
Abstract
Trypanosoma vivax (sub-genus Duttonella) is largely responsible for non profitable livestock production in sub-Sahara Africa. In Nigeria, no study has addressed the molecular characteristic of T. vivax except Y486. Hence, we characterized and assessed the genetic diversity among T. vivax detected in naturally infected cattle in Nigeria using internal transcribed spacer 1 (ITS1) of ribosoma DNA (rDNA) and diagnostic antigen gene (DAG) sequences. The length of ITS1 and DAG sequences range from 215–220 to 257–338 bp, respectively and the mean G–C contents were 60 and 61.5 %. Homology search revealed 93–99 and 95–100 % homologies to T. vivax DAG and ITS1 sequences from GenBank. Aligned sequences revealed both ITS1 rDNA and DAG to be less polymorphic but DAG sequences of the Y486 strain and its clone showed marked variation from autochthonous strains. Phylogenetic analysis yielded tree that grouped T. vivax ITS1rDNA gene and DAG sequences into two main clades each. Considering the ITI1 rDNA sequences, clade A contained autochthonous T. vivax within which the South American sequences clustered, clade B contained the sequences of T. vivax from East Africa. Analysis of DAG revealed that the clade A contains autochthonous T. vivax sequences but clade B contained the Y486 and its clones. In conclusion, the diagnostic antigen gene sequences of the T. vivax detected in this study may have undergone considerable gene recombination through time and suggests that more than one strain of T. vivax exist among cattle population in Nigeria.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Trypanosomes are haemoprotozoan parasites that cause different disease syndromes in animals and humans. These parasites cause trypanosomosis (nagana) in domestic animals especially, ruminants and it has been a major impediment to profitable livestock production in sub-Sahara African (Ng’ayo et al. 2005). Among the three species of trypanosomes (T. brucei, T. congolense and T. vivax) that are pathogenic in cattle in Nigeria, T. vivax is the most pathogenic (Takeet et al. 2013), though other strains with varying pathogenicities have been reported in East Africa, Central and South America. In Nigeria, transmission of T. vivax is both cyclical by tsetse flies and mechanical by tabanids, whereas in Central and South America, transmission is only mechanical by tabanids and other biting flies (Jones and Davila 2001; Chamond et al. 2010).
It is widely accepted that pathogenicity of T. vivax is strain dependent and vary from region to region (Osorio et al. 2008; Adams et al. 2010; Nakayima et al. 2013). For instance strains of this species detected in naturally infected cattle in Nigeria shows severe anemia that is significantly different from the anemia elicited by T. brucei and T. cogolense (Takeet et al. 2013). In East Africa, another strain has been reported to cause severe haemorhagic syndrome in infected cattle (Gardiner and Mahmoud 1992) but in South America, the infection by T. vivax has been reported to range from chronic to asymptomatic with rare outbreak (Osorio et al. 2008).
The genetic diversity of T. vivax has been studied to some extent in Tanzania (Adams et al. 2010), Zambia and Ghana (Nakayima et al. 2013) and South America (Cortez et al. 2006; Garcia et al. 2014) and compared with T. vivax Y486 isolated from Nigeria (Leeflang et al. 1976) using Flourescent Fragment Length Barcoding (FFLB) of RNA locus, Cathepsin L-like gene sequences analysis and Microsatellite Multiloci Genotyping (MLGs). Up till date, the sequence of T. vivax isolate that has been reported from Nigeria is Y486 strain that was isolated from infected cattle in 1976 in Zaria, Nigeria. Shortly after this period, Dirie et al. (1986) reported morphological changes of the blood stream form of Y486 strain in mice. Aside the fact that this may be pointing to great genetic endowment on this strain that others strains lack, it is apparent that studies involving only T. vivax Y486 and its clone (Tvi ILDat1.2) is not a balanced representation of other strains of the same species and their genetic diversity study in Nigeria. Thus, in this study, we assessed the genetic diversity within the T. vivax detected in naturally infected cattle in Nigeria by sequencing and analysis of the sequences of diagnostic antigen gene (DAG) and internal transcribed spacer 1 (ITS-1) of ribosomal DNA (rDNA) and compared it with those from East Africa and the Americas.
Materials and methods
The study area
The animals sampled were predominantly from two locations (Ogun and Kaduna States) in Nigeria (Fig. 1). Ogun state, a transitional zone between the tropical rain forest and derived savannah zone in the south-west of Nigeria lies between latitude 7°10′N and 3°21′E and it has two pronounced seasons, the dry season (November to March) and the wet season (April–October) while Kaduna state, is located within the Northern Guinea Savannah zone of Nigeria and it lies between 11°10′N and 7°38′E.

Map of Nigeria, showing towns and states where samples were collected. Legends describe the towns where samples were collected
Study population and sample collection
Four hundred and eleven samples were collected between September and December 2010 from two locations of about 1000 km apart with different vegetations that could significantly affect the breeding of the obligate vector, tsetse flies. The cattle kept under traditional management system of free grazing (nomadic) and various abattoirs and slaughter slabs were randomly selected for sampling. Blood samples were collected from the jugular vein of each cattle into 5 ml tubes containing 5 mg of ethylenediaminetetraacetic acid (EDTA) as anticoagulant and samples were transported in mobile refrigerator to the laboratory within 3 h of collection. The blood samples were stored at 4 °C prior to DNA extraction. The protocol for this research was approved by the Institutional Animal Use and Care Committee of the Federal University of Agriculture, Abeokuta, Ogun State, Nigeria.
DNA extraction and Trypanosoma vivax detection
DNA was extracted from the blood in EDTA bottle using Quick-gDNA™ MiniPrep (Zymo Research Corporation, Irvine, CA 92614, USA) as described by the manufacturer. Quantification of DNA yield and assessment of quality were done using Nanodrop ND-1000 UV/Vis spectrophotometer (Nanodrop Technologies, Inc., DE, USA). The eluted DNA was stored at −20° until use.
T. vivax species specific primer set (ILO1264: 5′-CAGCTCGCCGAAGGCCACTTGGCTGGG-3′ and ILO1265: 5′-TCGCTACCACAGTCGCAATCGTCGTCTCAAGG-3′) for a 400 bp diagnostic antigen gene fragment (Masake et al. 1997) and generic primers set (ITS CF: 5′-CCGGAAGTTCACCGATATTG-3′ and ITS BR: 5′-TTGCTGCGTTCTTCAACGAA-3′) for 250 bp internal transcribed spacer 1 fragment (Njiru et al. 2005) were used. The PCR amplifications were carried out in 20 µl final volume containing equivalent of 20 ng of genomic DNA, 10 mM Tris–HCl, pH 8.3, 1.5 mM MgCl2, 50 µM KCl, 200 µM each of dNTPs, 40 ng of each of the primers and 1 unit of Taq DNA polymerase (Bioneer, Inc. Alameda, CA USA). The reactions were placed in a C-1000 series thermocycler (Biorad, Hercules, CA, USA). The reaction conditions were as follows for: initial denaturation at 94 °C for 4 min followed by 35 cycles of 94 °C for 30 s, 58/60 °C (ITS1/DAG) for 45 s and 72 °C for 30 s followed by final extension at 72 °C for 5 min. Ten microliters of the PCR products were electrophoresed through 1 % agarose gel in 1× TBE (89 mM Tris, 89 mM boric acid 1 mM EDTA) at 90 V for 80 min along with 10 µl of GENE Mate Quanti-Marker 100 bp DNA ladder (BioExpress, Kaysville, UT, USA). After electrophoresis, PCR products were visualized on a UV transilluminator and were photographed using an AlphaImager HP System (Protein Simple, Santa Clara, CA, USA). Positive and negative (no DNA) samples were used as controls in each run.
Sequencing and sequences analysis
To confirm and validate our results, 48 PCR products of T. vivax positive samples were selected and sequenced directly using Big Dye Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) with the forward amplification PCR primers and AmpliTaq-FS DNA Polymerase. The sequences obtained were viewed and compared on Finch TV and Sequence Scanner (Applied Bioscience), manually cleaned, before they were aligned with each other and published gene sequences of various Trypanosoma species using the Molecular Evolutionary Genetic Analysis (MEGA 5.05) software. Phylogenetic trees were constructed using the un-weighted pair group method using average linkage (UPGMA) algorithm of the phylogeny program of MEGA 5.05 (Tamura et al. 2011) which included sequences of South America and East Africa species obtained from GenBank. The phylogenetic robustness was assessed by undertaking bootstrap resampling using 1000 replicate.
Results
Trypanosoma vivax DNA detection
The electrophoresed PCR product revealed band sizes of 250 and 400 bp corresponding to expected band size of T. vivax ITS1 and diagnostic antigen gene, respectively (Fig. 2a, b). Among the 411 animals sampled, 106 (26 %) were positive for T. vivax infection as reported by Takeet et al. (2013) and ITS1 primers detected 74 (18.01 %). The ILO 1264 and 1265 primers set did not amplify the DNA from known positive sample of T. brucei and T. congolense.

Gel electrophoresis pictures. a Gel electrophoresis picture showing Trypanosoma species DNA amplified by ITS1-primers. M 100 bp molecular marker, 1 negative control, 2 and 3 T. brucei, 4 T. congolense, 5 T. vivax, 6 positive control containing DNA from blood sample that tested positive for T. brucei, T. congolense and T. vivax; b gel electrophoresis showing T. vivax DNA amplified by ILO-primers. M 100 bp molecular marker, 1 negative control, 2–5 T. vivax, 6 positive control
Sequences analysis
Out of the 48 PCR products sent for sequencing, 22 and 28 of diagnostic antigen gene and ITS1, respectively were of good quality and were included in our analysis. The obtained diagnostic antigen gene sequences lengths range from 257 to 338 bp with mean G–C content of 61.5 % while the ITS1 sequences range from 215 to 220 bp with mean G/C content of 61 %. BLAST search for homology of the diagnostic antigen sequences from this study revealed homologies ranging from 93–99 % to T. vivax Y486 sequences and its clones (Accession nos: HE573027, U43183 and L25129) whereas, the ITS1 of the rDNA sequences range from 95–100 % to T. vivax Y486 strain and TviBfL 445 clone 1 (Accession nos: HE573019 and KC196671). The aligned sequences of DAG revealed minimal points of deletions and insertion without any point of alteration, but when aligned with Y486 strain and its clones, complete points of deletions were observed at points 201–217 (Fig. 3b). The evolutionary distance within the sequences from this study ranges from 0.01 to 0.09 but 0.01–1.09 between the sequences from this study and those available in GenBank. The aligned sequences of ITS1, including those obtained from the GenBank revealed that those sequences from this study are not different from those from the South America but different from those from the East Africa. The difference was seen as complete deletions at points 154–166 (Fig. 3a).

Alignment of autochthonous T. vivax with those obtained from GenBank. a Aligned ITS1 rDNA sequences with those obtained from GenBank including AB569250, DQ316051, DQ316041, HE573019, JN673392, JN673393 and U22316; b aligned DAG sequences with those obtained from GenBank including HE73027, L25129 and U43183
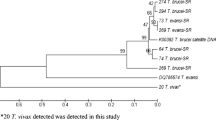
Phylogenetic trees generated from the alignment of the autochthonous T. vivax sequences using UPGMA yielded a tree that grouped T. vivax ITS1 rDNA gene and DAG sequences into two main clades each, designated A and B (Figs. 4, 5). Considering the ITS1 rDNA sequences, clade A contained autochthonous T. vivax within which the South American sequences (DQ316041) and Y486 (HE573019) clustered tightly, clade B contained the sequences of T. vivax from Kenya (DQ316041) and Tanzania (JN673392 and JN673393). The out group, T. theileri (AB569250) was clearly separated from the rest of the sequences. Analysis of DAG revealed that the clade A contains autochthonous T. vivax sequences alone but clade B contained the Y486 and its clones. But within the autochthonous T. vivax sequences, both trees showed sample number 17 to present the most distant phylogenetic relationship with other member of the groups with bootstraps of 99 and 100 % for ITS1 and DAG, respectively.

Evolutionary relationships of strains of T. vivax detected in Nigerian cattle and compared to other sequences from the GenBank, using ITS-1 rDNA sequences analyzed by the UPGMA method. The percentage of replicate trees above 50 % in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree

Evolutionary relationships of strains of T. vivax detected in Nigerian cattle compared to other sequences from the GenBank, using Diagnostic antigen gene sequences analyzed by the UPGMA method. The percentage of replicate trees above 50 % in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree
Discussion
This is the first study that attempted to shed light on the molecular characteristic and genetic diversity among T. vivax strains from naturally infected cattle in Nigeria. Diagnostic antigen gene (DAG) and ITS1 of rDNA were amplified and the bands sizes of about 400 and 250 bp, respectively obtained were supported by the work of other researchers (Masake et al. 1997; Garcia et al. 2005; Adams et al. 2010; Sanni et al. 2013). The ITS1 sequences lengths of the T. vivax detected in this study were not significantly different from those T. vivax obtained and sequenced from Nigeria and Kenya by Njiru et al. (2005). This may indicate the less polymorphic nature of T. vivax. The G–C content of the subgenus duttonella is generally believed to be high (Stevens and Rambaut, 2001; Cortez et al. 2006), which is consistent with the 61.5 and 61 % G–C contents of the DAG and ITS1 partial sequences reported in this study.
The alignment of the T. vivax sequences detected in naturally infected cattle in Nigeria and those from South America (Venezuela) revealed that they are less polymorphic in the two gene regions amplified. It is important to note that when DAG sequences of T. vivax Y485 strain and its clones (ILdat 1.2) were aligned with the sequences of autochthonous T. vivax, the differences noticed in the alignment indicate that the T. vivax in Nigeria has undergone moderate genetic modification or recombination over time. It is generally believed that persistent genetic recombination is responsible for the difficulty encountered in the control of animal trypanosomosis in Africa (Osorio et al. 2008; Gibson et al. 2015). Since it is difficult to ascertain the frequency of genetic recombination among trypanosomes, there is need for regular appraisal of genetic characteristic to understand the dynamics of genetic variation among the T. vivax strains in Nigeria. This will help in proper diagnosis and therapeutic planning against trypanosomosis.
The available reports on phylogenetic analysis of T. vivax from Africa and the Americas placed the Americas strains of T. vivax in the same clade with those strains from West Africa (Dirie et al. 1993a, b; Cortez et al. 2006; Rodrigues et al. 2008; Nakayima et al. 2013) and well separated from the clade formed by the East African strains (Fasogbon et al. 1990; Malele et al. 2003; Rodrigues et al. 2008; Cortez et al. 2006). These reports are consistent with the findings in this study. Rodrigues et al. (2008) posited that the T. vivax strain (Y486) that has been widely used to study West African isolate has been for long time maintained in mice, and as such could have led to selection of homogenous clone. Based on this observation, in addition to sequences divergent and separation of T. vivax Y486 into a separate clade on the phylogenetic tree inferred from DAG, one may suggest that more than one strain of T. vivax are in circulation among cattle population in Nigeria. Though we did not amplify the diagnostic antigen gene of T. vivax from the East Africa and the America to compare with present study, we believe that further study that analyze this gene and compare with West African isolate will increase the understanding of genetic diversity of T. vivax.
In conclusion, the diagnostic antigen gene sequences of the T. vivax detected in this study may have undergone considerable gene recombination through time and suggests that more than one strain of T. vivax exist among cattle population in Nigeria.
References
Adams ER, Hamilton PB, Rodrigues AC, Malele II, Delespaux V, Teixeira MMG, Gibson W (2010) New Trypanosoma (Duttonella) vivax genotypes from tsetse flies in East Africa. Parasitology 137:641–650
Chamond N, Cosson A, Blom-Potar MC, Jouvion G, D’Archivio S, Medina M, Droin-Bergère S, Huerre M, Goyard S, Minoprio P (2010) Trypanosoma vivax infections: pushing ahead with mouse models for the study of Nagana. Parasitological, hematological and pathological parameters. PLoS Neg Trop Dis 10:e792
Cortez AP, Ventura RM, Rodrigues AC, Batista JS, Paiva F, Añez N, Machado RZ, Gibson WC, Teixeira MMG (2006) The taxonomic and phylogenetic relationships of Trypanosoma vivax from South America and Africa. Parasitology 133:159–169
Dirie EF, Croft SL, Molyneux DH (1986) Morphological changes of Trypanosomavivax in mice. Vet Parasitol 19:23–27
Dirie MF, Otte MJ, Thatthi R, Gardner PR (1993a) Comparative studies of Trypanosoma (Duttonella) vivax isolates from Colombia. Parasitology 106:21–29
Dirie MF, Murphy NB, Gardiner PR (1993b) DNA finger printing of Trypanosoma vivax isolates rapidly identifies intraspecific relationships. J Eukaryot Microbiol 40:132–134
Fasogbon AI, Knowles G, Gardiner PR (1990) A comparison of the isoenzymes of Trypanosoma (Duttonella) vivax isolates from East and West Africa. Int J Parasitol 20:389–394
Garcia H, Garcia ME, Perez H, Mendoza-Leon A (2005) The detection and PCR-based characterization of the parasites causing trypanosomiasis in water-buffalo herds in Venezuela. Ann Trop Med Parasitol 99(4):1–12
Garcia HA, Rodrigues AC, Rodrigues CMF, Bengaly Z, Minervino AHH, Riet-Correa F, Machado RZ, Paiva F, Batista JS, Neves L, Hamilton PB, Teixeira MMG (2014) Microsatellite analysis supports clonal propagation and reduced divergence of Trypanosoma vivax from asymptomatic to fatally infected livestock in South America compared to West Africa. Parasites Vectors 7:210
Gardiner PR, Mahmoud MM (1992) Salivarian trypanosomes causing disease in livestock outside sub-saharan Africa. In: Kreier JP, Baker JR (eds) Parasitic protozoa. Academic Press, London, pp 277–313
Gibson W, Peacock L, Ferris V, Fischer K, Livingstone J, Thomas J, Bailey M (2015) Genetic recombination between human and animal parasites creates novel strains of human pathogen. PLoS Negl Trop Dis 9(3):e0003665. doi:10.1371/journal.pntd.0003665
Jones TW, Davila AM (2001) Trypanosoma vivax-out of Africa. Trends Parasitol 17:99–101
Leeflang P, Buys J, Blockamp C (1976) Study on Trypanosoma vivax: infectivity and serial maintenance of natural bovine isolates in mice. Int J Parasitol 6:413–417
Malele I, Craske L, Knight C, Ferris V, Njiru Z, Hamilton P, Lehane S, Lehane M, Gibson WC (2003) The use of specific and generic primers to identify trypanosome infections of wild tsetse flies in Tanzania by PCR. Inf Gen Evol 3:271–279
Masake RA, Majiwa PAO, Moloo SK, Makau JM, Njuguna JT, Maina M, Kabata J, ole-MoiYoi OK, Nantulya VM (1997) Sensitive and specific detection of Trypanosoma vivax using the polymerase chain reaction. Exp Parasitol 85:193–205
Nakayima J, Nakao R, Alhassan A, Hayashida K, Namangala B, Mahama C, Afakye K, Sugimoto C (2013) Genetic diversity among Trypanosoma (Duttonella) vivax strains from Zambia and Ghana, based on cathepsin L-like gene. Parasite 20:24
Ng’ayo MO, Njiru ZK, Kenya EU, Muluvi GM, Osir EO, Masiga DK (2005) Detection of trypanosomes in small ruminants and pigs in western Kenya: important reservoirs in the epidemiology of sleeping sickness? Kinetoplastid Biol Dis 4:5. doi:10.1186/1475-9292-4-5
Njiru ZK, Constantine CC, Guya S, Crowther J, Kiragu JM, Thompson RCA, Davila AMR (2005) The use of ITS1 rDNA PCR in detecting pathogenic African trypanosomes. Parasitol Res 95:186–192
Osorio A, Madruga CR, Desquesnes M, Soares CO, Ribeiro LRR, da Costa SCG (2008) Trypanosoma (Duttonella) vivax: its biology, epidemiology, pathogenesis, and introduction in the new world—a review. Mem Inst Oswaldo Cruz 103:1–13
Rodrigues AC, Neves L, Garcia HA, Viola LB, Marcili A, Maia Da Silva F, Sigauque I, Batista JS, Paiva F, Teixeira MMG (2008) Phylogenetic analysis of Trypanosoma vivax supports the separation of South American/West African from East African isolates and a new T. vivax-like genotype infecting a nyala antelope from Mozambique. Parasitology 135:1317–1328
Sanni T, Onasanya G, Adefenwa MA, Yakubu A, Ikeobi CON, Adebambo OA, Talabi AO, Ozoje O, Wheto M, Takeet MI, Peters SO, De Donato M, Thomas B, Imumorin IG (2013) Molecular diagnosis of subclinical African Trypanosoma vivax infection and association with physiological indices and serum metabolites in extensively managed goats in the tropics. Open J Vet Med 3:39–45
Stevens J, Rambaut A (2001) Evolutionary rate differences in trypanosomes. Inf Gen Evol 1:143–150
Takeet MI, Fagbemi BO, De Donato M, Yakubu A, Rodulfo HE, Peters SO, Wheto M, Imumorin IG (2013) Molecular survey of pathogenic trypanosomes in naturally infected Nigerian cattle. Res Vet Sci 94(3):555–561
Tamura K, Peterson D, Peterson N, Stecher G, Nei M (2011) Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739
Acknowledgments
Financial support provided by the Educational Trust Fund of the Federal Republic of Nigeria (Grant No. TETFUND-2010) and also thankful for the financial support by the College of Agriculture and Life Sciences, Cornell University, Ithaca, NY and Zoetis, Inc. Additional support by National Research Initiative Competitive Grant Program (Grant No. 2006-35205-16864) from the USDA National Institute of Food and Agriculture; USDA-NIFA Research Agreements (Nos. 2009-65205-05635, 2010-34444-20729). We also thank the entire staff of the Department of Parasitology and Entomology, Faculty of Veterinary Medicine, Ahmadu Bello University, Zaria, Nigeria for permission granted to use their laboratory facilities for part of this study, with special gratitude to Prof. I. A. Lawal and Dr. O.O. Okubanjo.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Takeet, M.I., Fagbemi, B.O., Peters, S.O. et al. Genetic diversity among Trypanosoma vivax strains detected in naturally infected cattle in Nigeria based on ITS1 of rDNA and diagnostic antigen gene sequences. J Parasit Dis 41, 433–441 (2017). https://doi.org/10.1007/s12639-016-0822-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12639-016-0822-1