
Abstract
The lipid droplet (LD) is a highly dynamic organelle that maintains cellular lipid homeostasis in addition to storing energy sources. Current research suggests LDs are responsible for the transportation, storage and lipolysis-driven mobilization of lipids within cells. Here, we review the landscape of evidence for LD involvement in regulating lipid homeostasis. LD interactions with other organelles, particularly the endoplasmic reticulum, mitochondria, lysosomes (or vacuoles in yeast), and peroxisomes, highlight their importance for lipid transfer and metabolism.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
For many organisms, the most efficient form of energy storage is fat. At the cellular level, the ability to store neutral lipids in cytoplasmic lipid droplets (LD) is essential for cellular and organismal survival. LDs, also termed lipid bodies, oil bodies or adiposomes, are multi-functional organelle that consists of a core of neutral lipids, such as sterol esters (SE) and triacylglycerols (TAG), and a coating of a monolayer of phospholipids decorated by LD-associated proteins that are variable with organisms and tissues [1, 2].
LDs appear to be conserved, as they are found in nearly all organisms. The organelles have been isolated from many different organisms, including bacteria (Rhodococcus opacus and Rhodococcus ruber) [3], green algae (Chlamydomonas reinhardtii) [4], yeast (Saccharomyces cerevisiae) [5, 6], nematodes (Caenorhabditis elegans) [7], insects (Drosophila melanogaster) [8, 9], plants (Arabidopsis thaliana, Sesamum indicum L., Brassica napus) [10–12], fish (Ctenopharyngodon idella) [13], mammals such as mice, rat, macaque monkey and human cells [14–17]. LD-associated proteins have been investigated through the proteomic analysis of LDs isolated from the aforementioned species [3–17].
LDs are highly dynamic and are controlled by a constant cycle of recruitment and disassociation of proteins from the LD surface. As key players in lipid metabolism, LDs frequently communicate with other organelles via direct membrane contact sites or vesicle transport in response to stress responses and cellular signaling. In order to manage lipid storage and lipid homeostasis, LDs interact with organelles including endoplasmic reticulum (ER), mitochondria, lysosome, and peroxidases [18–20]. A large subset of the research into LD biogenesis and metabolism has been conducted in yeast. Research into LDs in human cells generally is focused on the role of LDs in human lipid-accumulation diseases. In this article, we review recent findings on the formation of LDs from the ER, followed by LD growth and breakdown. In particular, we focus on how LDs regulate lipid transport and metabolism in yeast and human cells.
Lipid droplet biogenesis
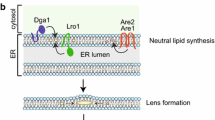
Most models suggest that LDs originate from the ER bilayer and then either remain connected to the ER, or bud off to become independent organelles [21]. ER membranes are often found close to LDs [22]. Enzymes (e.g. diacylglycerol O-acyltransferase [DGAT] and sterol O-acyltransferase [ACAT]) that synthesize neutral lipids for the cores of LDs are localized primarily in the ER [22]. Electron microscopy data reveal close apposition between LDs and the ER [23–25], further supporting the ER model of LD biogenesis. After budding off, LDs continue to maintain a relationship with the ER throughout their lifespan (Fig. 1).
The first step of ER biogenesis involves synthesis of neutral lipids. Neutral lipids are initially generated by enzymes in the ER lumen, and then disperse into the leaflets of the ER bilayer. As their concentration increases, neutral lipids eventually coalesce and form lens-like structures. The growing droplet progressively distends the ER membrane and eventually buds off into the cytoplasm to form a nascent LD [26–28].
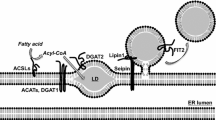
Nascent LDs, which may be connected to the ER or separated completely from it, reengage with the ER during expansion. In yeast, the lipid monolayer of LDs remains contiguous with the ER membrane [29]. The existence of ER–LD membranous bridges was supported by high-pressure freezing electron microscopy and tomography analyses, which showed direct connections between the LD phospholipid monolayer and the adjacent ER [24, 30]. Such bridges permit the exchange of the membrane proteins and lipids between the two compartments [29]. At the contact sites and membrane extensions, neutral lipids synthesized in the ER are transported to LDs [27, 31]. TG synthesis enzymes [e.g. glycerol-3-phosphate acyltransferase (GPAT4) and diacylglycerol O-acyltransferase 2 (DGAT2)] also partially or fully migrate from the ER to LDs via membrane bridges [24]. In higher order eukaryotes, a population of LDs eventually detaches from the ER, but may reattach later.
Re-attachment of LDs to the ER requires the COPI coatomer complex. The COPI machinery promotes the formation of membrane bridges between LDs and ER [32, 33]. The COPI complex is recruited to LDs by the small GTPase ARF1 and its guanine nucleotide exchange factor (GEF) GBF1, which promotes the budding of very small droplets called “nano-droplets” from the existing LDs. Nano-droplets remove phospholipids from the LD surface, which in turn increases LD surface tension and facilitates LD fusion with the ER bilayer. The mechanisms behind COPI facilitation of LD fusion to the ER remain under investigation. Upon fusion, integral membrane proteins such as DGAT2 and GPAT4 are able to diffuse from the ER to LDs, indicating the re-establishment of membrane bridges between the two organelles.
The second step of LD biogenesis is the recruitment of biogenesis factors such as seipin to the lens structure in order to facilitate the growth of the nascent LD. Seipin, an integral ER membrane protein, was originally identified in congenital generalized lipodystrophy [34] and has been increasingly identified as a major player in LD growth. Seipin is localized to ER–LD contact sites and may directly transfer neutral lipids and/or modulate phospholipid transfer at the contact site [35, 36]. Seipin may also act as a diffusion barrier for proteins between the ER and LDs [36]. In accordance with its proposed gatekeeper function, seipin deletion mutations lead to the abnormal relocalization of GPAT4 to LDs instead of the ER [35, 37]. Seipin is also necessary to maintain a population of appropriately sized LDs. Experimental depletion of seipin causes defects in LD morphology, ranging from the accumulation of small LDs with both heterogeneous size and shape, to the creation of supersized LDs [27, 38]. However, the exact cause of this LD size heterogeneity remains elusive. Another study suggested seipin could control the sites at which LDs develop, facilitate continuous delivery of triglycerides from the ER to LDs, and prevent their shrinkage by ripening [39]. Another important regulator of LD growth and maturation is Rab18, a Rab guanosine triphosphatase. Recent discoveries revealed the Rab18-NRZ-SNARE complex is critical for tethering ER–LD and establishing the ER–LD contact site to promote LD growth [40]. Finally, the ER-localized protein DFCP1 acts as a Rab18 effector for LD localization, and mediates LD expansion by interacting with the Rab18-ZW10 complex and thus controlling ER–LD contact formation [41]. Through the interactions of these proteins, the LD grows.
LDs may continue to grow by local synthesis of neutral lipids or via fusion with other LDs [42, 43]. The translocation of TAG synthetic enzymes GPAT4, AGPAT3 (1-acyl-sn-glycerol-3-phosphate acyltransferase 3), and DGAT2 to the LD is one way that LDs may grow in size [24]. Phospholipids are required to stabilize the LD neutral lipid core, which are replenished by CTP:phosphocholine cytidylyltransferase (CCT), a rate-limiting enzyme in the phospholipid synthesis pathway. Newly synthesized phosphatidylcholine (PC) may be transferred from ER to LDs by LD-ER conduits, but other pathways remain to be discovered. LD growth can also result from fusion of LDs, which rarely occurs under normal conditions [44]. However, LD fusion frequently occurs under conditions of PC deficiency or accumulation of phosphatidic acid (PA) [45, 46], which permit the exchange of lipids between LDs.
An additional pathway is found in adipocytes, where cell death-inducing DNA fragmentation factor-alpha-like effector (CIDE) proteins can induce droplet fusion [47]. LD–LD contact sites are established by CIDE proteins (e.g. CIDEC/Fsp27) [48, 49]. Gong et al. show that Fsp27 localizes to membrane contact sites between adjacent LDs, where the protein appears to form stable “contacts” between the LD and mediates net unidirectional transfer of neutral lipids to the larger LD [50]. Another possibility is that SNARE proteins mediate LD fusion [50], similar to their roles in the fusion of vesicle bilayers. Triacylglycerol transfer invariably occurs from the smaller LD to the larger LD, which is hypothesized to be driven by differences in internal pressure and by surface tension [48].

Steps in lipid droplet formation. Lipid droplets (LDs) emerge from the endoplasmic reticulum (ER). Step 1: Neutral lipids are synthesized and accumulate within the ER bilayer. Beyond a certain concentration, the neutral lipids demix and coalesce into a lens. Step 2: As the lens accumulates additional neutral lipids, the bilayer deforms and a nascent LD buds into the cytoplasm. Seipins, as an important LD biogenesis factor, are recruited to the lens structure and facilitate the growth of the nascent LD. The nascent LD may remain attached to the ER or separate completely. Step 3: LDs bud from the ER and grow through fusion or local lipid synthesis. In adipocytes, FSP27 is involved during fusion.
Lipid Droplet Breakdown
The breakdown of LDs is mediated by the degradation of neutral lipids under energetically demanding conditions. During nutrient stress, cells shift their metabolism from reliance on glucose metabolism to mitochondrial fatty acids (FAs) oxidation [51]. FAs are important sources of cellular energy after being released from neutral lipids. FAs transported intracellularly to mitochondria for oxidation and ATP generation. Cells use two primary mechanisms for mobilizing FAs: lipolysis of LDs, and autophagy of membrane-bound organelles (e.g. the ER) or LDs [52–56].
When cells are starved, lipolysis can mobilize the lipids inside the LD through LD-associated neutral lipases. Adipose triglyceride lipase (ATGL), hormone sensitive lipase (HSL), and monoglyceride lipase (MGL) directly hydrolyze triacylglycerols on the LD surface in response to the cellular metabolic status and thus liberate fatty acids (FA) for the mitochondria [51, 57]. ATGL catalyzes the first step of lipolysis by hydrolyzing TAG and generating free FA and DAG. HSL then hydrolyzes DAG, which creates a free FA and monoacylglycerol (MAG). Finally, MGL converts MAG to glycerol and a free FA. This pathway can be regulated at the level of lipase activity, fine-tuned by the cell to its current energetic requirements [58, 59].
Lipolysis is modulated by the interaction of lipases present at the surface of the LD, with the structural proteins that surround LD and with inhibitory proteins in the cytosol [57]. Perilipins, for example, were proposed to slow lipolysis by shielding the triacylglycerol core of LDs from lipases [60]. Perilipin 1 was first identified as a LD-coating protein in mammalian cells [22], and its phosphorylation controls lipase access and anchorage to LDs [61]. Its relative, perilipin 5, normally serves as a barrier to lipolysis by regulating oxidative LD hydrolysis and controlling local FA flux to protect mitochondria against excessive exposure to FA during physiological stress. Perilipin 5 also regulates lipolysis through its interactions with ATGL and its activator ABHD5 (1-acylglycerol-3-phosphate O-acyltransferase) [62].
A second mechanism for mobilizing FAs during starvation is autophagy. Autophagy is an evolutionarily conserved physiological process for the degradation of proteins and organelles in the lysosome or vacuole of the cell to maintain homeostasis [63]. LDs are either sequestered into the autophagosome (macrolipohagy), or directly engulfed into the lysosome/vacuole lumen (microlipophagy) [64]. Both prolonged and brief kiss-and-run interactions between LDs and lysosomes have been observed in mammalian cells [65–67]. Recent data implicate these contacts as sites for the degradation of perilipin 2 and perilipin 3 via chaperone-mediated autophagy (CMA) [66]. In addition to the activity of cytosolic lipases, FAs can be mobilized from LDs through macroautophagy in mammalian cells. During macroautophagy, also known as macrolipophagy, a LD is engulfed into an autophagosome that subsequently fuses with a lysosome, which will digest the LD and release free FAs that move into the cytoplasm [55]. Microlipophagy is the engulfment of a LD in an invaginated vacuole or lysosome. In yeast, the vacuole is major catabolic organelle equivalent to the metazoan lysosome. Recent studies highlight a complex interplay between autophagy and LDs: LDs are degraded by autophagy via lipophagy, but LDs have also been implicated in regulation of the autophagic process in mammals.
Although it may seem contradictory, the number of LDs increases during prolonged periods of nutrient deprivation. The raw lipid metabolites generated in autolysosomes appear to directly contribute to droplet production [51, 68]. Master growth regulator mTOR complex 1 (mTORC1) was reported to control autophagy-dependent LD biogenesis during starvation [51]. mTORC1-regulated autophagy degrades membranous organelles, releasing FAs that are selectively channeled by DGAT1 into new LDs during nutrient deprivation, which in turn protects mitochondria from lipotoxic disruption [51]. Under these conditions, the biogenesis of LDs is necessary to sequester FAs in TAG-rich LDs, preventing accumulation of acyl-carnitine and subsequent mitochondrial dysfunction. However, autophagy-dependent LD biogenesis may not be limited to nutrient-limited conditions, acting instead as a general protective response to high levels of autophagy. These findings underscore the high degree of crosstalk between autophagy, the ER, LDs, and mitochondria that maintains lipid and energy homeostasis. Recent evidence suggests that, upon sensing a decline in nutrients, yeast exhibit a “bloom” of LDs organized at the periphery of the nuclear ER–vacuole junction (NVJ) [69, 70]. The NVJ-tethering protein Mdm1p appears to play a critical role in this LD accumulation. Analogous ER–autolysosomal contacts may be important for lipid droplet biogenesis during nutrient deprivation in mammalian cells.
In order to avoid FA toxicity, LD autophagy is regulated and works in parallel with other pathways, such as the coordinate of FA influx into mitochondria with storage of excess FAs in LDs [51]. FAs liberated from digestion of LDs can be used for energy production via mitochondrial (metazoans) or peroxisomal (yeasts and plants) beta-oxidation [71]. LDs provide an “on demand” source of FAs that can be mobilized in response to fluctuations in nutrient abundance. The autophagy-dependent LDs clustered in close proximity to mitochondria and LD-mitochondrial contacts have been proposed to function as sites for FA transfer [51, 72]. Such spatial positioning of LDs and mitochondria ensures oxidative tissues that require high FA influx have sufficient supply for their energy demands. Data from transmission electron microscopy, fluorescence imaging, and reconstitution assays in yeast demonstrated that LDs physically contact mitochondria in vivo and in vitro, and other experiments have described protein interactions between LDs and mitochondria or peroxisomes [73]. While exploring how FAs move into mitochondria in starved cells, Rambold et al. found that FAs used LDs as their conduit for delivery to organelles [51], instead of lingering in the cytoplasm.
Perilipin 5 is a key player in establishing the lipid droplet-mitochondrial junction, and is highly expressed in oxidative tissues (liver, skeletal muscle, heart and brown adipose tissue) [74, 75]. Overexpression of perilipin 5 is sufficient to induce a dramatic recruitment of mitochondria to the periphery of LDs [74, 75]. Other experiments have suggested perilipin 5 recruits mitochondria to the LD surface through a C-terminal region and induces the physical contact between LD and mitochondria [75]. In addition to perilipin 5, the SNARE protein synaptosome associated protein 23 (SNAP23) was reported to mediate the interaction between LDs and mitochondria [76]. Knockdown of SNAP23 interrupts the interaction between mitochondria and LD [76].
Similar to LDs, peroxisomes are key to lipid and energy metabolism. In humans, most beta-oxidation of FAs occurs in mitochondria, but beta-oxidation of very-long-chain FAs and branched FA occurs in peroxisomes. Mice lacking peroxisomes accumulate enlarged LDs in the liver [77]. In yeast, peroxisomes are the sole sites of beta-oxidation. Delivery of lipases from peroxisomal extensions to LDs has been observed in Arabidopsis [78]. In yeast, peroxisome–droplet contacts are sites of FA transfer. Clear droplet–peroxisome contact sites have been observed in yeast undergoing stationary-phase growth [79]. In yeast exposed to FAs as their sole carbon source, LDs form prominent and stable associations with peroxisomes [79]. The LD membrane fuses with the outer phospholipid leaflet of the peroxisome bilayer, which may allow luminal peroxisome enzymes to directly access LD stored triacylglycerol or facilitate protein transfer [79]. In wild type cells, LD-peroxisome contacts may facilitate the coupling of lipolysis on the LD to FA oxidation in the peroxisome. LDs also sequester lipotoxic FAs through their incorporation into TAGs [68]. However, a portion of autophagy-released lipids is immediately re-esterified to form triacylglycerol, which is then packaged into new pools of LDs [51, 68].
The mechanism by which FAs are transported out of autolysosomes and trafficked to the ER for triacylglycerol synthesis is not known. Membrane continuity between ER and LDs could also account for the reverse process i.e. the back transport of lipids from LDs to the ER during lipolysis. Under these conditions, LD-localized lipases hydrolyze TAG and steryl esters to liberate free FA, DAG and free sterols. These lipid intermediates are water insoluble, but could be released from LDs into the connecting ER membrane and thus enter the pool of lipids that are synthesized de novo by the ER-localized lipid biosynthetic enzymes. Lipolysis of LDs also induces ER–LD contacts for back transport of phospholipids and droplet proteins to the ER [80]. During lipolysis, for example, the diacylglycerols and FAs generated are directly transferred to the ER for phospholipid synthesis [81]. Similarly, a sterol transfer protein that was identified on the surface of LDs mediated the transport of released sterols back to the ER [81]. Early biochemical studies in yeast demonstrated the transport of membrane-bound LD marker proteins from the ER membrane to the newly made LDs. The transport is reversible and under lipolytic conditions, LD-localized membrane proteins will relocate to the ER [29]. Moreover, bidirectional transport of lipids between the ER and LDs must occur under lipogenic conditions, when LDs form and grow, and during lipolysis, when LDs regress [29].
Lipid droplet–nuclear envelope contacts also exist. LDs have been observed inside the nucleus in mammalian cells [82] and in yeast [83]. In yeast, LDs have been shown to bud directly from the inner nuclear membrane [83], as the lipid monolayer of the nuclear LDs remained contiguous with the inner nuclear membrane. Furthermore, seipin localizes to the inner nuclear membrane and is known to be necessary for the proper formation of the membrane bridge. Nuclear LDs may provide lipids for use in nuclear envelope expansion, store specific proteins, or sequester transcriptional factors [84, 85].
Conclusion
After budding off from the ER, LDs can associate with most cellular organelles through membrane contact sites. These contacts between LDs and other organelles are highly dynamic and coupled to the cycles of LD expansion and shrinkage. LD biogenesis and degradation, as well as their interactions with other organelles, are critical to cellular metabolism and to buffer the levels of toxic lipid species. LDs facilitate the coordination and communication between different organelles and act as vital hubs of cellular metabolism. Multiple mechanisms are involved in LD-mediated intracellular lipid trafficking. In view of evolution, these interactions appear less complex in yeast and become multifaceted, reflecting in protein–protein and lipid–protein interactions to accomplish diverging cellular functions in human cells. Further studies are needed to show detailed mechanisms that LDs are involved in various aspects of cellular physiology and pathology in human lipid-accumulated diseases.
The authors declare that they have no conflict of interest.
References
Farese, R.V., Jr. and Walther, T.C., Lipid droplets finally get a little R-E-S-P-E-C-T, Cell, 2009, vol. 139, pp. 855–860. doi:10.1016/j.cell.2009.11.005
Murphy, D.J., The biogenesis and functions of lipid bodies in animals, plants and microorganisms, Progress in Lipid Research, 2001, vol. 40, pp. 325–438. doi: 10.1016/S0163-7827(01)00013-3
Kalscheuer, R., Wältermann, M., Alvarez, M., and Steinbüchel, A., Preparative isolation of lipid inclusions from Rhodococcus opacus and Rhodococcus ruber and identification of granule-associated proteins, Arch. Microbiol., 2001 vol. 177, pp. 20–28. doi: 10.1007/s00203-001-0355-5
Moellering, E. and Benning, C., RNA interference silencing of a major lipid droplet protein affects lipid droplet size in Chlamydomonas reinhardtii, Eukaryot Cell, 2010, vol. 9, pp. 97–106. doi: 10.1128/EC.00203-09
Athenstaedt, K., Zweytick, D., Jandrositz, A., Kohlwein, S., et al., Identification and characterization of major lipid particle proteins of the yeast Saccharomyces cerevisiae, J. Bacteriol., 1999, vol. 181, pp. 6441–6448. doi: 10.1128/JB.181.20.6441-6448.1999
Grillitsch, K., Connerth, M., Köfeler, H., Arrey, T., et al., Lipid particles/droplets of the yeast Saccharomyces cerevisiae revisited: lipidome meets proteome, Biochim. Biophys. Acta, 2011, vol. 1811, pp. 1165–1176. doi: 10.1016/j.bbalip.2011.07.015
Zhang, P., Na, H., Liu, Z., Zhang, S., et al., Proteomic study and marker protein identification of Caenorhabditis elegans lipid droplets, Mol. Cell. Proteomics, 2012, vol. 11(8), pp. 317–328. doi: 10.1074/mcp.M111.016345
Beller, M., Riedel, D., Jänsch, L., Dieterich, G., et al., Characterization of the Drosophila lipid droplet subproteome, Mol. Cell. Proteomics, 2006, vol. 5, pp. 1082–1094. doi: 10.1074/mcp.M600011-MCP200
Cermelli, S., Guo, Y., Gross, S., and Welte, M., The lipid-droplet proteome reveals that droplets are a protein-storage depot, Curr. Biol., 2006, vol. 16, pp. 1783–1795. doi: 10.1016/j.cub.2006.07.062
Jolivet, P., Roux, E., D’Andrea, S., Davanture, M., et al., Protein composition of oil bodies in Arabidopsis thaliana ecotype WS, Plant Physiol. Biochem., 2004, vol. 42, pp. 501–509. doi: 10.1016/j.plaphy.2004.04.006
Lin, L., Liao, P., Yang, H., and Tzen, J., Determination and analyses of the N-termini of oil-body proteins, steroleosin, caleosin and oleosin, Plant Physiol. Biochem., 2005, vol. 43, pp. 770–776. doi: 10.1016/j.plaphy.2005.07.008
Katavic, V., Agrawal, G., Hajduch, M., Harris, S., et al., Protein and lipid composition analysis of oil bodies from two Brassica napus cultivars, Proteomics, 2006, vol. 6, pp. 4586–4598. doi: 10.1002/pmic.200600020
Tian, J., Zhang, J., Yu, E., Sun, J., et al., Identification and analysis of lipid droplet-related proteome in the adipose tissue of grass carp (Ctenopharyngodon idella) under fed and starved conditions, Comp. Biochem. Physiol. Part D Genomics Proteomics, 2020, vol. 36, pp. 100710. doi: 10.1016/j.cbd.2020.100710
Zhang, H., Wang, Y., Li, Y., Yu, J., et al., Proteome of skeletal muscle lipid droplet reveals association with mitochondria and apolipoprotein a-I, J. Proteome Res., 2011, vol. 10, pp. 4757–4768. doi: 10.1021/pr200553c
Yu, J., Zhang, L., Li, Y., Zhu, X., et al., The adrenal lipid droplet is a new site for steroid hormone metabolism, Proteomics, 2018, vol. 18, p. e1800136. doi: 10.1002/pmic.201800136
Wan, H., Melo, R., Jin, Z., Dvorak, A., et al., Roles and origins of leukocyte lipid bodies: proteomic and ultrastructural studies, FASEB J, 2007, vol. 21, pp. 167–178. doi: 10.1096/fj.06-6711com
Fujimoto, Y., Itabe, H., Sakai, J., Makita, M., et al., Identification of major proteins in the lipid droplet-enriched fraction isolated from the human hepatocyte cell line HuH7, Biochim. Biophys. Acta, 2004, vol. 1644, pp. 47–59. doi: 10.1016/j.bbamcr.2003.10.018
Murphy, S., Martin, S., and Parton, R.G., Lipid droplet-organelle interactions; sharing the fats, Biochim. Biophys. Acta, 2009, vol. 1791, pp. 441–447. doi: 10.1016/j.bbalip.2008.07.004
Zehmer, J.K., Huang, Y., Peng, G., Pu, J., et al., A role for lipid droplets in inter-membrane lipid traffic, Proteomics, 2009, vol. 9, pp. 914–921. doi: 10.1002/pmic.200800584
Barbosa, A.D., Savage, D.B., and Siniossoglou, S., Lipid droplet-organelle interactions: emerging roles in lipid metabolism, Curr. Opin. Cell. Biol., 2015, vol. 35, pp. 91–97. doi: 10.1016/j.ceb.2015.04.017
Walther, T.C. and Farese, R.V., Jr., Lipid droplets and cellular lipid metabolism, Annu Rev. Biochem., 2012, vol. 81, pp. 687–714. doi: 10.1146/annurev-biochem-061009-102430
Blanchette-Mackie, E.J., Dwyer, N.K., Barber, T., Coxey, R.A., et al., Perilipin is located on the surface layer of intracellular lipid droplets in adipocytes, J. Lipid Res., 1995, vol. 36, pp. 1211–1226.
Robenek, H., Robenek, M.J., and Troyer, D., PAT family proteins pervade lipid droplet cores, J. Lipid Res., 2005, vol. 46, pp. 1331–1338. doi: 10.1194/jlr.M400323-JLR200
Wilfling, F., Wang, H., Haas, J.T., Krahmer, N., et al., Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets, Dev. Cell., 2013, vol. 24, pp. 384–399. doi: 10.1016/j.devcel.2013.01.013
Waltermann, M., Hinz, A., Robenek, H., Troyer, D., et al., Mechanism of lipid-body formation in prokaryotes: how bacteria fatten up, Mol. Microbiol., 2005, vol. 55, pp. 750–763. doi: 10.1111/j.1365-2958.2004.04441.x
Choudhary, V., Ojha, N., Golden, A., and Prinz, W.A., A conserved family of proteins facilitates nascent lipid droplet budding from the ER, J. Cell. Biol., 2015, vol. 211, pp. 261–271. doi: 10.1083/jcb.201505067
Walther, T.C., Chung, J., and Farese, R.V., Jr., Lipid Droplet Biogenesis, Annu Rev. Cell. Dev Biol, 2017, vol. 33, pp. 491–510. doi: 10.1146/annurev-cellbio-100616-060608
Qi, Y., Sun, L., and Yang, H., Lipid droplet growth and adipocyte development: mechanistically distinct processes connected by phospholipids, Biochim. Biophys. Acta Mol. Cell. Biol. Lipids, 2017, vol. 1862, pp. 1273–1283. doi: 10.1016/j.bbalip.2017.06.016
Jacquier, N., Choudhary, V., Mari, M., Toulmay, A., et al., Lipid droplets are functionally connected to the endoplasmic reticulum in Saccharomyces cerevisiae, J. Cell. Sci., 2011, vol. 124, pp. 2424–2437. doi: 10.1242/jcs.076836
Kassan, A., Herms, A., Fernandez-Vidal, A., Bosch, M., et al., Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains, J. Cell. Biol., 2013, vol. 203, pp. 985–1001. doi: 10.1083/jcb.201305142
Joshi, A.S., Zhang, H., and Prinz, W.A., Organelle biogenesis in the endoplasmic reticulum, Nat. Cell. Biol., 2017, vol. 19, pp. 876–882. doi: 10.1038/ncb3579
Thiam, A.R., Antonny, B., Wang, J., Delacotte, J., et al., COPI buds 60-nm lipid droplets from reconstituted water–phospholipid–triacylglyceride interfaces, suggesting a tension clamp function, Proc. Natl. Acad. Sci. USA, 2013, vol. 110, pp. 13244–13249. doi: 10.1073/pnas.1307685110
Wilfling, F., Thiam, A.R., Olarte, M.J., Wang, J., et al., Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting, Elife, 2014, vol. 3, pp. e01607. doi: 10.7554/eLife.01607
Magre, J., Delepine, M., Khallouf, E., Gedde-Dahl, T., Jr., et al., Identification of the gene altered in Berardinelli–Seip congenital lipodystrophy on chromosome 11q13, Nat. Genet., 2001, vol. 28, pp. 365–370. doi: 10.1038/ng585
Wang, H., Becuwe, M., Housden, B.E., Chitraju, C., et al., Seipin is required for converting nascent to mature lipid droplets, Elife, 2016, vol. 5, p. e16582. doi: 10.7554/eLife.16582
Grippa, A., Buxo, L., Mora, G., Funaya, C., et al., The seipin complex Fld1/Ldb16 stabilizes ER–lipid droplet contact sites, J. Cell. Biol., 2015, vol. 211, pp. 829–844. doi:10.1083/jcb.201502070
Pagac, M., Cooper, D.E., Qi, Y., Lukmantara, I.E., et al., SEIPIN regulates lipid droplet expansion and adipocyte development by modulating the activity of glycerol-3-phosphate acyltransferase, Cell. Rep., 2016, vol. 17, pp. 1546–1559. doi:10.1016/j.celrep.2016.10.037
Henne, W.M., Reese, M.L., and Goodman, J.M., The assembly of lipid droplets and their roles in challenged cells, Embo j., 2019, vol. 38, p. e98947. doi:10.15252/embj.2019101816
Salo, V.T., Li, S., Vihinen, H., Holtta-Vuori, M., et al., Seipin facilitates triglyceride flow to lipid droplet and counteracts droplet ripening via endoplasmic reticulum contact, Dev. Cell., 2019, vol. 50, pp. 478–493.e479. doi: 10.1016/j.devcel.2019.05.016
Xu, D., Li, Y., Wu, L., Li, Y., et al., Rab18 promotes lipid droplet (LD) growth by tethering the ER to LDs through SNARE and NRZ interactions, J. Cell. Biol., 2018, vol. 217, pp. 975–995. doi: 10.1083/jcb.201704184
Li, D., Zhao, Y.G., Li, D., Zhao, H., et al., The ER-localized protein DFCP1 modulates ER–lipid droplet contact formation, Cell. Rep., 2019, vol. 27, pp. 343–358.e345. doi: 10.1016/j.celrep.2019.03.025
Wilfling, F., Haas, J.T., Walther, T.C., and Farese, R.V., Jr., Lipid droplet biogenesis, Curr. Opin. Cell. Biol., 2014, vol. 29, pp. 39–45. doi: 10.1016/j.ceb.2014.03.008
Yu, J. and Li, P., The size matters: regulation of lipid storage by lipid droplet dynamics, Sci. China Life. Sci., 2017, vol. 60, pp. 46–56. doi: 10.1007/s11427-016-0322-x
Murphy, S., Martin, S., and Parton, R.G., Quantitative analysis of lipid droplet fusion: inefficient steady state fusion but rapid stimulation by chemical fusogens, PLoS One, 2010, vol. 5, p. e15030. doi: 10.1371/journal.pone.0015030
Fei, W., Shui, G., Zhang, Y., Krahmer, N., et al., A role for phosphatidic acid in the formation of “supersized” lipid droplets, PLoS Genet., 2011, vol. 7, pp. e1002201. doi: 10.1371/journal.pgen.1002201
Guo, Y., Walther, T.C., Rao, M., Stuurman, N., et al., Functional genomic screen reveals genes involved in lipid-droplet formation and utilization, Nature, 2008, vol. 453, pp. 657–661. doi: 10.1038/nature06928
Xu, W., Wu, L., Yu, M., Chen, F.J., et al., Differential roles of cell death-inducing DNA fragmentation factor-alpha-like effector (CIDE) proteins in promoting lipid droplet fusion and growth in subpopulations of hepatocytes, J. Biol. Chem., 2016, vol. 291, pp. 4282–4293. doi:10.1074/jbc.M115.701094
Gong, J., Sun, Z., Wu, L., Xu, W., et al., Fsp27 promotes lipid droplet growth by lipid exchange and transfer at lipid droplet contact sites, J. Cell. Biol., 2011, vol. 195, pp. 953–963. doi:10.1083/jcb.201104142
Jambunathan, S., Yin, J., Khan, W., Tamori, Y., et al., FSP27 promotes lipid droplet clustering and then fusion to regulate triglyceride accumulation, PLoS One, 2011, vol. 6, pp. e28614. doi:10.1371/journal.pone.0028614
Bostrom, P., Andersson, L., Rutberg, M., Perman, J., et al., SNARE proteins mediate fusion between cytosolic lipid droplets and are implicated in insulin sensitivity, Nat. Cell. Biol., 2007, vol. 9, pp. 1286–1293. doi:10.1038/ncb1648
Rambold, A.S., Cohen, S., and Lippincott-Schwartz, J., Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics, Dev. Cell., 2015, vol. 32, pp. 678–692. doi:10.1016/j.devcel.2015.01.029
Axe, E.L., Walker, S.A., Manifava, M., Chandra, P., et al., Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum, J. Cell. Biol., 2008, vol. 182, pp. 685–701. doi:10.1083/jcb.200803137
Hayashi-Nishino, M., Fujita, N., Noda, T., Yamaguchi, A., et al., A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation, Nat. Cell. Biol., 2009, vol. 11, pp. 1433–1437. doi:10.1038/ncb1991
Kristensen, A.R., Schandorff, S., Hoyer-Hansen, M., Nielsen, M.O., et al., Ordered organelle degradation during starvation-induced autophagy, Mol. Cell. Proteomics, 2008, vol. 7, pp. 2419–2428. doi:10.1074/mcp.M800184-MCP200
Singh, R., Kaushik, S., Wang, Y., Xiang, Y., et al., Autophagy regulates lipid metabolism, Nature, 2009, vol. 458, pp. 1131–1135. doi:10.1038/nature07976
Yla-Anttila, P., Vihinen, H., Jokitalo, E., and Eskelinen, E.L., 3D tomography reveals connections between the phagophore and endoplasmic reticulum, Autophagy, 2009, vol. 5, pp. 1180–1185. doi:10.4161/auto.5.8.10274
Lass, A., Zimmermann, R., Oberer, M., and Zechner, R., Lipolysis—a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores, Prog. Lipid Res., 2011, vol. 50, pp. 14–27. doi:10.1016/j.plipres.2010.10.004
Wang, S., Soni, K.G., Semache, M., Casavant, S., et al., Lipolysis and the integrated physiology of lipid energy metabolism, Mol. Genet. Metab., 2008, vol. 95, pp. 117–126. doi:10.1016/j.ymgme.2008.06.012
Zechner, R., Zimmermann, R., Eichmann, T.O., Kohlwein, S.D., et al., FAT SIGNALS—lipases and lipolysis in lipid metabolism and signaling, Cell. Metab., 2012, vol. 15, pp. 279–291. doi:10.1016/j.cmet.2011.12.018
Brasaemle, D.L., Rubin, B., Harten, I.A., Gruia-Gray, J., et al., Perilipin A increases triacylglycerol storage by decreasing the rate of triacylglycerol hydrolysis, J. Biol. Chem., 2000, vol. 275, pp. 38486–38493. doi:10.1074/jbc.M007322200
Londos, C., Brasaemle, D.L., Schultz, C.J., Adler-Wailes, D.C., et al., On the control of lipolysis in adipocytes, Ann. NY Acad. Sci., 1999, vol. 892, pp. 155–168. doi:10.1111/j.1749-6632.1999.tb07794.x
Granneman, J.G., Moore, H.P., Mottillo, E.P., Zhu, Z., et al., Interactions of perilipin-5 (Plin5) with adipose triglyceride lipase, J. Biol. Chem., 2011, vol. 286, pp. 5126–5135. doi:10.1074/jbc.M110.180711
Weidberg, H., Shvets, E., and Elazar, Z., Biogenesis and cargo selectivity of autophagosomes, Annu. Rev. Biochem., 2011, vol. 80, pp. 125–156. doi:10.1146/annurev-biochem-052709-094552
Zechner, R., Madeo, F., and Kratky, D., Cytosolic lipolysis and lipophagy: two sides of the same coin, Nat. Rev. Mol. Cell. Biol., 2017, vol. 18, pp. 671–684. doi:10.1038/nrm.2017.76
Valm, A.M., Cohen, S., Legant, W.R., Melunis, J., et al., Applying systems-level spectral imaging and analysis to reveal the organelle interactome, Nature, 2017, vol. 546, pp. 162–167. doi:10.1038/nature22369
Kaushik, S. and Cuervo, A.M., Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis, Nat. Cell. Biol., 2015, vol. 17, pp. 759–770. doi:10.1038/ncb3166
Schroeder, B., Schulze, R.J., Weller, S.G., Sletten, A.C., et al., The small GTPase Rab7 as a central regulator of hepatocellular lipophagy, Hepatology, 2015, vol. 61, pp. 1896–1907. doi:10.1002/hep.27667
Nguyen, T.B., Louie, S.M., Daniele, J.R., Tran, Q., et al., DGAT1-dependent lipid droplet biogenesis protects mitochondrial function during starvation-induced autophagy, Dev. Cell., 2017, vol. 42, pp. 9–21. e25. doi:10.1016/j.devcel.2017.06.003
Barbosa, A.D. and Siniossoglou, S., Spatial distribution of lipid droplets during starvation: Implications for lipophagy, Commun. Integr. Biol., 2016, vol. 9, p. e1183854. doi:10.1080/19420889.2016.1183854
Hariri, H., Rogers, S., Ugrankar, R., Liu, Y.L., et al., Lipid droplet biogenesis is spatially coordinated at ER-vacuole contacts under nutritional stress, EMBO Rep., 2018, vol. 19, pp. 57–72. doi:10.15252/embr.201744815
Finn, P.F. and Dice, J.F., Proteolytic and lipolytic responses to starvation, Nutrition, 2006, vol. 22, pp. 830–844. doi:10.1016/j.nut.2006.04.008
Herms, A., Bosch, M., Reddy, B.J., Schieber, N.L., et al., AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation, Nat. Commun., 2015, vol. 6, pp. 7176. doi:10.1038/ncomms8176
Pu, J., Ha, C.W., Zhang, S., Jung, J.P., et al., Interactomic study on interaction between lipid droplets and mitochondria, Protein Cell, 2011, vol. 2, pp. 487–496. doi:10.1007/s13238-011-1061-y
Benador, I.Y., Veliova, M., Mahdaviani, K., Petcherski, A., et al., Mitochondria bound to lipid droplets have unique bioenergetics, composition, and dynamics that support lipid droplet expansion, Cell Metab., 2018, vol. 27, pp. 869–885. e6. doi:10.1016/j.cmet.2018.03.003
Wang, H., Sreenivasan, U., Hu, H., Saladino, A., et al., Perilipin 5, a lipid droplet associated protein, provides physical and metabolic linkage to mitochondria, J. Lipid Res., 2011, vol. 52, pp. 2159–2168. doi:10.1194/jlr.M017939
Jagerstrom, S., Polesie, S., Wickstrom, Y., Johansson, B.R., et al., Lipid droplets interact with mitochondria using SNAP23, Cell. Biol. Int., 2009, vol. 33, pp. 934–940. doi:10.1016/j.cellbi.2009.06.011
Dirkx, R., Vanhorebeek, I., Martens, K., Schad, A., et al., Absence of peroxisomes in mouse hepatocytes causes mitochondrial and ER abnormalities, Hepatology, 2005, vol. 41, pp. 868–878. doi:10.1002/hep.20628
Thazar-Poulot, N., Miquel, M., Fobis-Loisy, I., and Gaude, T., Peroxisome extensions deliver the Arabidopsis SDP1 lipase to oil bodies, Proc. Natl. Acad. Sci. USA, 2015, vol. 112, pp. 4158–4163. doi:10.1073/pnas.1403322112
Binns, D., Januszewski, T., Chen, Y., Hill, J., et al., An intimate collaboration between peroxisomes and lipid bodies, J. Cell. Biol., 2006, vol. 173, pp. 719–731. doi:10.1083/jcb.200511125
Kory, N., Farese, R.V., Jr., and Walther, T.C., Targeting Fat: Mechanisms of protein localization to lipid droplets, Trends Cell. Biol., 2016, vol. 26, pp. 535–546. doi:10.1016/j.tcb.2016.02.007
Hynynen, R., Suchanek, M., Spandl, J., Back, N., et al., OSBP-related protein 2 is a sterol receptor on lipid droplets that regulates the metabolism of neutral lipids, J. Lipid Res., 2009, vol. 50, pp. 1305–1315. doi:10.1194/jlr.M800661-JLR200
Ohsaki, Y., Kawai, T., Yoshikawa, Y., Cheng, J., et al., PML isoform II plays a critical role in nuclear lipid droplet formation, J. Cell. Biol., 2016, vol. 212, pp. 29–38. doi:10.1083/jcb.201507122
Romanauska, A. and Kohler, A., The inner nuclear membrane is a metabolically active territory that generates nuclear lipid droplets, Cell, 2018, vol. 174, pp. 700–715. e718. doi:10.1016/j.cell.2018.05.047
Farese, R.V., Jr. and Walther, T.C., Lipid droplets go nuclear, J. Cell. Biol., 2016, vol. 212, pp. 7–8. doi:10.1083/jcb.201512056
Welte, M.A., Expanding roles for lipid droplets, Curr. Biol., 2015, vol. 25, pp. R470–481. doi:10.1016/j.cub.2015.04.004
Funding
This work was supported by the Natural Science Foundation of Guangdong Province, China (Grant Number 202015150224).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Huang, J., Chen, X., Zhang, F. et al. Lipid Droplet Metabolism Across Eukaryotes: Evidence from Yeast to Humans. J Evol Biochem Phys 56, 396–405 (2020). https://doi.org/10.1134/S0022093020050026
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0022093020050026