
Abstract
There are 11 different pathogenic trypanosomes in trypanosomiasis endemic regions of Africa. Their detection and characterisation by molecular methods relies on species-specific primers; consequently several PCR tests have to be made on each sample. Primers ITS1 CF and ITS1 BR, previously designed to amplify the internal transcribed spacer (ITS1) of rDNA, have been evaluated for use in a universal diagnostic test for all pathogenic trypanosomes. Blood was collected from 373 cattle and 185 camels. The primers gave constant PCR products with the stocks of each taxon tested. Members of subgenus Trypanozoon (T. brucei brucei, T. evansi, T. b. rhodesiense and T. b. gambiense) gave a constant product of approximately 480 bp; T. congolense, savannah 700 bp, T. congolense kilifi 620 bp and T. congolense forest 710 bp: T. simiae 400 bp, T. simiae tsavo 370 bp, T. godfreyi 300 bp and T. vivax 250 bp. The sensitivity of the test ranged from 10 pg for Trypanozoon, T. congolense clade and T. vivax to 100 pg for T. simiae and T. godfreyi. The primers detected cases of multi-taxa samples, although the sensitivity was reduced with an increase in the combinations. A better detection rate of trypanosome DNA was recorded with buffy coats than from direct blood. With the field samples, the diagnostic sensitivity was close to the sensitivity obtained using single reactions with species-specific primers for Trypanozoon 38/40 (95%) and T. congolense savannah 30/33 (90.9%) but was lower with T. vivax 25/31 (77.4%). The primers offer promise as a routine diagnostic tool through the use of a single PCR; however, further evaluation is recommended.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Trypanosomiasis in animals (nagana) and humans (sleeping sickness) continues to cause immeasurable losses in tropical Africa. Accurate detection of trypanosomes both in vectors and in host blood relies heavily on molecular techniques (polymerase chain reaction, PCR). Such techniques have proved effective in the characterisation of trypanosomes (Masiga et al. 1992; Wuyts et al. 1994; Majiwa et al. 1994; Lehane et al. 2000; Malele et al. 2003), typing new trypanosomes (Gibson et al. 2001), in the collection of epidemiological data (Solano et al. 1999), and in animal treatment.
In Kenya, ten pathogenic trypanosomes occur and overlap in most of the tsetse belt (Njiru et al. 2004), such that multiple infection of both host and vector is to be expected. This implies that several PCR tests have to be made to ascertain whether a collected sample is positive, negative or even of mixed infection. For instance a bovine sample from an endemic area in Kenya will require at least five species-specific PCR tests to cover the possibilities of Trypanosoma vivax, T. congolense savannah, T. congolense kilifi, T. congolense forest and T. brucei spp, three PCRs for suids (T. simiae, T. godfreyi and T. simiae tsavo) and eight for every tsetse sample collected. This translates into an enormous cost which normally prohibits such studies. Efforts to combine already available primers (multiplex) in a single PCR have been discouraging due to lower sensitivity compared to individual species-specific tests and the appearance of non-specific PCR products. The development of a universal single PCR based on ribosomal genes by McLaughlin et al. (1996) and further evaluated by Desquesnes et al. (2001) detected combined trypanosome infections. However, extensive evaluation of these primers (KIN1 and KIN2) was not done.
The internal transcribed spacer (ITS) region of rDNA is a preferred target for a universal test because of its highly conserved flanking regions and size variability among trypanosomes species and subgroups. This locus has 100–200 copies and each transcribed unit is composed of 18S, 5.8S and 28S rRNA genes separated by two ITS regions, (Hernandez et al. 1993; Desquesnes and Davila 2002). New ITS-based primers were designed by Davila (unpublished) specifically to amplify ITS1. This study reports the evaluation of these new primers (ITS1 CF and ITS1 BR) both in the laboratory using trypanosome reference DNA and in the field. Further, the test was compared with earlier ITS-based primers KIN1 and KIN2 (McLaughlin et al. 1996). It is anticipated that a successful universal PCR-based test would add value to the collection of epidemiological data on trypanosomiasis, while easing the cost of running several PCRs, especially in the endemic zones of Africa.
Materials and methods
Field sample collection
Samples were collected from trypanosomiasis endemic zones. Cattle samples (357) were taken from Nguruman in southern Kenya and 185 camel blood samples from northern Kenya. The animals were bled from the jugular vein (5 ml) into heparinised Vacutainer tubes. Two capillaries were filled three-quarters full for parasite investigations using BCT (Murray et al. 1977) and HCT (Woo 1970) and determination of packed cell volume (PCV). Approximately 0.5 ml of blood was transferred into cryovials, mixed with phosphate buffered saline (PBS) and 10% glycerol, and stored in liquid nitrogen for DNA extraction. All parasitologically positive animals, and those judged infected using clinical symptoms by the field veterinarian, were treated with trypanocides. Sixteen positive cattle samples from Lambwe valley were collected separately and analysed during the sensitisation and launching of a sterile insect technique project.
Trypanosomes DNA
Well-characterised reference DNA for the subgenera Nannomonas and Duttonella used in this study were obtained from Wendy Gibson, University of Bristol, while the members of the subgenusTrypanozoon, T. lewisi and T. theileri, were from well-characterised isolates from the KETRI cryobank. Further, characterised trypanosome DNA from tsetse samples that had been analysed previously were included in this study (Njiru et al. 2004).
Template preparation and PCR cycling
DNA from blood samples was extracted using commercially available QIAamp DNA mini kit (Qiagen, Australia). The purified DNA templates were stored at −20°C for later use. For tsetse samples, DNA was initially extracted using the proteinase K method as detailed by Masiga et al (1992) and later re-extracted using the Qiagen kit for uniformity purposes. PCR was carried out in 25-µl reaction mixtures containing 10× reaction buffer (670 mM Tris-HCl pH 8.8, 166 mM (NH4)2SO4, 4.5% Triton X-100, 2 mg/ml gelatin) (Fisher Biotech), 2 mM MgCl2, 200 µM of each of the four deoxynucleoside triphosphates (dNTPs), primers at 1 µM and 0.5 U of Taq DNA polymerase (Fisher Biotech). PCR cycles for ITS1 CF and BR primers were: initial step at 94°C for 5 min, followed by 35 cycles of 94°C for 40 s, 58°C for 40 s, 72°C for 90 s, and final extension at 72°C for 5 min, and for KIN primers as described by Desquesnes et al. (2001). Species-specific primers and their PCR conditions were those described by Masiga et al. (1992) for Nannomonas, Wuyts et al. (1994) for Trypanozoon and Duttonella by Masake et al. (1997) and Morlais et al. (2001). KIN1, 5′GCGTTCAAAGATTGGGCAAT and KIN2, 5′CGCCCGAAAGTTCACC (McLaughlin et al. 1996) and those developed for this study ITS1 CF, 5′CCGGAAGTTCACCGATATTG and ITS1 BR, 5′TTGCTGCGTTCTTCAACGAA (Davila, unpublished). The ITS1 CF and BR primers were tested for sensitivity and ability to detect known spiked trypanosome DNA. The amount of DNA template analysed for reference DNA was 50 ng and 2 µl of the template was used for field samples. Amplification products were resolved in 1.5% molecular grade agarose (Fisher Biotech) stained with ethidium bromide.
Sequencing
PCR products were purified from the agarose gel using a freeze-squeeze method. Briefly, the PCR band was cut from the gel and frozen at −20°C for 4 h. The frozen gel slice was placed between Parafilm, squeezed and approximately 50 µl of liquid collected. The collected volume was mixed with 1 µl of 3 M sodium acetate and 100 µl of 70% ethanol, and then left on ice for 20 min. The mix was then centrifuged at 14,000 rpm for 10 min, the supernatant discarded and the resulting pellet vacuum-dried and resuspended in 10 µl PCR water. A total of 1 µl of this final sample was used in running the sequencing PCR reaction using either forward or reverse ITS1 primers. Sequencing was done in an ABI automatic DNA sequencer (Applied Biosystems). For T. godfreyi, the resulting PCR product was cloned into a TOPO vector (Invitrogen) and sequenced (GenBank accession no. AY661891).
Alignment
The sequences obtained were ‘blasted’ using the basic BLAST search tool (http://www.ncbi.nlm.nih.gov/BLAST/) and alignment made with specific ribosomal products found in the GenBank using DNAman (Lynnon Biosoft, Canada).
Results
Sensitivity
The analytical sensitivity of the ITS1 CF and BR primers was tested using serially diluted trypanosome DNA. The resulting DNA dilutions were also analysed for analytical sensitivity using species-specific primers to check dilution accuracy. We recorded sensitivities of between 10 pg and 100 pg with ITS1 CF and BR primers, while with the species-specific ones, the sensitivities were equivalent to those already published by Masiga et al. (1992) and Wuyts et al. (1994) (Table 1 and Fig. 1). The ITS1 CF and BR based primers could detect all spiked DNA samples with double and triple combinations. However, the analytical sensitivity was very low in quadruple combinations. Use of >50 ng of reference DNA gave rise to non-specific PCR products while PCR reactions with ammonium sulphate buffers showed a higher sensitivity than those without.

The sensitivity of ITS1 CF and BR (A) and KIN1 and KIN2 (B) for Trypanosoma vivax (KETRI 2619), T. brucei rhodesiense (KETRI 2522) and T. congolense savannah (Gam 2)
PCR size products with ITS1 CF and BR
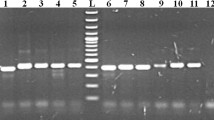
All members of the subgenus Trypanozoon gave an approximate band size of 480 bp; Nannomonas: T. congolense savannah 700 bp; T. congolense kilifi 620; T. congolense forest 700; T. simiae 400; T. simiae tsavo 370; T. godfreyi 300 and Duttonella; T. vivax 250 bp as shown in Table 2 and Fig. 2.

Gel electrophoresis of reference DNA samples (50 ng) amplified with ITS1 CF and BR. Lane M1 100 bp marker, lane 2 T. brucei brucei, lane 3 T. evansi, lane 4 T. congolense savannah, lane 5 T. congolense kilifi, lane 6 T. congolense forest, lane 7 T. simiae tsavo, lane 8 T. godfreyi, lane 9 T. simiae, lane 10 T. vivax (Y58), lane 11 T. lewisi, NC negative control, M2 1 kb marker
Field analyses
Analyses of field samples showed a trypanosome prevalence of 86/357 (24.1%) and 48/185 (25.9%) in cattle and camels, respectively, using subgenus and species-specific primers. For bovine samples, 71 (27 Trypanozoon, 33 Nannomonas and 11 T. vivax) were of single infections and 15 double infections (7 T. b. rhodesiense/T. congolense savannah, 4 T. b. rhodesiense/T. vivax and 4 T. congolense savannah/T. vivax). The ITS1 CF and BR primers detected 73 (84.9%), while KIN1 and KIN2 detected 58 (67.4%) of the samples positive using species-specific primers. The major differences between the KIN and ITS1 CF and BR primers were the inability of the KIN primers to detect T. vivax from field samples, although they detected reference T. vivax DNA (Y58) isolated in Nigeria, West Africa. For camel samples, ITS1 CF and BR primers detected 35/48 (72.9%) and KIN primers 31/48 (64.5%) of trypanosome DNA from direct blood isolation (Table 3). However, with PCR done on 48 positive buffy coat, detection rates improved to 44/48 (91.6%) using ITS1 CF and BR and 43/48 (89.9%) using KIN primers while on the 35 bovine aparasitaemic samples that were species-specific positive, detection improved from 22/35 (62.9%) to 29/35 (82.9%) with ITS1 CF and BR and 17/35 (48.6%) to 26/35 (74.3%) with KIN primers. Similar better results in detection and the characterisation of mixed infections were also recorded with 16 microscopy-positive bovine samples from the Lambwe valley in Kenya (Fig. 3B).

Gel electrophoresis of PCR products from direct blood DNA isolation (A) and their respective buffy coats (B) from 16 MHCT positive bovine samples collected in Lambwe valley in Kenya. Identification with species-specific primers are: Lane 1 T. vivax, lane 2 T. brucei, lane 3 T. congolense savannah/T. vivax, lane 4 T. brucei, lane 5 T. congolense savannah, lane 6 T. brucei/T. vivax, lane 7 T. congolense savannah, lane 8 T. vivax, lane 9 T. congolense savannah, lane 10 T. brucei, lane 11 T. brucei, lane 12 T. vivax, lane 13 T. congolense savannah/T. brucei/T. vivax, lane 14 T. brucei, lane 15 T. brucei/T. congolense savannah, lane 16 T. vivax, lane 17 T. congolense savannah-positive control, lane 18 T. brucei-positive control for B and negative control for print A, lane 19 negative control for B
Discussion
The ITS1 CF and BR based primers showed a higher diagnostic sensitivity than that previously described using the KIN primers. The major observation was the ability of the ITS1 CF and ITS1 BR primers to detect more T. vivax samples than the KIN primers. Their specificity (no amplification with host and vector DNA) and capability of detecting all pathogenic trypanosomes in a single PCR indicates a greater potential for this system as a universal test for pathogenic trypanosomes. Furthermore, the primers did not amplify the non-pathogenic T. lewisi and T. theileri used in this study. ITS1 CF and ITS1 BR are designed from the conserved region of rDNA genes. Specifically ITS1 CF anneals to 18S and ITS1 BR in 5.8S regions of rDNA allowing the amplification of (ITS)1 which varies in size within trypanosomes species and in principle should differentiate trypanosomes by their ITS1 sizes.
The ITS1 region of trypanosomes varies in size (Desquesnes et al. 2001; Desquesnes and Davila 2002), except for members of the subgenus Trypanozoon which are thought to be closely related, and their divergence may be a recent occurrence. Sequencing of PCR products in this study showed almost perfect homology with the respective sequences in GenBank. The observed single PCR products for each species were approximately the same size as their sequenced product and were also in agreement with estimations from GenBank sequences. Lack of amplification with non-pathogenic trypanosomes was supported by T. theileri GenBank sequence AB007814 which shows only 25% homology with the ITS1 CF forward primer. This is expected because the primers were designed to improve homology with pathogenic trypanosomes whilst minimising homology with non-pathogenic trypanosomes (Davila unpublished). Observed sequence differences in ITS1 region for pathogenic and non-pathogenic trypanosomes may indicate evolutionary divergence of the Kinetoplastida rDNA region (Stevens and Rambaut 2001).
Using serially diluted DNA in this study, the analytical sensitivity of the ITS1 CF and BR primers ranged between 10 pg (100 trypanosomes) for Trypanozoon, T. vivax and T. congolense clades to 100 pg (1,000 trypanosomes) for T. simiae and T. godfreyi. The primers detected all parasitologically positive samples from direct blood isolation. With 36 aparasitaemic bovine field samples, sensitivity improved by between 20% to 30% when the DNA was extracted from buffy coats (Table 3). This is expected because centrifugation concentrates the parasites. Under field conditions, the most challenging cases are when parasites are not demonstrable from the peripheral blood, and this is where PCR becomes useful. Apparently, low parasitaemia in host blood is a characteristic of most trypanosome infections, and especially T. evansi and T. vivax which may run chronic forms with no parasites demonstrable in the peripheral blood. In such cases, the concentration of trypanosomes is important for improving the detection and the characterisation of mixed infections (Fig. 3B).
ITS1 CF and BR primers show 100% homology with the available T. vivax sequence (accession no. U22316) while KIN1 and KIN2 show 75–90% homology, which may explain the observed differences in T. vivax detection rates between the two tests. Desquesnes et al. (2001) observed low detection rates of T. vivax with KIN primers and proposed redesigning other ITS based primers to improve detection rates, an objective Davila (unpublished) took into account. Despite the improved detection rates of T. vivax with the new primers, we could only detect 25/31 (77.4%) of T. vivax from field samples in Kenya (all data not shown), a fact that may be related to low target DNA and/or existence of T. vivax genetic variants with changes in primer annealing regions. This fact is supported by the observation that some T. vivax from Kenya analysed in this study gave non-specific PCR products with KIN primers (Fig. 1B) and differing sizes of the ITS region (Table 2). T. vivax genetic variants have been reported in the field (Masake et al. 1997; Malele et al. 2003). Further, this has been supported by the work of Stevens and Rambaut (2001), who noted that T. vivax 18 rRNA is evolving at 7 to 10 times the rate of non-salivarian trypanosomes, and also significantly faster than all other trypanosomes.
The usefulness of the new test was demonstrated by its ability to detect mixed trypanosome infections in cryopreserved samples and from field samples. In our analysis, we picked out four isolates that had been previously categorised as single infections to be mixed infections. Direct sequencing of these products showed that KETRI 2729 contained a T. vivax [not the West African type because it was negative with TVW1 and TVW2, Masiga et al. (1992) but positive with universal T. vivax primers (Masake et al. 1997; Morlais et al. 2001] and T. congolense savannah. Camel sample MG1 from a tsetse-infested area had T. brucei ssp and T. vivax, bovine samples GJ4 T. brucei ssp and T. congolense savannah, and GJ3 T. brucei ssp, T. vivax and T. congolense savannah. The test was not able to differentiate T. congolense savannah (697 bp) and T. congolense forest (714 bp) mixed infections based on the size of their PCR product in a normal agarose gel. However, for routine treatment purposes, the veterinarian would only be interested in knowing the infection status of the animal rather than infecting species, in which case the test fulfils the purpose. With mixed spiked DNA, the new test detected all of the expected double and triple combinations. However, with the quadruple combination the sensitivity was low. Double infections in animals are a normal occurrence in the field (Mugittu et al. 2000), while triple and quadruple infections may be a rare occurrence in animals, although common in tsetse flies (Njiru et al. 2004).
The use of universal tests for pathogenic trypanosomes would reduce the cost of PCR three to five times as the number of reactions required per sample would be reduced to one (Desquesnes and Davila 2002). Further, this would be useful in picking out any new trypanosome prevailing in the field. However, the greatest challenge that will face ITS-based tests will be diagnostic sensitivity, because ITS1 has approximately 100–200 copies as compared to species-specific tests which target satellite DNA with over 10,000 copies (Desquesnes and Davila 2002). In summary, our results show that the test has the ability to detect multiple species in a single PCR from field samples collected in Kenya. To further improve detection of trypanosomes, DNA extraction from buffy coats is recommended.
References
Desquesnes M, Davila AM (2002) Applications of PCR-based tools for detection and identification of animal trypanosomes: a review and perspectives. Vet Parasitol 11:213–231
Desquesnes M, McLaughlin G, Zoungrana A, Davila AM (2001) Detection and identification of Trypanosoma of African livestock through a single PCR based on internal transcribed spacer 1 of rDNA. Int J Parasitol 31:610–614
Gibson WC, Stevens JR, Mwendia CMT, Makumi JM, Ngotho JM, Ndung’u JM (2001) Unraveling the phylogenetic relationships of African trypanosomes of suids. Parasitology 122:625–631
Hernandez P, Martin-Parras L, Martinez-Robles ML, Schvartzman JB (1993) Conserved features in the mode of replication of eukaryotic ribosomal RNA genes. EMBO J 12:1475–1485
Lehane MJ, Msangi AR, Whitaker CJ, Lehane SM (2000) Grouping of trypanosomes species in mixed infections in Glossina pallidipes. Parasitology 120:583–592
Majiwa PAO, Thatti R, Moloo SK, Nyeko JHP, Otieno LH, Moloo S (1994) Detection of trypanosome infections in the saliva of tsetse flies and buffy coat samples from antigenic but aparasitaemic cattle. Parasitology 108:313–322
Malele I, Craske L, Knight C, Ferris V, Njiru ZK, Hamilton P, Stella L, Lehane M, Gibson WC (2003) The use of specific and generic primers to identify trypanosome infections of wild tsetse flies in Tanzania by PCR. Infect Genet Evol 3:271–279
Masake RA, Majiwa PAO, Moloo SK, Makau JM, Njuguna JT, Maina M, Kabata J, Ole-MoiYoi, OK, Nantulya VM (1997) Sensitive and specific detection of Trypanosoma vivax using the polymerase chain reaction. Exp Parasitol 85:193–205
Masiga DK, Smyth AJ, Hayes P, Bromidge TJ, Gibson WC (1992) Sensitive detection of trypanosomes in tsetse flies by DNA amplification. Int J Parasitol 22:909–918
McLaughlin Gl, Ssenyonga SS, Nanteza E, Rubaire-Akiki, Wafula O, Hansen RD, Vodkin MH, Novak RJ, Gordon VR, Montenegro-James S, James M, Aviles H, Armijos R, Santrich C, Weigle K, Saravia N, Wozniak E, Gaye O, Mdachi R, Shapiro SZ, Chang KP, Kakoma I (1996) PCR based detection and typing of parasites. In: Zcel MA, Alkan MZ (eds) Parasitology for the 20th century. CAB International, Wallingford, pp 261–287
Morlais I, Ravel S, Grebaut P, Dumas V, Cuny G (2001) New molecular marker for Trypanosoma (Duttonella) vivax identification. Acta Trop 80:207–213
Mugittu KN, Silayo RS, Majiwa PAO, Kimbita EK, Mutayoba BM, Maselle R (2000) Application of PCR and DNA probes in the characterization of trypanosomes in the blood of cattle in farms in Morogoro Tanzania. Vet Parasitol 94:177–189
Murray M, Murray PK, McIntyre IM (1977) An improved parasitological technique for the diagnosis of African trypanosomiasis. Trans R Soc Trop Med Hyg 71:325–326
Njiru ZK, Makumi JN, Okoth S, Ndungu JM, Gibson WC (2004) Identification of trypanosomes in Glossina pallidipes and G. longipennis in Kenya. Infect Genet Evol 4:29–35
Stevens J, Rambaut A (2001) Evolutionary rate differences in trypanosomes. Infect Genet Evol 1:143–150
Solano P, Michel JF, Lefrancois T, De La Rocque S, Sidibe I, Zoungrana A, Cuisance D (1999) Polymerase chain reaction as a diagnosis tool for detecting trypanosomes in naturally infected cattle in Burkina Faso. Vet Parasitol 30:95–103
Woo PTR (1970) The haematocrit centrifuge technique for the diagnosis of African trypanosomes. Acta Trop 27:384–386
Wuyts N, Chokesajjawatee N, Panyim S (1994) A simplified and highly sensitive detection of Trypanosoma evansi by DNA amplification. S E Asian J Trop Med Public Health 25:266–271
Acknowledgements
This work received financial support from the International Atomic Energy Agency (IAEA) under grant no. KEN 11414 to Z.K. Njiru and supplementary funds from the Kenyan Government. Z.K. also thanks Dr. Wendy Gibson for supplying some reference DNA and the staff of Biochemistry Division, KETRI for technical support. The work reported here complies with the current laws of Kenya where the work was done. Nucleotide sequence data reported in this paper is available in the GenBank and DDBJ data bases under accession number AY661891.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Njiru, Z.K., Constantine, C.C., Guya, S. et al. The use of ITS1 rDNA PCR in detecting pathogenic African trypanosomes. Parasitol Res 95, 186–192 (2005). https://doi.org/10.1007/s00436-004-1267-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-004-1267-5