
Abstract
Cancer can be identified as a chaotic cell state, which breaks the rules that govern growth and reproduction, with main characteristics such as uncontrolled division, invading other tissues, usurping resources, and eventually killing its host. It was once believed that cancer is caused by a progressive series of genetic aberrations, and certain mutations of genes, including oncogenes and tumor suppressor genes, have been identified as the cause of cancer. However, piling evidence suggests that epigenetic modifications working in concert with genetic mechanisms to regulate transcriptional activity are dysregulated in many diseases, including cancer. Cancer epigenetics explain a wide range of heritable changes in gene expression, which do not come from any alteration in DNA sequences. Aberrant DNA methylation, histone modifications, and expression of long non-coding RNAs (lncRNAs) are key epigenetic mechanisms associated with tumor initiation, cancer progression, and metastasis. Within the past decade, cancer epigenetics have enabled us to develop novel biomarkers and therapeutic target for many types of cancers. In this review, we will summarize the major epigenetic changes involved in cancer biology along with clinical and preclinical results developed as novel cancer therapeutics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
According to the Centers for Disease Control and Prevention (CDC), cancer is the second leading cause of death in the top ten diseases, next to heart disease (Heron et al. 2012). Although we have accumulated vast knowledge about cancer, the statistics show that we are still far from overcoming cancer. What makes cancer so hard to overcome and how much do we know about cancer? Until early 2000s, cancer was considered as a set of diseases caused by the accumulation of genetic mutations that control normal cellular homeostasis (Vogelstein et al. 2013). Oncogenes and tumor suppressor genes (TSGs) are the most well-known classes of genes implicated in cancer (Zhu et al. 2015). Proto-oncogenes, which normally help to regulate cell growth or differentiation, can become oncogenic by genetic mutation. Point mutation, chromosomal mutation, or copy number variation can lead to oncogene activation through amplified expression or gain-of-function from protein structural rearrangement. Translocation of the Philadelphia (Ph) chromosome in chronic myeloid leukemia (CML) was discovered in 1960 (Nowell 2007). Translocation of proto-oncogene ABL at 9q34 to BCR on chromosome 22 can produce a fusion gene called BCR-ABL1, coding for a hybrid oncoprotein (Rowley 2001; Imbach 2014). BCR-ABL1 fusion oncoprotein is a constitutively active tyrosine kinase signaling protein, causing the cell to divide uncontrollably and therefore develop CML. Ras mutation is another most well-known gain-of-function mutation identified in human cancer (Bos 1989; Fernández-Medarde and Santos 2011; Prior et al. 2012). RAS proteins (KRAS, NRAS, and HRAS) function as GDP–GTP-regulated binary on–off switches, which regulate cytoplasmic signaling networks that are responsible for proliferation and cell survival (Bos 1989). Mutation of RAS proteins at 12, 13, or 61 codon enhances the binding of GTP to the Ras protein, resulting in constitutive activation of Ras, which is associated with hyperproliferative developmental disorders and cancer. Among three isoforms, K-Ras has been shown to be the most frequently mutated isoform in most cancers. K-Ras gene is found to be mutated in 22% of all tumors, especially 90% of pancreatic tumors (Forbes et al. 2011).
In contrast to oncogenes that are activated mainly by gain-of-function mutations, tumor suppressors lose their functions (loss-of-function mutation) through deletions or point mutations. The retinoblastoma protein (RB) is a tumor suppressor protein, which mutation was originally identified in a rare childhood cancer retinoblastoma (Knudson 1971). Rb can suppress cellular proliferation by regulating the E2F transcription factor, and the Rb/E2F pathway plays a critical role in the initiation of DNA replication (Nevins 2001). Later studies have identified complex molecular functions of Rb through interactions with various proteins, and the Rb/E2F pathway was found to be functionally inactivated in virtually all human cancers (Chinnam and Goodrich 2011; Dyson 2016).
The functions of proto-oncogene proteins are to enhance cell division or inhibit cell death, while the functions of tumor suppressors are normally to prevent cell division or cause cell death. Therefore, either gain-of-function mutations of proto-oncogenes or loss-of-function mutations of tumor suppressors could initiate cancer through uncontrolled cell growth and defective apoptosis (Zhu et al. 2015). After the Human Genome Project (HGP) was completed, we achieved a great deal in human genetics, and it became the starting point for human genomics (Gonzaga 2012; Hood and Rowen 2013). Moreover, large-scale cancer genome projects, such as The Cancer Genome Atlas (TCGA), the Wellcome Trust Sanger Institute’s Cancer Genome Project, and the International Cancer Genome Consortium (ICGC), have shed light on cancer genomics. In addition, somatic mutations from thousands of tumors have provided insights into cancer development processes along with available therapeutic targets for cancer (McLendon et al. 2008; Hudson et al. 2010; Pleasance et al. 2010).
Achievements of HGP and other big studies have been powerful; however, the sequence itself does not explain how the genome is packaged into chromatin and provide differential expression of genes for proliferation, development, and differentiation. Therefore, the current paradigm to explain cancer development has now expanded to cancer genetics and epigenetics. While cancer genetics focus on abnormal gene expression, including altered protein expression by either deletion or amplification mutations, cancer epigenetics focus on the regulation of gene expression without changing the genome sequence. Altered gene expression in cancer through epigenetic pathways is very complex and is determined by chromatin structure changes, including DNA methylation, histone variants and various modifications, nucleosome remodeling, and small non-coding RNAs (Dawson and Kouzarides 2012). This review highlights the basic principles of epigenetic pathways involved in cancer development along with recent progress in clinical and preclinical studies targeting cancer epigenetics.
DNA methylation
Epigenetic control is the way to determine which genes should be turned on or off for normal development and in response to the environment. They are mostly regulated by groups of proteins called ‘epigenetic writers’, ‘epigenetic readers’, and ‘epigenetic erasers.’ The writer is the enzyme that creates modifications around the genome. This change is recognized by the reader. Finally, when the epigenetic change is no longer needed, erasers can remove it.
DNA methylation was the first epigenetic modification found in humans in the early 1980s (Cooper 1983; Doerfler 1983). DNA methylation occurs in cytosines of CpG (Cytosine-phosphate-Guanine) dinucleotide sequences to create 5-methylcytosine (5mC), which is catalyzed by DNA methyltransferases (DNMTs) using S-adenyl methionine (SAM) as the methyl donor. Promoter regions containing higher GC content are called CpG islands (CGIs). Hypermethylation of CGIs occurs in heterochromatin regions, while hypomethylation commonly occurs in actively expressed genes (Ohm et al. 2007; Meissner et al. 2008). DNA methylation of CGIs can be found at many different locations within the genome, including centromeres, telomeres, and inactive X-chromosomes (Vera et al. 2008; Pasque et al. 2018; Skakkebæk et al. 2018). There are three identified DNMT enzymes, which are DNMT1, DNMT3A, and DNMT3B. DNMT3A and DNMT3B are de novo methyltransferases that are responsible for the initial CpG methylation during embryogenesis (Okano et al. 1998). DNMT1 maintains the methylation pattern during chromosome replication by preferential methylation on hemimethylated CpGs. After CpG methylation, 5mC can become a platform for several methyl-CpG-binding domain (MBD) proteins, such as MBD1, MBD2, MBD3, MBD4, and MeCP2, for further chromatin-templated processes (Mashimo et al. 2013). There are other MBD-containing proteins, such as MBD5/6, SETDB1/2, and BAZ2A/B. The MBD proteins cooperate with other epigenetic proteins like histone modifying enzymes or chromatin remodeling complexes at the 5mC region and facilitate transcriptional repression (Du et al. 2015a).
Although direct removal of DNA methylation has not been detected so far, there are a few ways to remove DNA methylation. First, passive DNA demethylation through steady dilution of methylation patterns can happen by replication (Kriukiene et al. 2012). Secondly, ten–eleven translocation (TET 1–3) enzymes can oxidize 5mCs to create 5-hydroxymethylcytosine (5hmC), and subsequently formyl-(5-fc) and carboxyl-(5caC) derivatives are formed. The derivatives finally can be excised by the DNA repair protein thymine glycosylase (TDG) to be replaced by unmodified cytosine via the base excision repair (BER) pathway (Kohli and Zhang 2013).
Aberrant DNA methylation patterns, both hyper- and hypo-methylation, have been reported in many different types of cancer, including prostate, breast, gastric, liver, lung, glioblastoma, and leukemia (Sun et al. 2010; Barbano et al. 2013; Chao et al. 2013; Mehta et al. 2015; Liu and Brenner 2016; Cecotka and Polanska 2018; Klughammer et al. 2018). First cancer implication was the global hypomethylation at CpG sites of DNA repetitive elements identified in tumor cells (Bedford and van Helden 1987; Lin et al. 2001). Loss of imprinting at the insulin-like growth factor 2 (IGF2) gene locus is frequently observed in cancer and is provided as a colon cancer diagnosis (Cui et al. 2002). Conversely, hypermethylation of specific genes have also been identified to explain the role of DNMTs in tumorigenesis. Hypermethylation of CpG islands in TSG promoters, including Braca1, Rb, or p53 promoters, leads to inactivation of each protein and can enhance cancer development (Rideout et al. 1991; Sakai et al. 1991; Baldwin et al. 2000). Alteration of normal DNA methylation has been well profiled for over 25 years of epigenetic studies and provides its application for diagnostic and therapeutic targets (Heyn and Esteller 2012). Although the exact cause of deregulated DNA methylation patterns in cancer is not yet well established, an accumulation of data has shown that either mutation or overexpression of DNMT proteins and MBD protein is correlated with tumorigenesis (Du et al. 2015b; Spencer et al. 2017). In addition, several reports have emerged that mutations of TET family genes were found in numerous hematological malignancies (Cimmino et al. 2011; Nakajima and Kunimoto 2014).
Targeting of aberrant DNA methylation patterns has been attempted, and two cytidine analogs, 5-azacytidine/vidaza (AZA) and 5-aza-2′-deoxycytidine/dacogen (DAC), have been approved for the treatment of myelodysplastic syndromes (MDS) by the FDA (Raj and Mufti 2006; Santos et al. 2010). These two compounds form an irreversible covalent complex with DNMT1 and trigger proteasome-mediated DNMT1 degradation. Second-generation analog guadecitabine (SGI-110), which is an active metabolite of decitabine, is being tested in clinical trial for MDS and acute myeloid leukemia (AML) (Kantarjian et al. 2017). Although the role of the TET family in several cancers has been suggested from recent studies, a TET protein inhibitor has yet to be tested for cancer treatment.
Writers, readers, and eraser enzymes for DNA methylation and inhibitors are summarized in Table 1.
Histone modification-lysine acetylation
DNA within eukaryotic cells is packaged as chromatin, and the histone octamer is the central component of the nucleosomal subunit. The histone subunit in the nucleosome possesses a characteristic tail, which contains specific amino acid residues for covalent posttranslational modifications (PTMs), such as acetylation, methylation, phosphorylation, ubiquitylation, sumoylation, or ADP ribosylation. Each epigenetic PTM cooperates to regulate chromatin states.
Histone acetylation is crucial for active gene transcription to influence the compaction state of chromatin by neutralizing basic charges on unmodified lysine residues, decreasing the electrostatic interaction between negatively charged DNA and histones. Histone acetylation occurs on the lysine residue, balanced by two enzymes: histone acetyltransferase (HAT) and histone deacetylases (HDAC). There are primarily three families of HAT enzymes, including GNAT family (Gcn5, PCAF, Hat1), MYST family (MOZ/Morf, Ybf2, Sas2, Tip60), and CBP/P300 family (p300/CBP, Taf1) (Marmorstein and Roth 2001). These enzymes are also known to acetylate hundreds of other proteins besides histones, such as p53, sTAT3, GATA, etc., and have numerous biological functions, including regulation of protein stability, DNA binding affinity, and protein interactions (Spange et al. 2009). As epigenetic erasers, 18 HDAC isoforms have been identified in humans. Class I (HDACs 1, 2, 3, 8), Class IIa (HDACs 4, 5, 7, 9), Class IIb (HDACs 6, 10), and Class IV (HDAC11) are classical HDAC families that require a zinc ion (Zn2+) for their actions, whereas Class III HDACs (SIRT1 to 7) require NAD+ and are Zn2+-independent (Zhang et al. 2015). Aberrant histone lysine acetylation patterns, especially loss of histone H4 lysine (K) 16 acetylation, have been reported as a common hallmark of human cancer (Fraga et al. 2005). There are numerous reports showing involvement of HAT mutation or loss-of-function with many diseases, including cancer. Truncation mutations and in-frame insertion mutations of EP300 have been identified in several different cancers (Gayther et al. 2000). Further, it has been reported that the genes for p300, CBP, MOZ, and MORF are rearranged in recurrent leukemia-associated chromosomal abnormalities (Yang 2004). Although involvement of dysregulated HAT in many diseases is becoming clear, clinical application of the HAT inhibitor was not successful.
In addition to histone deacetylation, HDACs have other roles in association with several transcription factors, tumor suppressors, and oncogenes. For example, HDAC1 forms a complex with Rb and E2F transcription factors and regulates gene expression of the cell cycle (Brehm et al. 1998; Kennedy et al. 2001). Moreover, increased expression of HDAC family proteins has been observed in many cancers, including B cell acute lymphoblastic leukemia (ALL) and T cell ALL, indicating the role of histone acetylation in various leukemogenesis (Moreno et al. 2010; Tao et al. 2013). Although HAT inhibitors were not clinically successful, HDACs have become great targets for anticancer agents. Five classes of compounds—(I) hydroxamic acids; (II) short chain fatty acids; (III) benzamides; (IV) cyclic tetrapeptides; and (V) sirtuin inhibitors—are currently developed as anticancer reagent, and they are either isoform-selective or pan-inhibitors. Among hydroxamates, SAHA, Belinostat and Panobinostat are approved for T cell lymphoma. Romidespsin is a cyclicpeptide HDAC inhibitor which is approved for cutaneous T cell lymphoma (CTCL) and peripheral T-cell lymphoma (PTCL). The short chain fatty acid, Valproic acid, is approved for epilepsy. Many other classes of HDAC inhibitors are in different clinical stages for various cancers (Eckschlager et al. 2017).
Acetylated histone can serve as a binding site for regulatory factors, chromatin-remodeling complexes, and especially for bromodomain-containing proteins, which are known as histone acetylation readers. The human genome encodes 61 bromodomains present in 46 proteins, which are HATs, histone methyltransferases (HMTs), and transcription initiation factors. Among these BRD proteins, Bromodomain and extra-terminal (BET) proteins (BRD2, BRD3, BRD4, BRDT) are highly associated with several types of cancer (Padmanabhan et al. 2016). Small molecule triazolodiazepine-based inhibitors of the BET bromodomain, JQ1 and I-BET, were first developed (Pérez-Salvia and Esteller 2017). They selectively bind to bromodomains BD1 and BD2 of the BET family. BET inhibitors have shown great efficacy against human and murine MLL-fusion leukemic cell lines and mouse leukemia models. From further mechanistic studies, BET inhibition has been shown to suppress cancer through inhibition of Myc expression, targeting JAK-STAT, NF-κB pathway, and p53 acetylation (Chan et al. 2015; Huang et al. 2017; Xu and Vakoc 2017). Currently, many different BET inhibitors are in clinical trial phase I or II for various different types of cancers. Enzymes for histone acetylation, deacetylation, and readers are summarized in Table 2.
Histone modification-lysine and arginine methylation
Another well-known histone modification is histone methylation on arginine and lysine residues. Different from histone acetylation, methylation does not change the physical interaction between DNA and histone by neutralizing the histone charge. Further, methylation of specific lysine or arginine residues refers to either an active or repressive gene expression. Lysine methylation can exist in a mono-, di-, or tri-methylated state, implying the complexity of the regulatory mechanisms. Generally, H3 lysine 4 (H3K4), H3K36, and H3K79 methylation is correlated with active gene expression, while di- and tri-methylation of H3K9, H3K27, and H3K20 are linked to gene repression (Vermeulen et al. 2010).
Similar to other epigenetic modifications, histone methylation is also regulated by writer (lysine methyltransferases: KMTs), reader, and eraser (lysine demethylases: KDMs) proteins. KMTs are comprised of 51 SET (Su (var)3-9, Enhancer of Zeste, Trithorax) domain KMTs and one non-SET domain lysine HMT, known as DOT1L (Qian and Zhou 2006). DOT1L contains a catalytic domain, which is structurally related to the domains of protein arginine methyltransferases (Nguyen and Zhang 2011). SET-domain proteins transfer a methyl group from S-adenosyl-l-methionine (SAM) to the amino group of a lysine residue on the histone or other protein, leaving a methylated lysine residue and the cofactor byproduct S-adenosyl-l-homocysteine (SAH). Most KMTs can methylate several non-histone proteins, including p53, PCNA, STAT3, RARα, E2F1, FOXO3, DNMT1, and KMT1c (Moore and Gozani 2014). KDMs are comprised of two families of proteins based on the organization of their catalytic domains and the type of oxidative mechanisms for the demethylation reaction. The first group is the Jumonji (Jmjc) domain-containing KDM family, which utilizes 2-oxoglutarate (2-OG; α-ketoglutarate) as a co-factor. The second group is KDM1A (LSD1, BHC110, AOF2) and KDM1B (LSD2), which utilizes flavin adenine dinucleotide (FAD) as a co-factor for demethylation activity.
Aberrant histone lysine methylation patterns have been identified in various human cancers. For example, low levels of H3K4me2 correlated with low survival rates in both lung and kidney cancers and was also associated with adverse prognosis in non-small cell lung carcinomas (NSCLC), hepatocellular carcinomas (HCC), and breast cancers (Barlési et al. 2007; Elsheikh et al. 2009; Seligson et al. 2009). Either up- or down-regulated KMTs frequently found in cancer and KDMs are involved in tumorigenesis by several other mechanisms, including alteration of histone or non-histone protein methylation (Varier and Timmers 2011; Colón-Bolea and Crespo 2014). Dysregulation of H3K27me3 is frequently observed in many types of cancers, and overexpression of EZh2 or mutations in the SET domain of EZH2 have been reported in lymphomas, causing an increase of H3K27me3 (Pawlyn et al. 2017; Nienstedt et al. 2018). The histone demethylase LSD1 (KDM1A) is highly expressed in several cancers and is specifically required for terminal differentiation of hematopoietic cells (Sprüssel et al. 2012). By histone H3K4 1/2 demethylase activity, LSD1 (KDM1A) represses gene expression, but LSD1 can stimulate transcription through interaction with the androgen receptor (Metzger et al. 2005). Several LSD1 inhibitors, such as ORY-1001 or GSK2879552, have been developed and are under clinical trial for AML treatment (Maes et al. 2015).
Similar to any other PTMs, histone lysine methylation can serve as a recognition site for the ‘reader’ or effector proteins. The malignant brain tumor (MBT) domain protein, PHD (plant homeodomain) proteins, chromodomain proteins, PWWP domain, and WD40 repeat proteins are identified as histone lysine methylation readers (Herold et al. 2011). The inhibitor of growth (ING) family of tumor suppressor genes (ING1-5) contains a C-terminal PHD, which is known to preferentially bind di- and tri-methylated H3K4 and mediate many cellular processes (Champagne and Kutateladze 2009). Heterochromatin protein 1 (HP1) is another example of a methyl-lysine reader. Three isoforms of HP1 can interact with methylated H3K9 via its chromodomain. Much evidence has shown that not only alteration of histone modifying enzyme levels, but also alteration of methyl-lysine reader expression has cancer implications. For example, downregulation of HP1α has been linked to the higher invasive potential of breast cancer cells and papillary thyroid carcinoma (Wasenius et al. 2003; Norwood et al. 2006; De Koning et al. 2009).
Alterations in histone lysine methylation are tightly linked to the development of cancer and are suggested as potential cancer therapeutic targets. Many KMT inhibitors, such as DOT1L, EZH2, and SUV 39H1 inhibitors, are in preclinical or clinical trials. Many groups have developed EZH2 inhibitors, and among them, EPZ-6438 is in phase I/II trial for refractory B-cell lymphoma (Knutson et al. 2014). In addition, the DOT1L inhibitor EPZ-5676 is in phase I clinical trial for refractory AML and ALL (Lillico et al. 2018; Stein et al. 2018). However, the search for KMT or KDM inhibitors is still in its very first stages.
Histone arginine methylation is similar to lysine methylation in many ways. Protein arginine methyltransferases (PRMTs) also utilize SAM to transfer a methyl group to the guanidine nitrogen atoms of arginine to form methylarginines and SAH. There are three different forms of methylarginines: ω-NG-monomethylarginine (MMA), ω-NG,NG-asymmetric dimethylarginine (aDMA), and u-NG,N′G-symmetric dimethylarginine (sDMA). PRMTs can be subcategorized into three groups by their catalytic activity; type I (PRMT1, PRMT2, PRMT3, PRMT4, PRMT6, and PRMT8) and type II (PRMT5 and PRMT9) enzymes initially forms MMA as an intermediate before the establishment of aDMA or sDMA, respectively, while type III (PRMT7) enzymes only catalyze to form MMA (Yang and Bedford 2013). Generally, H4R3me2a, H3R2me2s, H3R17me2a, and H3R26me2a are correlated with active gene expression, while H3R2me2a, H3R8me2a, H3R8me2s, and H4R3me2s are linked to gene repression. PRMTs also can methylate many non-histone proteins that have arginine- and glycine-rich (GAR) motifs. RNA-binding proteins (RBPs), Tumor suppressor 53-binding protein 1 (53BP1), and many other proteins have been identified as substrates for PRMTs, and arginine methylation of these proteins is involved in various biological processes, such as transcription, cell signaling, mRNA translation, DNA damage signaling, receptor trafficking, protein stability, and pre-mRNA splicing (Wei et al. 2014).
Arginine methylation is a very stable modification; therefore, the existence of direct arginine demethylases is still controversial. Jumonji domain-containing protein, JmjD6, was reported to demethylate H3R2me2 and H4R3me2, but later it was identified as a lysine hydroxylase (Webby et al. 2009). Moreover, a recent study showed that one of peptidylarginine deiminases (PAIDs) protein PADI4 was recruited to the pS2 promoter region just prior to H3R17me2a loss, suggesting that it is responsible for removing this methyl mark (Denis et al. 2009). However, PADIs catalyze the deimination of arginine; therefore, they are not considered as “true” demethylases.
As epigenetic readers of arginine methylation, Tudor domain-containing proteins, such as SMN (Survival of motor neuron), SPF30 (Splicing factor 30), and TDRD1/2/3/6/9/11, have been identified to interact with methyl-arginine residues (Gayatri and Bedford 2014). However, the biological role of the interaction between these two is still unclear.
Aberrant expression of PRMT or dysregulation of PRMT activity are associated with several diseases, including many types of cancers. For example, PRMT1 is the major PRMT, which is responsible for 90% of arginine methylation, and it is upregulated in breast cancer, bladder cancer, pediatric ALL, etc. (Yoshimatsu et al. 2011; Zou et al. 2012). Most other PRMTs are also found to be upregulated in various types of cancers; as a result, PRMTs are attractive cancer targets. Recently, a few PRMT inhibitors, such as the PRMT5 selective inhibitor (EPZ015666), have been generated and demonstrate promising therapeutic results against specific cancer types in pre-clinical trials (Chen et al. 2017). Enzymes for histone methylation, demethylation, and readers are summarized in Tables 3 and 4.
Histone modification-phosphorylation, ubiquitination, and histone variant
Protein phosphorylation is a very important PTM involved in many cellular processes. Proteins with specific amino acid residues, such as serine, threonine, and tyrosine residues, are phosphorylated by a protein kinase by the addition of a covalently bound phosphate group. Phosphorylation alters the structural conformation of a protein, causing the target protein to become either activated or deactivated. Protein kinases and phosphatases work independently and balance modifications to regulate the function of proteins. The most well-characterized histone phosphorylation is H3S10. S28 and T11 phosphorylation are known for transcriptional regulation and mitosis. Aurora B kinase, mitogen and stress-activated protein kinases 1 and 2 (MSK1 and MSK2), Ribosomal s6 kinase 2 (RSK2) IκB kinase-α (IKK-α), or PIM1 kinase can phosphorylate H3S10 in a DNA-context manner upon different stimuli for immediate-early gene expression (Nowak and Corces 2004). In fact, several studies reported that Aurora B is overexpressed in a variety of human cancers, particularly in colorectal and breast cancer (Ota et al. 2002; Tanaka et al. 2008). Histone phosphorylation, in cooperation with other histone modifications, plays a crucial role in DNA damage response pathways and participates in recruitment of downstream DNA damage response and repair proteins, as well as in the amplification of DNA damage signals. The histone H2A variant, H2AX, is rapidly phosphorylated at S139 by ATM, DNA PK kinases, or ATR upon DNA damage stresses and spread over megabases from the break site, which serves as a platform for recruiting other DNA damage response proteins, including 53BP1 (p53-binding protein 1), BRCA1, and NBS1 (Turinetto and Giachino 2015). H2AX gene is frequently lost in cancer, and H2AX deficiency can lead to increased sensitivity to ionizing radiation, which exhibit genomic instability and enhanced susceptibility to cancer (Georgoulis et al. 2017). However, epigenetic drugs targeting histone phosphorylation have yet to be established.
Ubiquitin is a small (8.5 kDa) regulatory protein, and ubiquitination is the addition of ubiquitin to the lysine residue of a substrate protein. The most well-known role of protein ubiquitination is to degrade target protein primarily via the proteasomal degradation pathway (Swatek and Komander 2016). Histones, especially H2A and H2B, are well-known substrates for ubiquitination. All four histones and linker histone H1 can be ubiquitinated, and a single ubiquitin moiety conjugated to H2A-K119 (ubH2A) and H2B k120 (ubH2B) is the most dominant form. There are several histone ubiquitin ligases and deubiquitinating enzymes (DUBs) identified, and histone ubiquitination plays critical roles, including transcription, maintenance of chromatin structure, and DNA repair (Cao and Yan 2012). H2Bub is highly associated with active gene expression, while H2Aub plays a role in transcriptional silencing with other repressive histone modifying enzyme complexes, such as polycomb repressive complex 1 (PRC1) (Minsky et al. 2008; Zhou et al. 2008). Conversely, H2A DUBs are often required for gene activation, indicating the importance of histone ubiquitination in gene expression (Joo et al. 2007; Zhu et al. 2007). Histone ubiquitination also plays an important role in DNA damage. When DNA damage causes DNA double-strand breaks (DSB), histone variant H2AX is rapidly phosphorylated and recruits DNA damage response regulators followed by subsequent recruitment of histone ubiquitin ligases RNF8 and RNF168, which catalyze the K63-linked polyubiquitination chain formation of histone H2A and H2AX (Uckelmann and Sixma 2017). Aberrant histone ubiquitination, such as down-regulated H2Aub and H2Bub, was found in several cancers (Zhu et al. 2007; Prenzel et al. 2011). To date, there are no therapeutic reagents targeting ubiquitination or deubiquitination. Very little is known about histone modification through the small ubiquitin-related modifier (SUMO) or neddylation. SUMO shares 18% identity with ubiquitin, and NEDD8 is 90% homologous to ubiquitin. Histone H4 can be modified by SUMO family proteins and can associate with transcriptional repression through recruitment of HDAC1 and HP1 (Shiio and Eisenman 2003). Histone neddylation to H2A antagonizes H2A ubiquitination, which negatively regulates DNA damage repair pathways (Li et al. 2014). Although SUMO or Nedd8 share a similar structure with ubiquitin, they play distinctive epigenetic roles in cooperation with other modifiers, indicating the complexity of regulating the epigenetic process.
Histone variants are proteins that substitute for the core canonical histones (H3, H4, H2A, H2B) in nucleosomes in eukaryotes and often confer specific structural and functional features. Unlike epigenetic regulation of ‘canonical’ histones through posttranslational modification, histone variants work through specific deposition and removal machineries. They have important roles in early embryonic development, chromosome segregation, transcriptional regulation, DNA repair, and other processes. There are interesting reports for histone variants macroH2A1 association with cancer. Reduction of macroH2A1.1 protein is negatively associated with lung cancer recurrence, and later reports have shown that alternative splicing of macroH2A1 regulates cancer cell proliferation (Sporn et al. 2009; Novikov et al. 2011). There is a growing awareness that histone modifications and chromatin organization influence pre-mRNA splicing and its epigenetic role in cancer (Khan et al. 2012). It suggests that not only epigenetic modifying enzyme, but also the enzyme for pre-mRNA splicing could be epigenetic therapeutic targets.
Conclusion
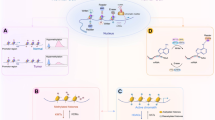
Involvement of epigenetic factors in cancer development is now widely accepted. We have accumulated vast knowledge on how epigenetic aberration can affect cancer initiation, progression, and metastasis. In this review, we have discussed basic epigenetics and its alteration in cancer as well as available drugs targeting epigenetic mechanisms. Major epigenetic modifications as a cancer target are summarized in Fig. 1. We focused on DNA methylation (Fig. 1a) and histone modification (Fig. 1b) among various other mechanisms and summarized current studies regarding how genetic alteration is linked to abnormal epigenetic changes. We should note that each epigenetic modification is not a separate or mutually exclusive event, but rather they are networking with each other to cause subsequent changes. For example, double-strand break from DNA damage rapidly enhances histone H2A and H2AX phosphorylation. In addition, other histone modifications, such as histone acetylation, and ubiquitination follow for further recruitment of DNA damage repair regulatory proteins.

Graphic summary of epigenetic alterations involved in cancer and available drugs targeting epigenetic mechanisms. a Tumorigenesis through aberrant methylation of CpG islands. DNA methylation can be written by DNMTs (in blue), recognized by MBD proteins (in green) and erased by TET proteins (in red). Epigenetic drugs targeting DNMT1 are approved by the FDA. b Tumorigenesis through aberrant histone modifications. Writers of each histone modification such as histone lysine methyltransferase (KMT), histone acetyltransferase (HAT), ubiquitin E3 ligases (E3 lig), protein arginine methyltransferase (PRMT), kinase are shown in blue. Readers such as methyl-lysine binding protein, tudor domain protein, bromodomain and extra terminal domain family member (BRD) are shown in green. Erasers such as histone deacetylase (HDAC), histone lysine demethylase (KDM), and deubiquitinating enzyme (DUB), phosphatase are shown in red. Canonical histone is shown in blue and histone variants is shown in brown. KMT inhibitors, KDM1 inhibitors, BET inhibitors, HDAC inhibitors are either approved or under clinical trials. Apart from the targets shown here other possible epigenetic targets for drug development are also available. AC acetylation, ME methylation, Ub ubiquitination, P phosphorylation
Along with the accumulation of knowledge about the biology and function of epigenetic modifications and their regulatory mechanisms in cancer, four anti-cancer drugs that target these mechanisms have been currently approved, and many others are in clinical trials. However, use of these drugs have a few limitations. As most of the histone modifying enzymes have several different substrates, use of enzyme inhibitors can have limitation in substrate specificity. Conversely, targeting non-histone proteins for cancer therapy can be another strategy for cancer drug development. As cancer results from a series of genetic and epigenetic molecular events, overcoming the disease would need the use of a combination of multiple genetic and epigenetic targets. To date, the only approved epigenetic anticancer agents are HDAC inhibitors and DNMT inhibitors. Our next challenge is to develop additional drugs targeting other classes of epigenetic enzymes and to attempt combinations with those developed to achieve better substrate and cancer specificity.
References
Baldwin RL, Nemeth E, Tran H, Shvartsman H, Cass I, Narod S, Karlan BY (2000) BRCA1 promoter region hypermethylation in ovarian carcinoma: a population-based study. Cancer Res 60:5329–5333
Barbano R, Muscarella LA, Pasculli B, Valori VM, Fontana A, Coco M, la Torre A, Balsamo T, Poeta ML, Marangi GF, Maiello E, Castelvetere M, Pellegrini F, Murgo R, Fazio VM, Parrella P (2013) Aberrant Keap1 methylation in breast cancer and association with clinicopathological features. Epigenetics 8:105–112
Barlési F, Giaccone G, Gallegos-Ruiz MI, Loundou A, Span SW, Lefesvre P, Kruyt FA, Rodriguez JA (2007) Global histone modifications predict prognosis of resected non-small-cell lung cancer. J Clin Oncol 25:4358–4364
Bedford MT, van Helden PD (1987) Hypomethylation of DNA in pathological conditions of the human prostate. Cancer Res 47:5274–5276
Bos JL (1989) ras oncogenes in human cancer: a review ras oncogenes in human cancer: a review. Cancer Res 49:4682–4689
Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T (1998) Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 391:597–601
Cao J, Yan Q (2012) Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front Oncol 2:1–9
Cecotka A, Polanska J (2018) Region-specific methylation profiling in acute myeloid leukemia. Interdiscip Sci Comput Life Sci 10:33–42
Champagne KS, Kutateladze TG (2009) Structural insight into histone recognition by the ING PHD fingers. Curr Drug Targets 10:432–441
Chan CH, Fang C, Yarilina A, Prinjha RK, Qiao Y, Ivashkiv LB (2015) BET bromodomain inhibition suppresses transcriptional responses to cytokine-Jak-STAT signaling in a gene-specific manner in human monocytes. Eur J Immunol 45:287–297
Chao C, Chi M, Preciado M, Black MH (2013) Methylation markers for prostate cancer prognosis: a systematic review. Cancer Causes Control 24:1615–1641
Chen H, Lorton B, Gupta V, Shechter D (2017) A TGFβ-PRMT5-MEP50 axis regulates cancer cell invasion through histone H3 and H4 arginine methylation coupled transcriptional activation and repression. Oncogene 36:373–386
Chinnam M, Goodrich DW (2011) RB1, development, and cancer. Curr Top Dev Biol 94:129–169
Cimmino L, Abdel-Wahab O, Levine RL, Aifantis I (2011) TET family proteins and their role in stem cell differentiation and transformation. Cell Stem Cell 9:193–204
Colón-Bolea P, Crespo P (2014) Lysine methylation in cancer: SMYD3-MAP3K2 teaches us new lessons in the Ras-ERK pathway. BioEssays 36:1162–1169
Cooper DN (1983) Eukaryotic DNA methylation. Hum Genet 64:315–333
Cui H, Onyango P, Brandenburg S, Wu Y, Hsieh CL, Feinberg AP (2002) Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res 62:6442–6446
Dawson MA, Kouzarides T (2012) Cancer epigenetics: from mechanism to therapy. Cell 150:12–27
De Koning L, Sauignoni A, Boumendil C, Rehman H, Asselain B, Sastre-Garau X, Almouzni G (2009) Heterochromatin protein lα: a hallmark of cell proliferation relevant to clinical oncology. EMBO Mol Med 1:178–191
Denis H, Deplus R, Putmans P, Yamada M, Métivier R, Fuks F (2009) Functional connection between deimination and deacetylation of histones. Mol Cell Biol 29:4982–4993
Doerfler W (1983) DNA methylation and gene activity. Annu Rev Biochem 52:93–124
Du Q, Luu P-L, Stirzaker C, Clark SJ (2015) Methyl-CpG-binding domain proteins: readers of the epigenome. Epigenomics 7:1051–1073
Dyson NJ (2016) RB1: a prototype tumor suppressor and an enigma. Genes Dev 30:1492–1502
Eckschlager T, Plch J, Stiborova M, Hrabeta J (2017) Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci 18:1–25
Elsheikh SE, Green AR, Rakha EA, Powe DG, Ahmed RA, Collins HM, Soria D, Garibaldi JM, Paish CE, Ammar AA, Grainge MJ, Ball GR, Abdelghany MK, Martinez-Pomares L, Heery DM, Ellis IO (2009) Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res 69:3802–3809
Fernández-Medarde A, Santos E (2011) Ras in cancer and developmental diseases. Genes and Cancer 2:344–358
Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR, Futreal PA (2011) COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res 39:945–950
Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Pérez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M (2005) Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37:391–400
Gayatri S, Bedford MT (2014) Readers of histone methylarginine marks. Biochim Biophys Acta 1839:702–710
Gayther SA, Batley SJ, Linger L, Bannister A, Thorpe K, Chin SF, Daigo Y, Russell P, Wilson A, Sowter HM, Delhanty JD, Ponder BA, Kouzarides T, Caldas C (2000) Mutations truncating the EP300 acetylase in human cancers. Nat Genet 24:300–303
Georgoulis A, Vorgias CE, Chrousos GP, Rogakou EP (2017) Genome instability and γH2AX. Int J Mol Sci 18:1–10
Gonzaga C, Lupski JR, Gibbs RA (2012) Human genome sequencing in health and disease. Annu Rev Med 63:35–61
Herold JM, Ingerman LA, Gao C, Frye SV (2011) Drug discovery toward antagonists of methyl-lysine binding proteins. Curr Chem Genomics 5:51–61
Heron M, Statistics V (2012) Deaths: leading causes for 2009. Natl Vital Stat Rep 61:1–96
Heyn H, Esteller M (2012) DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet 13:679–692
Hood L, Rowen L (2013) The human genome project: big science transforms biology and medicine. Genome Med 5:1
Huang M, Zeng S, Zou Y, Shi M, Qiu Q, Xiao Y, Chen G, Yang X, Liang L, Xu H (2017) The suppression of bromodomain and extra-terminal domain inhibits vascular inflammation by blocking NF-κB and MAPK activation. Br J Pharmacol 174:101–115
Hudson TJ, Anderson W, Artez A, International Cancer Genome Consortium (2010) International network of cancer genome projects. Nature 464:993–998
Imbach P (2014) Acute lymphoblastic leukemia. Pediatric oncology. Springer, New York, pp 5–20
Joo HY, Zhai L, Yang C, Nie S, Erdjument-Bromage H, Tempst P, Chang C, Wang H (2007) Regulation of cell cycle progression and gene expression by H2A deubiquitination. Nature 449:1068–1072
Kantarjian HM, Roboz GJ, Kropf PL, Yee KWL, O’Connell CL, Tibes R, Walsh KJ, Podoltsev NA, Griffiths EA, Jabbour E, Garcia-Manero G, Rizzieri D, Stock W, Savona MR, Rosenblat TL, Berdeja JG, Ravandi F, Rock EP, Hao Y, Azab M, Issa JJ (2017) Guadecitabine (SGI-110) in treatment-naive patients with acute myeloid leukaemia: phase 2 results from a multicentre, randomised, phase 1/2 trial. Lancet Oncol 18:1317–1326
Kennedy BK, Liu OW, Dick FA, Dyson N, Harlow E, Vidal M (2001) Histone deacetylase-dependent transcriptional repression by pRB in yeast occurs independently of interaction through the LXCXE binding cleft. Proc Natl Acad Sci USA 98:8720–8725
Khan DH, Jahan S, Davie JR (2012) Pre-mRNA splicing: role of epigenetics and implications in disease. Adv Biol Regul 52:377–388
Klughammer J, Kiesel B, Roetzer T, Fortelny N, Nemc A, Nenning KH, Furtner J, Sheffield NC, Datlinger P, Peter N, Nowosielski M, Augustin M, Mischkulnig M, Ströbel T, Alpar D, Ergüner B, Senekowitsch M, Moser P, Freyschlag CF, Kerschbaumer J, Thomé C, Grams AE, Stockhammer G, Kitzwoegerer M, Oberndorfer S, Marhold F, Weis S, Trenkler J, Buchroithner J, Pichler J, Haybaeck J, Krassnig S, Mahdy Ali K, von Campe G, Payer F, Sherif C, Preiser J, Hauser T, Winkler PA, Kleindienst W, Würtz F, Brandner-Kokalj T, Stultschnig M, Schweiger S, Dieckmann K, Preusser M, Langs G, Baumann B, Knosp E, Widhalm G, Marosi C, Hainfellner JA, Woehrer A, Bock C (2018) The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat Med 24:1611
Knudson AG (1971) Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 68:820–823
Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Waters NJ, Smith JJ, Porter-Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Uenaka T, Pollock RM, Kuntz KW, Yokoi A, Keilhack H (2014) Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther 13:842–854
Kohli RM, Zhang Y (2013) TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502:472–479
Kriukiene E, Liutkevičiute Z, Klimašauskas S (2012) 5-Hydroxymethylcytosine—the elusive epigenetic mark in mammalian DNA. Chem Soc Rev 41:6916–6930
Li T, Guan J, Huang Z, Hu X, Zheng X (2014) RNF168-mediated H2A neddylation antagonizes ubiquitylation of H2A and regulates DNA damage repair. J Cell Sci 127:2238–2248
Lillico R, Lawrence CK, Lakowski TM (2018) Selective DOT1L, LSD1, and HDAC Class i inhibitors reduce HOXA9 expression in MLL-AF9 rearranged leukemia cells, but dysregulate the expression of many histone-modifying enzymes. J Proteome Res 17:2657–2667
Lin CH, Hsieh SY, Sheen IS, Lee WC, Chen TC, Shyu WC, Liaw YF (2001) Genome-wide hypomethylation in hepatocellular carcinogenesis. Cancer Res 61:4238–4243
Liu X, Brenner DA (2016) Liver: DNA methylation controls liver fibrogenesis. Nat Rev Gastroenterol Hepatol 13:126–128
Maes T, Carceller E, Salas J, Ortega A, Buesa C (2015) Advances in the development of histone lysine demethylase inhibitors. Curr Opin Pharmacol 23:52–60
Marmorstein R, Roth SY (2001) Histone acetyltransferases: function, structure, and catalysis. Curr Opin Genet Dev 11:155–161
Mashimo M, Kato J, Moss J (2013) ADP-ribosyl-acceptor hydrolase 3 regulates poly (ADP-ribose) degradation and cell death during oxidative stress. Proc Natl Acad Sci USA 110:18964–18969
McLendon R, Friedman A, Bigner D, Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068
Mehta A, Dobersch S, Romero-Olmedo AJ, Barreto G (2015) Epigenetics in lung cancer diagnosis and therapy. Cancer Metastasis Rev 34:229–241
Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, Gnirke A, Jaenisch R, Lander ES (2008) Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454:766–770
Metzger E, Wissmann M, Yin N, Müller JM, Schneider R, Peters AH, Günther T, Buettner R, Schüle R (2005) LSD1 demethylates repressive histone marks to promote androgen-receptor- dependent transcription. Nature 437:436–439
Minsky N, Shema E, Field Y, Schuster M, Segal E, Oren M (2008) Monoubiquitinated H2B is associated with the transcribed region of highly expressed genes in human cells. Nat Cell Biol 10:483–488
Moore KE, Gozani O (2014) An unexpected journey: lysine methylation across the proteome. Biochim Biophys Acta 1839:1395–1403
Moreno DA, Scrideli CA, Cortez MA, de Paula Queiroz R, Valera ET, da Silva Silveira V, Yunes JA, Brandalise SR, Tone LG (2010) Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia: research paper. Br J Haematol 150:665–673
Nakajima H, Kunimoto H (2014) TET2 as an epigenetic master regulator for normal and malignant hematopoiesis. Cancer Sci 105:1093–1099
Nevins JR (2001) The Rb/E2F pathway and cancer. Hum Mol Genet 10:699–703
Nguyen AT, Zhang Y (2011) The diverse functions of Dot1 and H3K79 methylation. Genes Dev 3:1345–1358
Nienstedt JC, Schroeder C, Clauditz T, Simon R, Sauter G, Muenscher A, Blessmann M, Hanken H, Pflug C (2018) EZH2 overexpression in head and neck cancer is related to lymph node metastasis. J Oral Pathol Med 47:240–245
Norwood LE, Moss TJ, Margaryan NV, Cook SL, Wright L, Seftor EA, Hendrix MJ, Kirschmann DA, Wallrath LL (2006) A requirement for dimerization of HP1Hsαin suppression of breast cancer invasion. J Biol Chem 281:18668–18676
Novikov L, Park JW, Chen H, Klerman H, Jalloh AS, Gamble MJ (2011) QKI-mediated alternative splicing of the histone variant MacroH2A1 regulates cancer cell proliferation. Mol Cell Biol 31:4244–4255
Nowak SJ, Corces VG (2004) Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet 20:214–220
Nowell PC (2007) Review series personal perspective Discovery of the Philadelphia chromosome: a personal perspective. J Clin Invest 117:2033–2035
Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, Berman DM, Jenuwein T, Pruitt K, Sharkis SJ, Watkins DN, Herman JG, Baylin SB (2007) A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 39:237–242
Okano M, Xie S, Li E (1998) Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases Non-invasive sexing of preimplantation stage mammalian embryos. Nat Am 19:219–220
Ota T, Suto S, Katayama H, Han ZB, Suzuki F, Maeda M, Tanino M, Terada Y, Tatsuka M (2002) Increased mitotic phosphorylation of histone H3 attributable to AIM-1/aurora-B overexpression contributes to chromosome number instability. Cancer Res 62:5168–5177
Padmanabhan B, Mathur S, Manjula R, Tripathi S (2016) Bromodomain and extra-terminal (BET) family proteins: new therapeutic targets in major diseases. J Biosci 41:295–311
Pasque V, Karnik R, Chronis C, Petrella P, Langerman J, Bonora G, Song J, Vanheer L, Sadhu Dimashkie A, Meissner A, Plath K (2018) X chromosome dosage influences DNA methylation dynamics during reprogramming to mouse iPSCs. Stem Cell Reports 10:1537–1550
Pawlyn C, Bright MD, Buros AF, Stein CK, Walters Z, Aronson LI, Mirabella F, Jones JR, Kaiser MF, Walker BA, Jackson GH, Clarke PA, Bergsagel PL, Workman P, Chesi M, Morgan GJ, Davies FE (2017) Overexpression of EZH2 in multiple myeloma is associated with poor prognosis and dysregulation of cell cycle control. Blood Cancer J 7:e549-10
Pérez-Salvia M, Esteller M (2017) Bromodomain inhibitors and cancer therapy: from structures to applications. Epigenetics 12:323–339
Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordóñez GR, Bignell GR, Ye K, Alipaz J, Bauer MJ, Beare D, Butler A, Carter RJ, Chen L, Cox AJ, Edkins S, Kokko-Gonzales PI, Gormley NA, Grocock RJ, Haudenschild CD, Hims MM, James T, Jia M, Kingsbury Z, Leroy C, Marshall J, Menzies A, Mudie LJ, Ning Z, Royce T, Schulz-Trieglaff OB, Spiridou A, Stebbings LA, Szajkowski L, Teague J, Williamson D, Chin L, Ross MT, Campbell PJ, Bentley DR, Futreal PA, Stratton MR (2010) A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 463:191–196
Prenzel T, Begus-Nahrmann Y, Kramer F, Hennion M, Hsu C, Gorsler T, Hintermair C, Eick D, Kremmer E, Simons M, Beissbarth T, Johnsen SA (2011) Estrogen-dependent gene transcription in human breast cancer cells relies upon proteasome-dependent monoubiquitination of histone H2B. Cancer Res 71:5739–5753
Prior IA, Lewis PD, Mattos C (2012) A comprehensive survey of ras mutations in cancer. Cancer Res 72:2457–2467
Qian C, Zhou M (2006) SET domain protein lysine methyltransferases: Structure, specificity and catalysis. Cell Mol Sci 63:2755–2763
Raj K, Mufti GJ (2006) Azacytidine (Vidaza®) in the treatment of myelodysplastic syndromes. Ther Clin Risk Manag 2:377–388
Rideout WM, Coetzee GA, Olumi AF, Spruck CH, Jones PA (1991) 5-Methylcytosine as an endogenous mutagen in the p53 tumor suppressor gene. Science 22:207–219
Rowley JD (2001) Chromosome translocations: dangerous liaisons revisited. Nat Rev Cancer 1:245–250
Sakai T, Toguchida J, Ohtani N, Yandell DW, Rapaport JM, Dryja TP (1991) Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Am J Hum Genet 48:880–888
Santos FPS, Kantarjian H, Garcia-Manero G, Issa JP, Ravandi F (2010) Decitabine in the treatment of myelodysplastic syndromes. Expert Rev Anticancer Ther 10:9–22
Seligson DB, Horvath S, McBrian MA, Mah V, Yu H, Tze S, Wang Q, Chia D, Goodglick L, Kurdistani SK (2009) Global levels of histone modifications predict prognosis in different cancers. Am J Pathol 174:1619–1628
Shiio Y, Eisenman RN (2003) Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci USA 100:13225–13230
Skakkebæk A, Nielsen MM, Trolle C, Vang S, Hornshøj H, Hedegaard J, Wallentin M, Bojesen A, Hertz JM, Fedder J, Østergaard JR, Pedersen JS, Gravholt CH (2018) DNA hypermethylation and differential gene expression associated with Klinefelter syndrome. Sci Rep 8:13740
Spange S, Wagner T, Heinzel T, Krämer OH (2009) Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol 41:185–198
Spencer DH, Russler-Germain DA, Ketkar S, Helton NM, Lamprecht TL, Fulton RS, Fronick CC, O’Laughlin M, Heath SE, Shinawi M, Westervelt P, Payton JE, Wartman LD, Welch JS, Wilson RK, Walter MJ, Link DC, DiPersio JF, Ley TJ (2017) CpG Island hypermethylation mediated by DNMT3A is a consequence of AML progression. Cell 168:801–816.e13
Sporn JC, Kustatscher G, Hothorn T, Collado M, Serrano M, Muley T, Schnabel P, Ladurner AG (2009) Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene 28:3423–3428
Sprüssel A, Schulte JH, Weber S, Necke M, Händschke K, Thor T, Pajtler KW, Schramm A, König K, Diehl L, Mestdagh P, Vandesompele J, Speleman F, Jastrow H, Heukamp LC, Schüle R, Dührsen U, Buettner R, Eggert A, Göthert JR (2012) Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia 26:2039–2051
Stein EM, Garcia-Manero G, Rizzieri DA, Tibes R, Berdeja JG, Savona MR, Jongen-Lavrenic M, Altman JK, Thomson B, Blakemore SJ, Daigle SR, Waters NJ, Suttle AB, Clawson A, Pollock R, Krivtsov A, Armstrong SA, DiMartino J, Hedrick E, Löwenberg B, Tallman MS (2018) The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 131:2662–2669
Sun W, Liu Y, Glazer CA, Shao C, Bhan S, Demokan S, Zhao M, Rudek MA, Ha PK, Califano JA (2010) TKTL1 is activated by promoter hypomethylation and contributes to head and neck squamous cell carcinoma carcinogenesis through increased aerobic glycolysis and HIF1α stabilization. Clin Cancer Res 16:857–866
Swatek KN, Komander D (2016) Ubiquitin modifications. Cell Res 26:399–422
Tanaka S, Arii S, Yasen M, Mogushi K, Su NT, Zhao C, Imoto I, Eishi Y, Inazawa J, Miki Y, Tanaka H (2008) Aurora kinase B is a predictive factor for the aggressive recurrence of hepatocellular carcinoma after curative hepatectomy. Br J Surg 95:611–619
Tao YF, Li P, Du XJ, Sun LC, Hu SY, Lu J, Cao L, Zhao WL, Feng X, Wang J, Wu D, Wang N, Ni J, Pan J (2013) Differential mRNA expression levels of human histone-modifying enzymes in normal karyotype B cell pediatric acute lymphoblastic leukemia. Int J Mol Sci 14:3376–3394
Turinetto V, Giachino C (2015) Survey and summary multiple facets of histone variant H2AX: a DNA double-strand-break marker with several biological functions. Nucleic Acids Res 43:2489–2498
Uckelmann M, Sixma TK (2017) Histone ubiquitination in the DNA damage response. DNA Repair (Amst) 56:92–101
Varier RA, Timmers HTM (2011) Histone lysine methylation and demethylation pathways in cancer. Biochim Biophys Act 1815:75–89
Vera E, Canela A, Fraga MF, Esteller M, Blasco MA (2008) Epigenetic regulation of telomeres in human cancer. Oncogene 27:6817–6833
Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, Mann M (2010) Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 142:967–980
Vogelstein B, Papadopoulos N, Velculescu V, Zhou S, Diaz LA Jr, Kinzler KW (2013) Cancer genome landscapes. Science 339(6127):1546–1558
Wasenius VM, Hemmer S, Kettunen E, Knuutila S, Franssila K, Joensuu H (2003) Hepatocyte growth factor receptor, matrix metalloproteinase-11, tissue inhibitor of metalloproteinase-1, and fibronectin are up-regulated in papillary thyroid carcinoma: a cDNA and tissue microarray study. Clin Cancer Res 9:68–75
Webby CJ, Wolf A, Gromak N, Dreger M, Kramer H, Kessler B, Nielsen ML, Schmitz C, Butler DS, Yates JR 3rd, Delahunty CM, Hahn P, Lengeling A, Mann M, Proudfoot NJ, Schofield CJ, Böttger A (2009) Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science 325:90–94
Wei H, Mundade R, Lange KC, Lu T (2014) Protein arginine methylation of non-histone proteins and its role in diseases. Cell Cycle 13:32–41
Xu Y, Vakoc CR (2017) Targeting cancer cells with BET bromodomain inhibitors. Cold Spring Harb Perspect Med 7:1–17
Yang XJ (2004) The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res 32:959–976
Yang Y, Bedford MT (2013) Protein arginine methyltransferases and cancer. Nat Rev Cancer 13:37–50
Yoshimatsu M, Toyokawa G, Hayami S, Unoki M, Tsunoda T, Field HI, Kelly JD, Neal DE, Maehara Y, Ponder BA, Nakamura Y, Hamamoto R (2011) Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int J Cancer 128:562–573
Zhang C, Zhong JF, Stucky A, Chen XL, Press MF, Zhang X (2015) Histone acetylation: novel target for the treatment of acute lymphoblastic leukemia. Clin Epigenetics 7:1–10
Zhou W, Zhu P, Wang J, Pascual G, Ohgi KA, Lozach J, Glass CK, Rosenfeld MG (2008) Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell 29:69–80
Zhu P, Zhou W, Wang J, Puc J, Ohgi KA, Erdjument-Bromage H, Tempst P, Glass CK, Rosenfeld MG (2007) A histone H2A deubiquitinase complex coordinating histone acetylation and H1 dissociation in transcriptional regulation. Mol Cell 27:609–621
Zhu K, Liu Q, Zhou Y, Tao C, Zhao Z, Sun J, Xu H (2015) Oncogenes and tumor suppressor genes: comparative genomics and network perspectives. BMC Genomics 16:S8
Zou L, Zhang H, Du C, Liu X, Zhu S, Zhang W, Li Z, Gao C, Zhao X, Mei M, Bao S, Zheng H (2012) Correlation of SRSF1 and PRMT1 expression with clinical status of pediatric acute lymphoblastic leukemia. J Hematol Oncol 5:1–11
Acknowledgements
This work was supported by NRF-2015R1C1A1A01053803 and NRF-2018R1D1A1B07044230.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Park, J.W., Han, JW. Targeting epigenetics for cancer therapy. Arch. Pharm. Res. 42, 159–170 (2019). https://doi.org/10.1007/s12272-019-01126-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-019-01126-z