
Abstract
Purpose
Surra is a zoonotic disease caused by Trypanosoma evansi (Trypanozoon), a salivary trypanosome native to Africa which affects a wide range of mammals worldwide and causes mortality and significant economic loss. The present study was devoted to the molecular characterization of T. evansi derived from naturally infected dromedary camels in Algeria.
Methods
A total of 148 blood samples were collected from mixed age camels living in one of four geographic regions (Ouargla, El Oued, Biskra and Ghardaia) of Algeria. Samples underwent PCR amplification and sequencing of the internal transcribed spacer 1 (ITS1) complete sequence.
Results
DNA of Trypanosoma spp. was found in 19 camels (12.84%). Trypanosoma spp. molecular positivity was not affected by sex (p = 0.50), age (p = 0.08), or geographic location (p = 0.12). Based on multiple sequence alignment of the obtained DNA sequences with representative T. evansi ITS1 sequences available globally, the Algerian sequences were grouped within four different haplotypes including two which were original.
Conclusion
Results of this study provide preliminary data on which future studies of genetic diversity and molecular epidemiology of T. evansi can be based.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Trypanosoma evansi is a single-celled eukaryotic parasite which is closely related to T. brucei, the causative agent of sleeping sickness and nagana disease in human and animals, respectively [1]. T. evansi is the etiological agent of “Surra”, or mal de cadeiras, characterized by anemia, immunosuppression and damage to the central nervous system causing ocular and reproductive disorders [2]. It affects a wide range of domestic and wild mammalian species [2], and it is one of the most pathogenic trypanosomes transmitted mechanically by different blood-sucking fly species such as Tabanus spp. and Stomoxys spp. [3] as well as by vampire bats Desmodus rotundus [4]. Due to the high mortality and morbidity rates affecting livestock productivity, this disease is responsible for disastrous economic losses estimated at $404,630 USD per year [5, 6]. Although some authors have suggested that T. evansi originated and mainly affected camels in Africa [7], all domestic mammals studied to date are susceptible [8]. The disease results in a high rate of mortality in horses, camels, and dogs [2, 8]. Outbreaks of animal surra have been reported in North Africa, the Middle East, South America, Asia, and recently Europe [9]. T. evansi infection in humans was not only reported initially in India [10] but also confirmed recently in Vietnam [11]. Other probable cases in humans have been reported worldwide, but molecular parasite detection was not performed [2].
Many diagnostic methods (with varying degrees of sensitivity and specificity) are available to detect T. evansi infections including parasitological, serological, and molecular assays [12, 13]. Some methods detect trypanosome infection by microscopical examination of fresh or stained bloodsmears [14], whereas others identify different T. evansi strains, being classified as Type A or Type B, according to their Kinetoplast DNA minicircle sequences [15] and the presence of the RoTat1.2 variant surface glycoprotein (VSG) gene [16, 17] or antigen [18]. Other molecular-based methods target sequences within ribosomal genes of Trypanosoma spp., such as the small sub-unit ribosomal gene (18S) [19] or the internal transcribed spacers (ITS) [20, 21], and have been used for species identification.
In Algeria, a huge number of dromedary camels (Camelus dromedarius) is concentrated in the dry desert of the Sahara which occupies 87% of the total area of Algeria (2,381,741 km2). In this region, camels constitute the major source of animal protein and transport for nomads; however, there is a paucity of information regarding camel diseases—especially those concerning hemoparasites [22]. A variety of diagnostic tests has been used to assist in the detection of T. evansi infections in Algeria, and these tests have demonstrated species differences in infection susceptibility among camels, dogs, horses, and donkeys [23,24,25,26]. However, those previous studies did not extensively study the genetic structure and/or the genetic diversity of T. evansi, particularity in camels from Algeria. Therefore, the present study was devoted to the molecular characterization of T. evansi derived from naturally infected camels in eastern Algeria and comparison between isolates from different parts of the world in order to describe distinct T. evansi haplotypes.
Materials and Methods
Study Area
This study was conducted in four provinces (Ouargla, El Oued, Biskra and Ghardaia) located at 2°04′–7°35′ E and 28°32′–34°56′ N (Fig. 2). These areas are known for extreme aridity and extreme heat; they are among the hottest places on Earth during the height of summer. These regions comprise one of the most significant camel rearing areas in Algeria and play a key role in food security and transportation of Saharan and steppe community people [22].
Camel Blood Sampling
Between July 2018 and June 2019, a total of 12 camel herds ranging in size from 10 to 70 head were randomly selected for this study. Blood samples (n = 148) were collected from a random set of animals which included both sexes (20 males and 128 females) and animals in various age groups: < 1 year (n = 7), 1–3 years (n = 33), 4–9 years (n = 53), 10–15 years (n = 45), and > 15 years (n = 10) based on dental wear and owner information. Whole blood was collected from each camel via jugular venipuncture into a 5-mL Vacutainer®tube containing the anticoagulant ethylene diamine tetra acetic acid (EDTA). Samples were stored (in the blood collection tubes) at − 20 °C until DNA extraction was performed.
Molecular Characterization of T. evansi
DNA Extraction from Camel Blood
Total genomic DNA was extracted from 200 µL of EDTA-preserved whole blood of each of the 148 camel blood samples using the DNeasy Blood and Tissue Kit (Qiagen®, Hilden, Germany), according to the manufacturer's instructions. DNA was eluted in a final volume of 200 μL and stored at − 20 °C until used for molecular characterization.
DNA Amplification and Sequencing of ITS Ribosomal Spacer
Genomic DNA samples were subjected to a standard conventional PCR assay to amplify DNA. In order to obtain the ITS1 complete sequence, it was necessary to incorporate the partial 18S and 5.8S sequences so that they could act as anchors for the ITS1 sequence using the two previously described primers ITS1 CF (5′CCG-GAA-GTT-CAC-CGA-TAT-TG3′) and ITS1 BR (5′TTG-CTG-CGT-TCT-TCA-ACG-AA3′) amplifying African Trypanosomal species [21]. Standard PCR reaction was performed in a final volume of 25 μL containing 3.5 mM of MgCl2, 0.2 mM of each deoxyribonucleoside triphosphate (dNTPs) mixture, 1 × PCR buffer, 1 U of Taq DNA Polymerase (Roche, Germany), 0.2 μM of each primer (Eurogentec, Belgium), and 5 μL of genomic DNA. DNA-free distilled water was used as negative control. The PCR reactions were performed in an automated DNA thermal cycler (Biometra TRIO Thermoblock Heat Cycler, Germany) using the following reaction conditions: 95 °C for 10 min for initial denaturation, 35 cycles at 95 °C for 30 s, 58 °C for 30 s, and 72 °C for 1 min, and a final extension at 72 °C for 5 min. The PCR products were subject to electrophoresis in a 1.5% agarose gel stained with ethidium bromide and then visualized using UV light. Six positive PCR product samples were subsequently purified using QIAquick PCR Purification Kit (QIAGEN, Germany) according to manufacturer’s recommendation. Purified PCR products were sequenced in both directions with the same primers used in the PCR amplification. Sequencing reactions were performed by an ABI 3100 Genetic Analyzer automated sequencer (Applied Biosystems, USA) using an ABI PRISM BigDye™ terminator cycle sequencing kits (Applied Biosystems, Foster City, USA).
Phylogenetic and Phylogeographic Analyses
The ChromasPro software (Technelysium PTY, Australia) was used to analyze, assembl and edit the DNA sequences obtained in this study. A nucleotide BLAST was made to compare the identity of the sequences with the NCBI database. Multiple sequence alignment was performed based on ITS1 complete sequences after ligation of the amplicons products using ClustalW. In order to determine the potential novelty of haplotypes identified in our experimental animals, an exhaustive search on GenBank was performed to obtain all sequences of T. evansi for which the ITS1 region was complete. Altogether 96 available sequences were included in the analysis: 69 from Asia, 21 from Africa, and six from South America. A ClustalW alignment was performed on sequences belonging to the same country, and representative haplotypes from different countries, including haplotypes of the present study, were subsequently aligned. According to previous studies on gene diversity of different parasites, each sequence containing an original nucleotide signature is considered a new haplotype [27,28,29].
The phylogeny reconstruction was performed with the DNA sequences of our 6 T. evansi isolates and 39 other representative haplotypes of T. evansi (previously identified in Africa, Asia and South America and deposited in GenBank). The tree was inferred with complete deletion using the maximum likelihood (ML) method, with 500 bootstrap replicates, using Molecular and Evolution Genetic Analysis (MEGA v5 software) [30]. For the latter, the best model was the Tamura 3-parameter with five rate categories and assuming that a certain fraction of sites are evolutionarily invariable (+ I). Genetic distances [Kimura 3-parameter distance] were estimated with MEGA v5 software [30]. All codon positions were used, and all positions containing gaps or missing data were eliminated. Haplotype diversity < Hd > and nucleotide diversity (π) were calculated using DNA Sequence Polymorphism software (DNASP 5.10.01) [31].
Nucleotide Sequence Accession Numbers
The ITS 1 complete sequences generated from this study were deposited in GenBank through accession numbers MT539996 to MT540001.
Results
Descriptive Epidemiology
Of 148 camel blood samples, 19 (12.84%) were PCR-positive for Trypanosoma species. All ITS1 PCR products were approximately 480 bp [specific size of all members of the subgenus Trypanozoon [21]]. The result revealed a non-significant difference between localities (χ2 = 5.83, df = 3, P = 0.12); the prevalence rate was highest in Ghardaia (40.0%), followed by 17.1% in El Oued, 10.0% in Biskra, and 0% in Ouargla (Table 1). Similarly, there was no association of sex (χ2 = 0.45, df = 1, P = 0.50) or age (χ2 = 8.33, df = 4, P = 0.08) with recorded prevalence rate. Prevalence of trypanosome detection was higher in females (13.3%) than in males (10.0%) and higher in camels between 4 and 9 years of age (18.9%) than in other age groups (Table 1).
Molecular Characterization and Phylogenetic Analysis
Sequencing of the rDNA complete internal transcribed spacer (ITS 1) region, including the partial sequences of 18S and 5.8S, generated six sequences corresponding to Trypanosoma evansi with BLAST. Multiple sequence alignment revealed limited heterogeneity among the six sequences (nucleotide diversity π = 0.00550), in the form of five polymorphic or segregating sites and two alignment gaps or missing data, leading to the exhibition of four different haplotypes with a rate of gene diversity Hd corresponding to 0.800. This heterogeneity was localized at the 3′ region of the fragment corresponding to ITS1 (336 bp). Overall, there is one Singleton variable site at position 207 and four Parsimony informative sites at positions 212, 232, 233 and 341(Fig. 1).

Alignment of the four Trypanosoma evansi ITS1 sequence haplotypes. The single nucleotides polymorphisms are marked with colors in the frame.
The first haplotype is represented by isolate 23 from Biskra. The second haplotype is represented by isolate 74 from El Oued. Three identical sequences corresponding to isolates 81, 99 and 119 from El Oued represent the third haplotype, and isolate 139 from Ghardaia represents the fourth haplotype (Fig. 1).
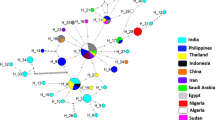
To determine the phylogeographic relationships of T. evansi isolates in camels from Algeria, ITS1 complete sequences of different haplotypes previously reported in Africa, Asia and South America were used. The resulting alignment of the complete ITS1 region exhibited at least 41 different haplotypes confirmed by DNASP (Table 2), with a high rate of gene diversity Hd corresponding to 0.993. The first 30 haplotypes (H1-H30) were found in Asia, two haplotypes (H32 and H33) were found in South America and the remainder of the haplotypes (H34 to H41) originated in Africa. Some haplotypes are shared across continents; for example, H3 was found in China and Kenya, H8 was found in China, Thailand and Egypt, and H10 which has a worldwide distribution. No host specificity was observed.
The new Algerian sequences of T. evansi from camel blood samples are grouped into four different haplotypes: H28 is identical to the isolate found in Saudi Arabia (Genbank N°MN625864; unknown source); H10 is identical to a haplotype found in a wide range of domestic and wild mammalian species and having a worldwide distribution; H40 and H41 are unique and do not match any known haplotypes. Pairwise genetic distances (computed using the Tamura 3-parameter model) between our new haplotypes and other representative haplotypes of the dataset showed a genetic divergence between 0.3 and 14% for H40 and between 0.3 and 15% for H41 (Table 3). Phylogenic analyses were performed with all different representative haplotypes, including our Algerian sequences. The unrooted tree topology shows that each haplotype clustered separately, including the two new haplotypes from Algeria (Fig. 2).

Phylogenetic (a) and geographic (b) distribution of Trypanosoma evansi haplotypes identified in dromedary camel populations in eastern Algeria in relation to T. evansi haplotypes reported worldwide
Discussion
In the present study, Trypanosoma spp. DNA was isolated from 12.84% of camel blood samples collected in eastern Algeria. Six sequences based on ITS1 complete sequence amplification were obtained and identified as T. evansi. Two previous studies concerning T. evansi in camels using molecular tools have been reported in Algeria. Overall molecular prevalence of T. evansi in 1056 camels from four provinces of southern Algeria (El Bayadh, Bechar, Ouargla, Tamanrasset) was 11.2% with RoTat 1.2 PCR [26], and rates of 13% were recorded with the 18S qPCR and 6.2% with the ITS1 PCR in 161 camels from Ghardaia [24], However, those PCR positivity rates were lower compared with results from other countries. For example, T. evansi was detected in 42% of camels from Sudan [32], 32% of camels in Egypt, 31% of camels in Pakistan, 26% of camels in Kenya and 25% of camels in Saudi Arabia [8], as well as in 16.8% of camels in Iran [33]. The lower PCR positivity rate in our study may reflect the fact that the majority of our Algerian samples were collected from apparently healthy camels intended for slaughter (and not from animals showing clinical signs or symptoms of disease). There was a huge difference of sampling among males and females and, independent of previous studies, this difference made analysis concerning the influence of sex on T. evansi infection in the present study difficult. Moreover, the lack of sex or age effects on the prevalence of infection is in agreement with previous studies which revealed that sex and age groups are equally exposed to trypanosomiasis in Pakistan [13] and in Algeria [34].
There are ample reports of characterization studies on trypanosomes in general and T. evansi in particular, among which, DNA targets were used to distinguish T. evansi interspecifically and intraspecifically including 18S rDNA, kinetoplast DNA, microsatellite sequences and the expression-site-associated genes (ESAGs) [35, 36]. Amongst those studies, the molecular targets of variable surface glycoprotein (VSG) RoTat 1.2 gene of T. evansi are widely employed [37] because it is an integral part of the parasite’s surface coat and is known to be expressed in early, middle and late stages of infection [38]. The heterogeneity of T. evansi cannot be demonstrated using those targets; however, studies of ITS regions are more efficient for differentiation and may reflect geographical and/or host range effects [5, 35].
To increase knowledge of genetic diversity of T. evansi in Algeria, and to infer phylogenies and relatedness of the Algerian isolates, molecular analysis based on ITS1 complete sequences was performed on naturally infected dromedary camels. The obtained sequences revealed limited nucleotide variability leading to identification of four different haplotypes. However, in a recent study, identical sequences of T. evansi were reported from partial ITS1 belonging to different dogs in northeastern Algeria [39]. The phylogenetic analysis of our isolates, including representative sequences of the ITS1 complete sequences from Africa, Asia and South America, showed that Algerian sequences isolated from camels belong to four different haplotypes: (i) H28, identical to a sequence isolated from Saudi Arabia;(ii) H10, present worldwide in a different host and (iii) two novel sequences representing new haplotypes H40 and H41.
Phylogenetic analyses of reported T. evansi sequences from various species revealed neither host nor geographic specificity. However, due to lack of nucleotide sequences from previous cases of T. evansi in Algeria, history of these haplotypes could not be determined. For this reason, future population and phylogeographic analyses involving a larger number of samples from different Algerian regions and North African countries are needed to evaluate this evolutionary process.
In conclusion, genetic diversity of T. evansi among dromedary camels from Algeria has been demonstrated and two new haplotypes have been identified. These novel findings can serve as the basis for future studies of T.evansi genetic diversity. Nevertheless, additional studies including more samples from different hosts and geographic areas are needed to fully understand the extent of distribution of T.evansi haplotypes in Algeria.
Availability of data and material
Data transparency.
References
Lai DH, Hashimi H, Lun ZR, Ayala FJ, Lukes J (2008) Adaptations of Trypanosoma brucei to gradual loss of kinetoplast DNA: Trypanosoma equiperdum and Trypanosoma evansi are petite mutants of T. brucei. Proc Natl Acad Sci USA 105:1999–2004. https://doi.org/10.1073/pnas.0711799105
Desquesnes M, Dargantes A, Lai DH, Lun ZR, Holzmuller P, Jittapalapong S (2013) Trypanosoma evansi and surra: a review and perspectives on transmission, epidemiology and control, impact, and zoonotic aspects. Biomed Res Int 2013:321237. https://doi.org/10.1155/2013/321237
Nakayima J, Nakao R, Alhassan A, Mahama C, Afakye K, Sugimoto C (2012) Molecular epidemiological studies on animal trypanosomiases in Ghana. Parasit Vectors 5:217. https://doi.org/10.1186/1756-3305-5-217
Hoare CA (1965) Vampire bats as vectors and hosts of equine and bovine trypanosomes. Acta Trop 22:204–216. https://pubmed.ncbi.nlm.nih.gov/4379528
Amer S, Ryu O, Tada C, Fukuda Y, Inoue N, Nakai Y (2011) Molecular identification and phylogenetic analysis of Trypanosoma evansi from dromedary camels (Camelus dromedarius) in Egypt, a pilot study. Acta Trop 117:39–46. https://doi.org/10.1016/j.actatropica.2010.09.010
Salah AA, Robertson I, Mohamed A (2015) Estimating the economic impact of Trypanosoma evansi infection on production of camel herds in Somaliland. Trop Anim Health Prod 47:707–714. https://doi.org/10.1007/s11250-015-0780-0
Kamidi CM, Saarman NP, Dion K, Mireji PO, Ouma C, Murilla G, Aksoy S, Schnaufer A, Caccone A (2017) Multiple evolutionary origins of Trypanosoma evansi in Kenya. PLoS Negl Trop Dis 11:e0005895. https://doi.org/10.1371/journal.pntd.0005895
Aregawi WG, Agga GE, Abdi RD, Buscher P (2019) Systematic review and meta-analysis on the global distribution, host range, and prevalence of Trypanosoma evansi. Parasit Vectors 12:67. https://doi.org/10.1186/s13071-019-3311-4
Li Z, Pinto Torres JE, Goossens J, Stijlemans B, Sterckx YG, Magez S (2020) Development of a recombinase polymerase amplification lateral flow assay for the detection of active Trypanosoma evansi infections. PLoS Negl Trop Dis 14:e0008044. https://doi.org/10.1371/journal.pntd.0008044
Joshi PP, Shegokar VR, Powar RM, Herder S, Katti R, Salkar HR, Dani VS, Bhargava A, Jannin J, Truc P (2005) Human trypanosomiasis caused by Trypanosoma evansi in India: the first case report. Am J Trop Med Hyg 73:491–495. https://pubmed.ncbi.nlm.nih.gov/16172469.
Van Vinh CN, Buu CL, Desquesnes M, Herder S, Phu Huong LN, Campbell JI, Van CN, Yimming B, Chalermwong P, Jittapalapong S, Ramon FJ, Tri TN, Rabaa MA, Carrique-Mas J, Pham Thi TT, Tran Vu TN, Berto A, Thi HN, Van Minh HN, Canh TN, Khac CN, Wills B, Tinh HT, Thwaites GE, Yacoub S, Baker S (2016) A Clinical and Epidemiological Investigation of the First Reported Human Infection With the Zoonotic Parasite Trypanosoma evansi in Southeast Asia. Clin Infect Dis 62:1002–1008. https://doi.org/10.1093/cid/ciw052
Hassan-Kadle AA, Ibrahim AM, Nyingilili HS, Yusuf AA, Vieira TSWJ, Vieira RFC (2019) Parasitological, serological and molecular survey of camel trypanosomiasis in Somalia. Parasit Vectors 12:598. https://doi.org/10.1186/s13071-019-3853-5
Tehseen S, Jahan N, Qamar MF, Desquesnes M, Shahzad MI, Deborggraeve S, Buscher P (2015) Parasitological, serological and molecular survey of Trypanosoma evansi infection in dromedary camels from Cholistan Desert. Pakistan Parasit Vectors 8:415. https://doi.org/10.1186/s13071-015-1002-3
Chappuis F, Loutan L, Simarro P, Lejon V, Buscher P (2005) Options for field diagnosis of human African trypanosomiasis. Clin Microbiol Rev 18:133–146. https://doi.org/10.1128/CMR.18.1.133-146.2005
Masiga DK, Gibson WC (1990) Specific probes for Trypanosoma (Trypanozoon) evansi based on kinetoplast DNA minicircles. Mol Biochem Parasitol 40:279–283. https://doi.org/10.1016/0166-6851(90)90049-r
Birhanu H, Gebrehiwot T, Goddeeris BM, Buscher P, Van RN (2016) New Trypanosoma evansi Type B Isolates from Ethiopian Dromedary Camels. PLoS Negl Trop Dis 10:e0004556. https://doi.org/10.1371/journal.pntd.0004556
Ngaira JM, Njagi EN, Ngeranwa JJ, Olembo NK (2004) PCR amplification of RoTat 1.2 VSG gene in Trypanosoma evansi isolates in Kenya. Vet Parasitol 120:23–33. https://doi.org/10.1016/j.vetpar.2003.12.007
Songa BE, Hamers R (1988) A card agglutination test (CATT) for veterinary use based on an early VAT RoTat 1/2 of Trypanosoma evansi. Ann Soc Belg Med Trop 68: 233–240. https://pubmed.ncbi.nlm.nih.gov/3223785.
Geysen D, Delespaux V, Geerts S (2003) PCR-RFLP using Ssu-rDNA amplification as an easy method for species-specific diagnosis of Trypanosoma species in cattle. Vet Parasitol 110:171–180. https://doi.org/10.1016/s0304-4017(02)00313-8
da Silva FM, Noyes H, Campaner M, Junqueira AC, Coura JR, Anez N, Shaw JJ, Stevens JR, Teixeira MM (2004) Phylogeny, taxonomy and grouping of Trypanosoma rangeli isolates from man, triatomines and sylvatic mammals from widespread geographical origin based on SSU and ITS ribosomal sequences. Parasitology 129:549–561. https://doi.org/10.1017/s0031182004005931
Njiru ZK, Constantine CC, Guya S, Crowther J, Kiragu JM, Thompson RC, Davila AM (2005) The use of ITS1 rDNA PCR in detecting pathogenic African trypanosomes. Parasitol Res 95:186–192. https://doi.org/10.1007/s00436-004-1267-5
Bellabidi M, Benaissa MH, Bissati-Bouafia S, Harrat Z, Brahmi K, Kernif T (2020) Coxiella burnetii in camels (Camelus dromedarius) from Algeria: Seroprevalence, molecular characterization, and ticks (Acari: Ixodidae) vectors. Acta Trop 206:105443. https://doi.org/10.1016/j.actatropica.2020.105443
Benfodil K, Ansel S, Mohamed-Cherif A, Ait-Oudhia K (2019) Prevalence of Trypanosoma evansi in horses (Equus caballus) and donkeys (Equus asinus) in El-Bayadh district, southwestern Algeria. J Hellenic Veterinary Med Soc 70:1631–1638. https://doi.org/10.12681/jhvms.21786
Benfodil K, Buscher P, Abdelli A, Van RN, Mohamed-Herif A, Ansel S, Fettata S, Dehou S, Bebronne N, Geerts M, Balharbi F, Ait-Oudhia K (2020) Comparison of serological and molecular tests for detection of Trypanosoma evansi in domestic animals from Ghardaia district. South Algeria Vet Parasitol 280:109089. https://doi.org/10.1016/j.vetpar.2020.109089
Bennoune O, Adili N, Amri K, Bennecib L, Ayachi A (2013) Trypanosomiasis of camels (Camelus dromedarius) in Algeria: first report. Veterinary Res Forum 4: 273–275. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4279620/pdf/vrf-4-273.pdf.
Boushaki D, Adel A, Dia ML, Buscher P, Madani H, Brihoum BA, Sadaoui H, Bouayed N, Kechemir IN (2019) Epidemiological investigations on Trypanosoma evansi infection in dromedary camels in the South of Algeria. Heliyon 5:e02086. https://doi.org/10.1016/j.heliyon.2019.e02086
Jaimes-DuenezJ T-C, Valencia-Hernandez A, Sanchez-Arevalo D, Poche-Ceballos A, Ortiz-Alvarez J, Mejia-Jaramillo AM (2017) Molecular diagnosis and phylogeographic analysis of Trypanosoma evansi in dogs (Canis lupus familiaris) suggest an epidemiological importance of this species in Colombia. Prev Veterinary Med 139:82–89. https://doi.org/10.1016/j.prevetmed.2017.02.007
Martinez-Flores WA, Palma-Garcia JM, Caballero-Ortega H, Del Viento-Camacho A, Lopez-Escamilla E, Martinez-Hernandez F, Vinuesa P, Correa D, Maravilla P (2017) Genotyping Toxoplasma gondii with the B1 gene in naturally infected sheep from an endemic region in the Pacific coast of Mexico. Vector Borne Zoonotic Dis 17:495–502. https://doi.org/10.1089/vbz.2016.2085
Drali R, Abi-Rached L, Boutellis A, Djossou F, Barker SC, Raoult D (2016) Host switching of human lice to new world monkeys in South America. Infect Genet Evol 39:225–231. https://doi.org/10.1016/j.meegid.2016.02.008
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. https://doi.org/10.1093/molbev/msr121
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. https://doi.org/10.1093/bioinformatics/btp187
Salim B, Bakheit MA, Kamau J, Nakamura I, Sugimoto C (2011) Molecular epidemiology of camel trypanosomiasis based on ITS1 rDNA and RoTat 1.2 VSG gene in the Sudan. Parasit Vectors 4:31. https://doi.org/10.1186/1756-3305-4-31
Nazem MR, Karimidehkordi M, Farhoodi Moghadam M (2020) Detection of Trypanosoma evansi in camel abortions (Camelus dromedarius) in Iran using polymerase chain reaction. Turkish J Veterinary Anim Sci 44: 1–6. http://journals.tubitak.gov.tr/veterinary/issues/vet-20-44-2/vet-44-2-25-1908-76.pdf.
Benaissa MH, Mimoune N, Bentria Y, Kernif T, Boukhelkhal A, Youngs CR, Kaidi R, Faye B, Halis Y (2020) Seroprevalence and risk factors for Trypanosoma evansi, the causative agent of surra in the dromedary camel (Camelus dromedarius) population in southeastern Algeria. Onderstepoort J Vet Res 87(1):a1891. https://doi.org/10.4102/ojvr.v87i1.1891
Khuchareontaworn S, Singhaphan P, Viseshakul N, Chansiri K (2007) Genetic diversity of Trypanosoma evansi in buffalo based on internal transcribed spacer (ITS) regions. J Vet Med Sci 69:487–493. https://doi.org/10.1292/jvms.69.487
Konnai S, Mekata H, Mingala CN, Abes NS, Gutierrez CA, Herrera JR, Dargantes AP, Witola WH, Cruz LC, Inoue N, Onuma M, Ohashi K (2009) Development and application of a quantitative real-time PCR for the diagnosis of Surra in water buffaloes. Infect Genet Evol 9:449–452. https://doi.org/10.1016/j.meegid.2009.01.006
Sudan V, Jaiswal AK, Shanker D, Verma AK (2017) First report of molecular characterization and phylogenetic analysis of RoTat 1.2 VSG of Trypanosoma evansi from equine isolate. Trop Anim Health Prod 49:1793–1796. https://doi.org/10.1007/s11250-017-1384-7
Claes F, Verloo D, De Waal DT, Majiwa PA, Baltz T, Goddeeris BM, Buscher P (2003) The expression of RoTat 1.2 variable surface glycoprotein (VSG) in Trypanosoma evansi and T. equiperdum. Vet Parasitol 116:209–216. https://doi.org/10.1016/s0304-4017(02)00359-x
Medkour H, Laidoudi Y, Lafri I, Davoust B, Mekroud A, Bitam I, Mediannikov O (2019) Canine vector-borne protozoa: molecular and serological investigation for Leishmania spp., Trypanosoma spp., Babesia spp., and Hepatozoon spp. in dogs from Northern Algeria. Vet Parasitol Reg Stud Rep 19:100353. https://doi.org/10.1016/j.vprsr.2019.100353
OIE (2019) Use of animals in research and education. In: OIE terrestrial animal health code. http://www.oie.int/index.php?id=169&L=0&htmfile=chapitre_aw_research_education
Acknowledgements
We thank Professor Curtis R. Youngs, Animal Science Department, Iowa State University, Ames, IA 50011, United States of America for improvement of our English. We are also grateful to Nour el houda Ammani, Amina Arib and Amira Azzouz for their participation in the study.
Funding
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethics Approval
This study with camels was supervised by the division of biological resources of the Scientific and Technical Research Centre for Arid Areas (CRSTRA). It was conducted in accordance with the World Animal Health Organization (OIE) guiding principles on animal welfare included in the OIE Terrestrial Animal Health Code [40].Verbal consent of farm owners involved in this investigation was obtained prior to the collection of blood samples from their animals.
Rights and permissions
About this article

Cite this article
Boutellis, A., Bellabidi, M., Benaissa, M.H. et al. New Haplotypes of Trypanosoma evansi Identified in Dromedary Camels from Algeria. Acta Parasit. 66, 294–302 (2021). https://doi.org/10.1007/s11686-020-00316-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11686-020-00316-w