. Eur J Intern Med 19:598–601
Kelly DF, McArthur DL, Levin H, Swimmer S, Dusick JR, Cohan P, Wang C, Swerdloff R (2006) Neurobehavioral and quality of life changes associated with growth hormone insufficiency after complicated mild, moderate, or severe traumatic brain injury. J Neurotrauma 23:928–942
Kim YJ (2011) A systematic review of factors contributing to outcomes in patients with traumatic brain injury. J Clin Nurs 20:1518–1532
Kizaki T, Maegawa T, Sakurai T et al (2011) Voluntary exercise attenuates obesity-associated inflammation through ghrelin expressed in macrophages. Biochem Biophys Res Commun 413(3):454–459
Kleim J, Jones T, Schallert T (2003) Motor enrichment and the induction of plasticity before or after brain injury. Neurochem Res 28:1757–1769
Knaepen K, Goekint M, Heymann EM, Meeusen R (2010) Neuroplasticity—exercise-induced response of peripheral brain-derived neurotrophic factor: a systemic review of experimental studies in human subjects. Sports Med 40:765–801
Kobilo T, Liu QR, Gandhi K, Mughal M, Shaham Y, van Praag H (2011) Running is the neurogenic and neurotrophic stimulus in environmental enrichment. Learn Mem 18:605–609
Kramer AF, Hahn S, Cohen NJ et al (1999) Ageing, fitness and neurocognitive function. Nature 400:418–419
Kraus MF, Susmaras T, Caughlin BP, Walker CJ, Sweeney JA, Little DM et al (2007) White matter integrity and cognition in chronic traumatic brain injury: a diffusion tensor imaging study. Brain 130:2508–2519
Kreutzer J, Seel R, Gourley E (2001) The prevalence of symptom rates of depression after traumatic brain injury: a comprehensive examination. Brain Inj 15:563–576
Krogh J, Nordentoft M, Sterne JAC, Lawlor DA (2010) The effect of exercise in clinically-depressed adults: systematic review and meta-analysis of randomized controlled trials. J Clin Psychol. doi:10.4088/JCP.08r04913blu
Kruger K, Mooren FC (2007) T cell homing and exercise. Exerc Immunol Rev 13:37–54
Kruger K, Lechtermann A, Fobker M, Volker K, Mooren FC (2008) Exercise-induced redistribution of T lymphocytes is regulated by adrenergic mechanisms. Brain Behav Immun 22:324–338
Lai S, Studenski S, Richards L et al (2006) Therapeutic exercise and depressive symptoms after stroke. J Am Geriatr Soc 54:240–247
Lanctôt KL, Rapoport MJ, Chan F, Rajaram RD, Strauss J, Sicard T, McCullagh S, Feinstein A, Kiss A, Kennedy JL, Bassett AS, Herrmann N (2010) Genetic predictors of response to treatment with citalopram in depression secondary to traumatic brain injury. Brain Inj 24:959–969
Lawlor DA, Hopker SW (2001) The effectiveness of exercise as an intervention in the management of depression: systematic review and meta-regression analysis of randomized control trials. BMJ 322:1–8
Lee EJ, Lee MY, Chen HY, Hsu YS, Wu TS, Chen ST, Chang GL (2005) Melatonin attenuates gray and white matter damage in a mouse model of transient focal cerebral ischemia. J Pineal Res 38:42–52
Lewin B, Robertson IH, Cay EL, Irving JB, Campbell M (1992) Effects of self-help post-myocardial-infarction rehabilitation on psychological adjustment and use of health services. Lancet 339:1036–1040
Lin MR, Chiu WT, Chen YJ, Yu WY, Huang SJ, Tsai MD (2010) Longitudinal changes in the health-related quality of life during the first year after traumatic brain injury. Arch Phys Med Rehabil 91:474–480
Little DM, Kraus MF, Joseph J, Geary EK, Susmaras T, Zhou XJ, Pliskin N, Gorelick PB (2010) Thalamic integrity underlies executive dysfunction in traumatic brain injury. Neurology 74:558–564
Lombardi F (2008) Pharmacological treatment of neurobehaviouralsequelae of traumatic brain injury. Eur J Anaesthesiol Suppl 42:131–136
Lu B, Wang KH, Nose A (2009) Molecular mechanism underlying neural circuit formation. Curr Opin Neurobiol 19:162–167
Luft AR, McCombe-Waller S, Whitall J et al (2004) Repetitive bilateral arm training and motor cortex activation in chronic stroke: a randomized controlled trial. JAMA 292:1853–1861
Lustig C, Shah P, Seidler R, Reuter-Lorenz PA (2009) Aging, training, and the brain: a review and future directions. Neuropsychol Rev 19:504–522
Maisonpierre PC, Le Beau MM, Espinosa R 3rd, Ip NY, Belluscio L, de la Monte SM, Squinto S, Furth ME, Yancopoulos GD (1991) Human and rat brain-derived neurotrophic factor and neurotrophin-3: gene structures, distributions, and chromosomal localizations. Genomics 10:558–568
Marklund N, Morales D, Clausen F, Hånell A, Kiwanuka O, Pitkänen A, Gimbel DA, Philipson O, Lannfelt L, Hillered L, Strittmatter SM, McIntosh TK (2009) Functional outcome is impaired following traumatic brain injury in aging Nogo-A/B-deficient mice. Neuroscience 163:540–551
Marks BL, Katz L, Smith JK et al (2009) Exercise and the aging mind: buffing the baby boomer’s body and brain. Phys Sports Med 37:119–125
Marks BL, Katz L, Styner M, Smith JK (2010) Aerobic fitness and obesity: relationship to cerebral white matter integrity in the brain of active and sedentary older adults.Br J Sports Med 45:1208–1215
Martin EM, Lu WC, Helmick K, French L, Warden DL (2008) Traumatic brain injuries sustained in the Afghanistan and Iraq wars. Am J Nurs 108:40–47
Martin-Cordero L, Garcia JJ, Hinchado MD, Ortega E (2011) The interleukin-6 and noradrenaline mediated inflammation-stress feedback mechanism in metabolic syndrome: effect of exercise. Cardiovasc Diabetol 10. doi:10.1186/1475-2840-10-42
Mathur N, Pedersen BK (2011) Exercise as a means to control low-grade systemic inflammation. Mediators Inflamm. doi:10.1155/2008/109502
Max JE, Keatley E, Wilde EA, Bigler ED, Levin HS, Schachar RJ, Saunders A, Ewing-Cobbs L, Chapman SB, Dennis M, Yang TT (2011) Anxiety disorders in children and adolescents in the first six months after traumatic brain injury. J Neuropsychiatry Clin Neurosci 23:29–39
McAllister TW, Flashman LA, McDonald BC, Saykin AJ (2006) Mechanisms of working memory dysfunction after mild and moderate TBI: evidence from functional MRI and neurogenetics. J Neurotrauma 23:1450–1467
McArdle WD, Katch FI, Katch VI (1978) Appendix C. In: Lupash E (ed) Essentials of exercise physiology. Lippincott Williams and Wilkins, Baltimore, p 723
McDonnell MN, Smith AE, Mackintosh SF (2011a) Aerobic exercise to improve cognitive function in adults with neurologic disorders: a systematic review. Arch Phys Med Rehabil 92:1044–1052
McDonnell MN, Bryan J, Smith AE, Esterman AJ (2011b) Assessing cognitive impairment following stroke. J Clin Exp Neuropsychol 3:1–9
McMillan T, Robertson ICH, Brock D, Chorlton L (2002) Brief mindfulness training for attentional problems after traumatic brain injury: a randomised control treatment trial. Neuropsychol Rehabil 12:117–125
McMorris T, Sproule J, Turner A, Hale BJ (2011) Acute, intermediate intensity exercise, and speed and accuracy in working memory tasks: a meta-analytical comparison of effects. Physiol Behav 102:421–428
Mead GE, Morley W, Campbell P (2008) Exercise for depression. Cochrane Database Syst Rev 4:CD004366
Medina JA, Netto TL, Muszkat M, Medina AC, Botter D, Orbetelli R, Scaramuzza LF, Sinnes EG, Vilela M, Miranda MC (2010) Exercise impact on sustained attention of ADHD children, methylphenidate effects. Atten Defic Hyperact Disord 2:49–58
Menna E, Disanza A, Cagnoli C et al (2009) Eps8 regulates axonal filopodia in hippocampal neurons in response to brain-derived neurotrophic factor (BDNF). PLoS Biol 7:e1000138
Molloy C, Conroy RM, Cotter DR, Cannon M (2011) Is traumatic brain injury a risk factor for schizophrenia? A meta-analysis of case-controlled population-based studies.Schizophr Bull 37:1104–1110
Moppett IK (2007) Traumatic brain injury: assessment, resuscitation and early management. Br J Anaesth 99:18–31
Morris M, Schoo A (2004) Optimizing exercise and physical activity in older adults. Butterworth Heinemann, Edinburgh
Mota BC, Pereira L, Souza MA, Silva LF, Magni DV, Ferreira AP, Oliveira MS, Furian AF, Mazzardo-Martins L, Silva MD, Santos AR, Ferreira J, Fighera MR, Royes LF (2011) Exercise pre-conditioning reduces brain inflammation and protects against toxicity induced by traumatic brain injury: behavioral and neurochemical approach. Neurotox Res. doi:10.1007/s12640-011-9257-8
Mougios V (2010) Exercise metabolism. In: Bahrke MS (ed) Exercise biochemistry. Human Kinetics, Champaign, p 122
Nation DA, Hong S, Jak AJ, Delano-Wood L, Mills PJ, Bondi MW, Dimsdale JE (2011) Stress, exercise, and Alzheimer’s disease: a neurovascular pathway. Med Hypoth 76:847–854
Negrao A, Deuster P, Gold P et al (2000) Individual reactivity and physiology of the stress response. Biomed Pharmacother 54:122–128
Nieman DC, Henson DA, Smith LL et al (2001) Cytokine changes after a marathon race. J Appl Physiol 91:109–114
Nieman DC, Dumke CL, Henson DA et al (2005) Muscle damage is linked to cytokine changes following a 160-km race. Brain Behav Immun 19:398–403
Niizuma K, Endo H, Chan PH (2009) Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J Neurochem 109(Suppl 1):133–138
Niogi SN, Mukherjee P, Ghajar J et al (2008) Extent of microstructural white matter injury in postconcussive syndrome correlates with impaired cognitive reaction time: a 3T diffusion tensor imaging study of mild traumatic brain injury. AJNR Am J Neuroradiol 29:967–973
Ogoh S, Brothers RM, Jeschke M, Secher NH, Raven PB (2010) Estimation of cerebral vascular tone during exercise: evaluation by critical closing pressure in humans. Exp Physiol 95:678–685
Ogoh S, Fisher JP, Young CN, Fadel PJ (2011) Impact of age on critical closing pressure of the cerebral circulation during dynamic exercise in humans. Exp Physiol 96:417–425
Onyszchuk G, He YY, Berman NE, Brooks WM (2008) Detrimental effects of aging on outcome from traumatic brain injury: a behavioral, magnetic resonance imaging, and histological study in mice. J Neurotrauma 25:153–171
Ormsbee MJ, Choi MD, Medlin JK, Geyer GH, Trantham LH, Dubis GS, Hickner RC (2009) Regulation of fat metabolism during resistance exercise in sedentary lean and obese men. J Appl Physiol 106:1529–1537
Ortega E (2003) Neuroendocrine mediators in the modulation of phagocytosis by exercise: physiological implications. Exerc Immunol Rev 9:70–94
Park DC, Reuter-Lorentz P (2009) The adaptive brain: aging and neurocognitive scaffolding. Annu Rev Psychol 60:173–196
Pedersen BK (2006) The anti-inflammatory effect of exercise: its role in diabetes and cardiovascular disease. Essays Biochem 42:105–117
Peltz CB, Gratton G, Fabiani M (2011) Age-related changes in electrophysiological and neuropsychological indices of working memory, attention control, and cognitive flexibility. Front Psychol 2:190
Petersen AMW, Pedersen BK (2005) The anti-inflammatory effect of exercise. J Appl Physiol 98:1154–1162
Petrik D, Lagace DC, Eisch AJ (2012) The neurogenesis hypothesis of affective and anxiety disorders: are we mistaking the scaffolding for the building? Neuropharmacology 62:21–34
Pontifex M, Hillman C, Fernhall B, Thompson K, Valentini T (2009) The effect of acute aerobic and resistance exercise on working memory. Med Sci Sports Exerc 41:927–934
Powers SK, Talbert EE, Adhihetty PJ (2011) Reactive oxygen and nitrogen species as intracellular signals in skeletal muscle. J Physiol 589(Pt 9):2129–2138
Powner DJ, Boccalandro C, Alp MS, Vollmer DG (2006) Endocrine failure after traumatic brain injury in adults. Neurocrit Care 5:61–70
Qiu C, Xu W, Fratiglioni L (2010) Vascular and psychosocial factors in Alzheimer’s disease: epidemiological evidence toward intervention. J Alzheimers Dis 20:689–697
Raghupathi R (2004) Cell death mechanisms following traumatic brain injury. Brain Pathol 14:215–222
Rapoport MJ, Chan F, Lanctot K, Herrmann N, McCullagh S, Feinstein A (2008) An open-label study of citalopram for major depression following traumatic brain injury. J Psychopharmacol 22:860–864
Ratey JJ, Loehr JE (2011) The positive impact of physical activity on cognition during adulthood: a review of underlying mechanisms, evidence and recommendations. Rev Neurosci 22:171–185
Rethorst CD, Wipfli BM, Landers DM (2009) The antidepressive effects of exercise. A meta-analysis of randomized trials. Sports Med 39:491–511
Rice AC, Khaldi A, Harvey HB, Salman NJ, White F, Fillmore H, Bullock MR (2003) Proliferation and neuronal differentiation of mitotically active cells following traumatic brain injury. ExpNeurol 183:406–417
Richards L, Stweart K, Woodbury M, Senesac C, Cauraugh J (2008) Movement-dependent stroke recovery: a systematic review and meta-analysis of TMS and fMRI evidence. Neuropsychologia 46:3
Rimel RW, Jane JA, Tyson GW (1981) Emergency management of head injuries. Resuscitation 9:75–97
Rimel RW, Giordani B, Barth JT, Jane JA (1982) Moderate head injury: completing the clinical spectrum of brain trauma. Neurosurgery 11:344–351
Robertson CL (2004) Mitochondrial dysfunction contributes to cell death following traumatic brain injury in adult and immature animals. J Bioenerg Biomembr 36:363–368
Sallert M, Rantamäki T, Vesikansa A et al (2009) Brain-derived neurotrophic factor controls activity-dependent maturation of CA1 synapses by downregulating tonic activation of presynaptic kainate receptors. J Neurosci 29:11294–11303
Samorajski T, Delaney C, Durham L (1985) Effect of exercise on longevity, body weight, locomotor performance, and passive avoidance of C57Bl/6 mice. Neurobiol Aging 6:17–24
Satomura E, Baba H, Nakano Y, Maeshima H, Suzuki T, Arai H (2011) Correlations between brain-derived neurotrophic factor and clinical symptoms in medicated patients with major depression. J Affect Dis. doi:10.1016/j.jad.2011.06.041
Scheuer J, Tipton CM (1977) Cardiovascular adaptations to physical training. Annu Rev Physiol 39:221–251
Schneider-Garces NJ, Gordon BA, Brumback-Peltz CR, Shin E, Lee Y, Sutton BP, Maclin EL, Gratton G, Fabiani M (2010) Span, CRUNCH, and beyond: working memory capacity and the aging brain. J Cogn Neurosci 22:655–669
Schönberger M, Ponsford J, Reutens D, Beare R, Clarke D, O’Sullivan R (2011) The relationship between mood disorders and MRI findings following traumatic brain injury. Brain Inj 25:543–550
Schwandt M, Harris JE, Thomas S, Keightley M, Snaiderman A, Colantonio A (2011) Feasibility and effect of aerobic exercise for lowering depressive symptoms among individuals with traumatic brain injury: a pilot study. J Head Trauma Rehabil. doi:10.1097/HTR.0b013e31820e6858
Seo HG, Kim DY, Park HW, Lee SU, Park SH (2010) Early motor balance and coordination training increased synaptophysin in subcortical regions of the ischemic rat brain. J Korean Med Sci 25:1638–1645
Serra-Grabulosa JM, Junqué C, Verger K, Salgado-Pineda P, Mañeru C, Mercader JM (2005) Cerebral correlates of declarative memory dysfunctions in early traumatic brain injury. J Neurol Neurosurg Psychiatry 76:129–131
Sigurdardottir S, Jerstad T, Andelic N, Roe C, Schanke AK (2010) Olfactory dysfunction, gambling task performance and intracranial lesions after traumatic brain injury. Neuropsychology 24:504–513
Slobounov SM, Zhang K, Pennell D, Ray W, Johnson B, Sebastianelli W (2010) Functional abnormalities in normally appearing athletes following mild traumatic brain injury: a functional MRI study. Exp Brain Res 202:341–354
Smetanka RD, Lambert GB, Murray R (1999) Intestinal permeability in runners in the 1996 Chicago marathon. Int J Sport Nutr 9:426–433
Smith PJ, Blumenthal JA, Babyak MA, Georgiades A, Hinderliter A, Sherwood A (2007) Effects of exercise and weight loss on depressive symptoms among men and women with hypertension. J Psychosom Res 63:463–469
Stewart K, Cauraugh J, Summers J (2006) Bilateral movement training and stroke rehabilitation: a systematic review and meta-analysis. J Neurolog Sci 244:89
Ströhle A (2009) Physical activity, exercise, depression, and anxiety disorders. J Neural Transm 116:777–784
Stummer W, Weber K, Tranmer B (1994) Reduced mortality and brain damage after locomotor activity in gerbil forebrain ischemia. Stroke 25:1862–1869
Takada M, Suzuki A, Shima S, Inoue K, Kazukawa S, Hojoh M (2009) Associations between lifestyle factors, working environment, depressive symptoms and suicidal ideation: a large-scale study in Japan. Ind Health 47:649–655
Tanriverdi F, Senyurek H, Unluhizarci K et al (2006) High risk of hypopituitarism after traumatic brain injury: a prospective investigation of anterior pituitary function in the acute phase and 12 months after trauma. J Clin Endocrinol Metab 91:2105–2111
Tanriverdi F, Ulutabanca H, Unluhizarci K, Selcuklu A, Casanueva FF, Kelestimur F (2007) Pituitary functions in the acute phase of traumatic brain injury: are they related to severity of the injury or mortality? Brain Inj 21:433–439
Tanriverdi F, De Bellis A, Bizzarro A, Sinisi AA, Bellastella G, Pane E, Bellastella A, Unluhizarci K, Selcuklu A, Casanueva FF, Kelestimur F (2008a) Antipituitary antibodies after traumatic brain injury: is head trauma-induced pituitary dysfunction associated with autoimmunity? Eur J Endocrinol 159:7–13
Tanriverdi F, Ulutabanca H, Unluhizarci K, Selcuklu A, Casanueva FF, Kelestimur F (2008b) Three years prospective investigation of anterior pituitary function after traumatic brain injury: a pilot study. Clin Endocrinol 68:573–579
Tanriverdi F, Unluhizarci K, Karaca Z, Casanueva FF, Kelestimur F (2010a) Hypopituitarism due to sports related head trauma and the effects of growth hormone replacement in retired amateur boxers. Pituitary 13:111–114
Tanriverdi F, Unluhizarci K, Kelestrimur F (2010b) Persistent neuroinflammation may be involved in the pathogenesis of traumatic brain injury (TBI)-induced hypopituitarism: potential genetic and autoimmune factors. J Neurotrauma 27:301–302
TanriverdiF Unluhizarci K, Kelestrimur F (2010) Pituitary function in patients with mild traumatic brain injury: a review of literature and proposal of a screening strategy. Pituitary 13:146–153
Taylor RS, Brown A, Ebrahim S et al (2004) Exercise-based rehabilitation for patients with coronary heart disease: systematic review and meta-analysis of randomized controlled trials. Am J Med 116:682–692
Taylor AN, Rahman SU, Sanders NC (2008) Injury severity differentially affects short- and long-term neuroendocrine outcomes of traumatic brain injury. J Neurotrauma 25:311–323
Taylor AN, Rahman SU, Tio DL (2010) Injury severity differentially alters sensitivity to dexamethasone after traumatic brain injury. J Neurotrauma 27:1081–1089
Thaut MH, Leins AK, Rice RR et al (2007) Rhythmic auditory stimulation improves gait more than ndt/bobath training in near-ambulatory patients early poststroke: a single-blind, randomized trial. Neurorehabil Neural Repair 21:455–459
Tomkins O, Feintuch A, Benifla M, Cohen A, Friedman A, Shelef I (2011) Blood-brain barrier breakdown following traumatic brain injury: a possible role in posttraumatic epilepsy. Cardiovasc Psychiatry Neurol. doi:10.1155/2011/765923
Tsang HW, Chan EP, Cheung WM (2008) Effects of mindful and non-mindful exercises on people with depression: a systematic review. Br J Clin Psychol 47:303–322
Tseng CN, Gau BS, Lou MF (2011) The effectiveness of exercise on improving cognitive function in older people: a systematic review. J Nurs Res 19:119–131
Van Praag H, Christie BR, Sejnowski TJ, Gage FH (1999) Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad Sci USA 96:13427–13431
Viňa J, Gomez-Cabrera MC, Borras C, Froio T, Sanchis-Gomar F, Martinez-Bello VE, Pallardo FV (2009) Mitochondrial biogenesis in exercise and in aging. Adv Drug Deliv Rev 61:1369–1374
Voss MW, Prakash RS, Erickson KI et al (2010) Plasticity of brain networks in a randomized intervention trial of exercise training in older adults. Front Aging Neurosci 2:32. doi:10.3389/fnagi.2010.00032
Voss MW, Nagamatsu LS, Liu-Ambrose T, Kramer AF (2011) Exercise, brain, and cognition across the lifespan. J Appl Physiol 111:1505–1513
Voss MW, Chaddock L, Kim JS, Vanpatter M, Pontifex MB, Raine LB, Cohen NJ, Hillman CH, Kramer AF (2012) Aerobic fitness is associated with greater efficiency of the network underlying cognitive control in preadolescent children. Neuroscience. doi:10.1016/j.neuroscience.2011.10.009
Walsh NP, Gleeson M, Shepard RJ et al (2011) Position statement. Part one: immune function and exercise. Exerc Immunol Rev 17:6–63
Wang C, Bannuru R, Ramel J, Kupelnick B, Scott T, Schmid CH (2010) Tai chi and psychological well-being: systematic review and meta-analysis. BMC Compl Altern Med 10:23–39
Warden D (2006) Military TBI during the Iraq and Afghanistan wars. J Head Trauma Rehabil 21:398–402
Weltman A, Wideman L, Weltman JY, Veldhuis JD (2003) Neuroendocrine control of GH release during acute aerobic exercise. J Endocrinol Invest 26:843–850
Wilde EA, Newsome MR, Bigler ED, Pertab J, Merkley TL, Hanten G, Scheibel RS, Li X, Chu Z, Yallampalli R, Hunter JV, Levin HS (2011) Brain imaging correlates of verbal working memory in children following traumatic brain injury. Int J Psychophysiol 82:86–96
Wilson KR, Donders J, Nguyen L (2011) Self and parent ratings of executive functioning after adolescent traumatic brain injury. Rehabil Psychol 56:100–106
Wu A, Ying Z, Gomez-Pinilla F (2011) The salutary effects of DHA dietary supplementation on cognition, neuroplasticity, and membrane homeostasis after brain trauma. J Neurotrauma 28:2113–2122
Xiong Y, Gu Q, Peterson PL, Muizelaar JP, Lee CP (1997) Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. J Neurotrauma 14:23–34
Xuan AG, Long DH, Gu HG, Yang DD, Hong LP, Leng SL (2008) BDNF improves the effects of neural stem cells on the rat model of Alzheimer’s disease with unilateral lesion of fimbria-fornix. Neurosci Lett 440:331–335
Yacobian TA, Lo DC (2000) Truncated and full-length TrkB receptors regulate distinct modes of dendritic growth. Nat Neurosci 3:342–349
Zhang R, Zuckerman JH, Iwasaki K, Wilson TE, Crandall CG, Levine BD (2002) Autonomic neural control of dynamic cerebral autoregulation in humans. Circulation 106:1814–1820
Zhang F, Wu Y, Jia J (2011) Exercise preconditioning and brain ischemic tolerance. Neuroscience 177:170–176
Zhou XP, Wu KY, Liang B, Fu XQ, Luo ZG (2008) TrkB-mediated activation of geranylgeranyltransferase I promotes dendritic morphogenesis. Proc Natl Acad Sci USA 28:17181–17186
Zohar O, Rubovitch V, Milman A et al (2011) Behavioral consequences of minimal traumatic brain injury in mice. Acta Neurobiol Exp 71:36–45
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Archer, T. Influence of Physical Exercise on Traumatic Brain Injury Deficits: Scaffolding Effect. Neurotox Res 21, 418–434 (2012). https://doi.org/10.1007/s12640-011-9297-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-011-9297-0