
Abstract
Lipid droplets (LDs) are ubiquitous organelles that store and supply lipids to regulate cellular lipid homeostasis. Fatty acids are packaged as triglyceride and cholesterol ester into endoplasmic reticulum (ER) membranes to synthesize LDs. Cytosolic LDs move dynamically and interact with organelles, including other LDs. In this process, functional proteins for metabolism are also transferred to LDs. In this review, I focus on interactions between the ER and LDs related to lipid metabolism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lipid droplets (LDs) are ubiquitous organelles that store neutral lipids such as triglycerides and cholesterol esters in mammals. The number, size, and neutral lipid composition of LDs differs among cell types. Unlike other organelles, LDs include a phospholipid monolayer (Tauchi-Sato et al. 2002). Therefore, proteins localize on LDs by directly binding to the monolayer surface via amphipathic helices, by embedding in short hydrophobic regions, through lipid anchors, or by interacting with other proteins on LDs. These proteins are directly transferred from the cytoplasm to LDs or move to LDs from other organelles via membrane connections (Kory et al. 2016).
Proteomics studies with isolated LDs have revealed the presence of several proteins related to lipid metabolism, membrane trafficking, signaling, protein degradation, and so on. In mammals there are five PAT-family proteins that function in lipid droplet biogenesis and metabolism: PLIN1–5, which are also called perilipin, ADRP, TIP47, S3-12, and MLDP, respectively. PLIN1 and PLIN4 are expressed specifically in adipocytes and steroidogenic cells, whereas PLIN2 and PLIN3 are ubiquitously expressed (Bickel et al. 2009). In addition to the PAT protein family, several other proteins regulate LD metabolism. In this review, I will focus on the functions of LDs that are correlated with their connectivity to the endoplasmic reticulum (ER).
Biogenesis of LD in the ER
Formation of LDs is induced by several factors, such as long-chain fatty acids, oxidized low-density lipoproteins, oxidative stress, growth factors, inflammatory stimuli, and bacterial infections.
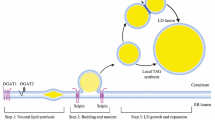
LD biogenesis is supported by several enzymes localized in the ER membrane. De novo triglyceride synthesis from glycerol-3-phosphate occurs through the activities of the following enzymes: glycerol-3-phosphate O-acyltransferase, 1-acylglycerol-3-phosphate O-acyltransferase, and the phosphatidic acid phosphatase/lipin and diacylglycerol acyltransferase (DGAT) families. Cholesterol ester is synthesized from cholesterol by acyl-CoA:cholesterol O-acyltransferase (ACAT). Acyl-CoAs made by acyl-CoA synthetase (ACS) are required in these esterification processes. Triacsin C is an inhibitor of ACSL1, ACSL3, and ACSL4, and inhibits LD biogenesis. In particular, ACSL3 is related to LD formation, and knockdown of ACSL3 alone significantly reduces the number of LDs (Kassan et al. 2013). Synthesized neutral lipids are accumulated between the leaflets of the ER bilayer, where they form a lens-like structure (a nascent LD) that grows and eventually becomes independent from the ER (Choudhary et al 2015) (Fig. 1).

LD biogenesis. Neutral lipids synthesized by ACATs and DGATs are accumulated into the phospholipid bilayer of the ER. After the regulation of the LD volume by lipin-1 and seipin, the LD buds off into the cytosol, as facilitated by FIT2
Other ER-localized proteins are also related to LD synthesis. Fat-storage-including transmembrane proteins 1 and 2 (FIT1 and FIT2) directly bind to triglyceride in vitro (Gross et al. 2011), and FIT1 and FIT2 overexpression in cultured cells increases triglyceride storage (Kadereit et al. 2008). Conversely, FIT2 knockdown in cultured adipocytes decreases triglyceride storage. Consistent with this, mice with FIT2 deleted in adipocytes show lipodystrophy (Miranda et al. 2014). Recently, it has been proposed that FIT2 is essential for the budding of LDs from the ER, because LDs remain in the ER in FIT2-depleted culture cells (Choudhary et al. 2015). Seipin is a transmembrane protein of the ER that localizes at the ER–LD contact sites and may be involved in LD size control (Wang et al. 2014). Lipin1, which generally localizes in the cytoplasm, also functions as a regulator of triglyceride synthesis via the generation of diacylglycerol (Reue and Zhang 2008). Interestingly, it has been observed that lipin-1 interacts with seipin (Sim et al. 2012). Despite these advances, details of the molecular mechanisms involved in LD biogenesis remain unclear.
Lipolysis of LDs via the ER–LD juncture
PLIN1 (called perilipin) is a major LD protein in adipocytes. Although perilipin normally binds to CGI-58, it is phosphorylated upon lipolysis stimulation and binds to the triglyceride lipase ATGL (called PNPLA2) instead of CGI-58, while CGI-58 binds to HSL. The ATGL/PLIN1 complex functions as a triglyceride lipase and the HSL/CGI-58 complex functions as a diacylglycerol lipase. These lipolysis processes in adipocytes occur on LDs.
On the other hand, the targeting of ATGL to LDs could be considered one of the most important processes involved in lipolysis in nonadipocytes, because PLIN1 is highly expressed in adipocytes while nonadipocytes express little PLIN1. The targeting of ATGL to LDs by COPI components and Arf1 regulates lipolysis (Guo et al. 2008) (Soni et al. 2009) (Suzuki et al. 2015). Two models have been proposed for this mechanism. One is a simple model in which COPI-coated vesicles originating in the Golgi and/or ER–Golgi intermediate compartment transport ATGL to LDs (Soni et al. 2009). In this case, LDs and vesicles should require a fusion system to transport ATGL, but such structures have not been reported. In the second model, the Arf1–COPI system acts directly on the LDs themselves to induce budding of small LDs called nanodroplets (Thiam et al. 2013) (Wilfling et al. 2014). The budding of nanodroplets from LDs should reduce the proportion of phospholipid in the LDs and hence increase the surface tension on the LD surfaces. This increased surface tension induces membrane bridges between the LDs and ER and recruits ATGL from the ER to the LDs (Wilfling et al. 2014) (Fig. 2). Interestingly, the targeting of ATGL to LDs is inhibited and the amount of triglycerides is increased in the knockdown of ELMOD2, which has an Arf1–GAP activity and is anchored on the LD by palmitoylation. Palmitoylated ELMOD2 may directly regulate the membrane-bridge-making activity of Arf1 on LDs (Suzuki et al. 2015).

ATGL dynamics from the ER to an LD via an ER–LD bridge. Nanodroplet budding prompted by activated Arf1 and COPI components subsequently leads to the generation of a bridge between the LD and the ER. The transfer of ATGL from the ER to the surface of the LD occurs via this bridge
Regulation of VLDL secretion in hepatocytes by the ER–LD juncture
In hepatocytes and enterocytes, neutral lipids are secreted as VLDL (very low density lipoprotein) and chylomicrons, respectively. Apolipoprotein B (ApoB) is a key molecule in the secretion of these lipoproteins by exocytosis. ApoB is lipidated by MTP at the luminal side of the ER for lipoprotein synthesis.
When the lipidation process is perturbed, nascent ApoB in the ER is subjected to the ER-associated degradation (ERAD) machinery; thus, nonlipidated ApoB is ubiquitinated, extracted from the Sec61 translocon, and degraded by the proteasome. Lipidated ApoB in the ER lumen is also degraded by the proteasome (Ohsaki et al. 2006). In hepatoma cells treated with proteasome inhibitor, lipidated ApoB is observed on the luminal side of LDs attached to the ER (Ohsaki et al. 2008). In this structure, the ER is extensively attached to LDs, forming the so-called ApoB-crescent structure (Fig. 3). The putative LD–ER bridge, Derlin-1, which is a component of the ERAD pathway, is located at the ER–LD juncture, and is thought to transport ApoB after lipidation from the ER lumen to the cytosol across the phospholipid bilayer. Ubiquitinated ApoB on the cytosolic side is captured by UBXD8 localized on the cytosolic surfaces of LDs in ApoB crescents via the ubiquitin-binding domain (UBA domain) at the N-terminus. UBXD8 is involved in the degradation of lipidated ApoB, and thereby plays a critical role in the regulation of lipid metabolism in hepatocytes (Suzuki et al. 2012; Imai et al. 2015). In UBXD8-knockdown hepatoma cells, the ApoB-crescent structure was increased and VLDL secretion was decreased (Suzuki et al. 2012). In accordance with these phenomena, a low VLDL serum level and facilitation of liver steatosis were observed in UBXD8-deficient mice on a high-fat diet (Imai et al. 2015).

Lipidated ApoB degradation via the ApoB-crescent structure. Lipidated ApoB accumulates in LDs attached to the ER lumen, forming the ApoB-crescent structure. The ApoB molecules are transferred from the luminal side of the ER to the cytosolic side through Derlin1. In the cytoplasm, the ApoB is ubiquitinated and binds to UBXD8 for degradation in the proteasome
Concluding remarks
ER is the major organelle for lipid metabolism, including neutral lipid synthesis and phospholipid synthesis. Phosphatidylcholine (PC) is a major phospholipid in the LD membrane. PC biosynthesis occurs via two pathways: the phosphatidylethanolamine N-methyltransferase (PEMT) pathway and the Kennedy pathway. In the PEMT pathway, the mitochondria associated membrane (MAM) is an important platform that transfers phosphatidylserine (PS) synthesized in the ER and phosphatidylethanolamine (PE) synthesized in mitochondria to facilitate PC synthesis. MAM was identified in electron microscopy studies over 50 years ago (Copeland and Dalton 1959). However, the functions of the MAM have only become clear in the last two decades due to technological advances in molecular biology (López-Crisosto et al. 2015). Similarly, there is increasing evidence that direct ER–LD interaction is important in LD metabolism. Morphological observations demonstrating physical organelle interactions may become more and more important for understanding their function.
References
Bickel PE, Tansey JT, Welte MA (2009) PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim Biophys Acta 1791(6):419–440. http://www.ncbi.nlm.nih.gov/pubmed/19375517. Accessed 4 Sep 2016
Choudhary V et al (2015) A conserved family of proteins facilitates nascent lipid droplet budding from the ER. J Cell Biol 211(2):261–71. http://www.ncbi.nlm.nih.gov/pubmed/26504167. Accessed 4 Sep 2016
Copeland DE, Dalton AJ (1959) An association between mitochondria and the endoplasmic reticulum in cells of the pseudobranch gland of a teleost. J Biophys Biochem Cytol 5(3):393–396. http://www.ncbi.nlm.nih.gov/pubmed/13664679. Accessed 4 Sep 2016
Gross DA, Zhan C, Silver DL (2011) Direct binding of triglyceride to fat storage-inducing transmembrane proteins 1 and 2 is important for lipid droplet formation. Proc Natl Acad Sci USA 108(49):19581–19586. http://www.ncbi.nlm.nih.gov/pubmed/22106267. Accessed 4 Sep 2016
Guo Y et al (2008) Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature 453(7195):657–661. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2734507&tool=pmcentrez&rendertype=abstract. Accessed 5 Aug 2011
Imai N et al (2015) Hepatocyte-specific depletion of ubxd8 induces periportal steatosis in mice fed a high-fat diet. PloS One 10(5):e0127114. http://www.ncbi.nlm.nih.gov/pubmed/25970332. Accessed 4 Sep 2016
Kadereit B et al (2008) Evolutionarily conserved gene family important for fat storage. Proc Natl Acad Sci USA 105(1):94–99. http://www.ncbi.nlm.nih.gov/pubmed/18160536. Accessed 4 Sep 2016
Kassan A et al (2013) Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. J Cell Biol 203(6):985–1001. http://www.ncbi.nlm.nih.gov/pubmed/24368806. Accessed 4 Sep 2016
Kory N, Farese RV, Walther TC (2016) Targeting fat: mechanisms of protein localization to lipid droplets. Trends Cell Biol 26(7):535–546. http://www.ncbi.nlm.nih.gov/pubmed/26995697. Accessed 4 Sep 2016
López-Crisosto C et al (2015) ER-to-mitochondria miscommunication and metabolic diseases. Biochim Biophys Acta 1852(10 Pt A):2096–2105. http://www.ncbi.nlm.nih.gov/pubmed/26171812. Accessed 4 Sep 2016
Miranda DA et al (2014) Fat storage-inducing transmembrane protein 2 is required for normal fat storage in adipose tissue. J Biol Chem 289(14):9560–9572. http://www.ncbi.nlm.nih.gov/pubmed/24519944. Accessed 4 Sep 2016
Ohsaki Y et al (2006) Cytoplasmic lipid droplets are sites of convergence of proteasomal and autophagic degradation of apolipoprotein B. Mol Biol Cell 17(6):2674–2683. http://www.ncbi.nlm.nih.gov/pubmed/16597703. Accessed 4 Sep 2016
Ohsaki Y et al (2008) Lipid droplets are arrested in the ER membrane by tight binding of lipidated apolipoprotein B-100. J Cell Sci 121(Pt 14):2415–2422. http://www.ncbi.nlm.nih.gov/pubmed/18577578. Accessed 12 Aug 2011
Reue K, Zhang P (2008) The lipin protein family: dual roles in lipid biosynthesis and gene expression. FEBS Lett 582(1):90–96. http://www.ncbi.nlm.nih.gov/pubmed/18023282. Accessed 4 Sep 2016
Sim MFM et al (2012) The human lipodystrophy protein seipin is an ER membrane adaptor for the adipogenic PA phosphatase lipin 1. Mol Metab 2(1):38–46. http://www.ncbi.nlm.nih.gov/pubmed/24024128. Accessed 4 Sep 2016
Soni KG et al (2009) Coatomer-dependent protein delivery to lipid droplets. J Cell Sci 122(Pt 11):1834–1841. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2684835&tool=pmcentrez&rendertype=abstract. Accessed 29 Sep 2011
Suzuki M et al (2012) Derlin-1 and UBXD8 are engaged in dislocation and degradation of lipidated ApoB-100 at lipid droplets. Mol Biol Cell 23(5):800–810. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3290640&tool=pmcentrez&rendertype=abstract. Accessed 7 Mar 2012
Suzuki M et al (2015) ELMOD2 is anchored to lipid droplets by palmitoylation and regulates adipocyte triglyceride lipase recruitment. Mol Biol Cell 26(12):2333–2342. http://www.ncbi.nlm.nih.gov/pubmed/25904333. Accessed 4 Sep 2016
Tauchi-Sato K et al (2002) The surface of lipid droplets is a phospholipid monolayer with a unique fatty acid composition. J Biol Chem 277(46):44507–44512. http://www.ncbi.nlm.nih.gov/pubmed/12221100. Accessed 4 Jun 2016
Thiam AR et al (2013) COPI buds 60-nm lipid droplets from reconstituted water–phospholipid–triacylglyceride interfaces, suggesting a tension clamp function. Proc Natl Acad Sci USA 110(33):13244–13249. http://www.ncbi.nlm.nih.gov/pubmed/23901109. Accessed 4 Sep 2016
Wang C-W, Miao Y-H, Chang Y-S (2014) Control of lipid droplet size in budding yeast requires the collaboration between Fld1 and Ldb16. J Cell Sci 127(Pt 6):1214–1228. http://www.ncbi.nlm.nih.gov/pubmed/24434579. Accessed 4 Sep 2016
Wilfling F et al (2014) Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting. Elife 3:e01607. http://www.ncbi.nlm.nih.gov/pubmed/24497546. Accessed 4 Sep 2016
Acknowledgements
I deeply thank Dr. Toyoshi Fujimoto (Nagoya University) for his assistance and mentorship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Rights and permissions
About this article

Cite this article
Suzuki, M. Regulation of lipid metabolism via a connection between the endoplasmic reticulum and lipid droplets. Anat Sci Int 92, 50–54 (2017). https://doi.org/10.1007/s12565-016-0378-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12565-016-0378-2