
Abstract
In recent years, many studies have focused on understanding the effects of genetic and epigenetic mechanisms on carcinogenesis, diagnosing the disease at an early stage, and determining personalized treatment strategies. Epigenetic and genetic alterations are effective in the initiation and progression of cancer, the second most common cause of death worldwide. Epigenetics is defined as heritable changes in gene expression without DNA sequence alterations. Epigenetic mechanisms include DNA methylation, histone modifications, and non-coding RNAs. Disruption of the balance in epigenetic processes, which are necessary for the normal maintenance of tissue-specific gene expression, may cause cancer formation and progression. The reversibility of epigenetic abnormalities is a promising feature for epigenetic cancer therapy studies. This chapter aims to summarize information about epigenetic mechanisms, their role in cancer initiation and progression, and their potential use in cancer therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Epigenetic mechanisms
- Cancer
- DNA methylation
- DNA demethylation
- 5-mC
- 5-hmC
- TET enzymes
- histone modifications
- non-coding RNAs
- miRNAs
1 Introduction
Cancer is the second leading cause of death in the world behind cardiovascular disease, understanding its etiology and identifying cancer hallmarks is of significant experimental and clinical importance. Although the process of carcinogenesis and the distinguishing features of cancer, mostly based on gene mutations, have been relatively detailed and some treatment approaches have been discovered, the number of cancer-related deaths is still increasing annually (Liang et al. 2019). The underlying reasons for this are the limitations of targeted clinical therapies due to intra-tumoral heterogeneity, plasticity, epigenomic structure and dormancy in tumor cells, and the inability to overcome the main obstacles to long-term therapeutic efficacy. In addition, the molecular pathologies involved in the metastatic progression of the tumor have yet to be fully elucidated (Marusyk et al. 2020; Hanahan 2022). It has been determined in the last decade that the epigenomic structure is significantly affected by the changes in the tumor microenvironment, leading to deregulation in gene expression control. Moreover, dormant cells are sustained by epigenetic mechanisms (Basu et al. 2021; Robinson et al. 2020). Since dormancy for cancer cells is essential to acquire new mutations, initiate metastasis, adapt to and survive in a new environment, develop resistance to cancer therapy, and avoid immune damage, understanding the mechanisms of dormancy cell cycle arrest is important for developing new targeted therapeutics (Recasens and Munoz 2019). In line with these developments, Hanahan (2022) has expanded cancer hallmarks by including cellular plasticity, non-mutational epigenetic reprogramming, and polymorphic variations in the tissue/organ microbiome. Since the number of cancer-related deaths is increasing annually, each newly discovered cancer feature is vital for understanding cancer development and metastatic progression mechanisms. These developments are also essential because of their potential to reflect on treatment (Liang et al. 2019).
Tumors consist of millions of cancer cells with neoplastic disruptions, which are embedded in a microenvironment. The startling molecular and cellular heterogeneity in tumors and tumor microenvironment heterogeneity are significantly correlated with the progression of the disease and development of resistance to therapy, consequently, clinical outcome.
The heterogeneity of cellular phenotype in tumors is a complicated and multifactorial phenomenon that combines environmental, epigenetic, and genetic features. Even though the genetic heterogeneity aspect of intratumoral heterogeneity has been studied in detail and understood well, there are still inadequacies in its reflection on clinical medicine (McGranahan and Swanton 2017; Marusyk et al. 2020).
In spite of improvements in understanding the complex molecular pathology of cancer, gene mutations continue to be at the center of molecular oncology, and Bert Vogelstein’s famous statement would remain valid for many researchers: “The revolution in cancer research can be summed up in a single sentence: cancer is, in essence, a genetic disease” (Vogelstein and Kinzler 2004). The primary goal of cancer research over the past few decades has been identifying tumor-associated genetic alterations and evaluating their functional and clinical implications (Garraway and Lander 2013; Cheng et al. 2021; Marei et al. 2021; Vogelstein et al. 2013). Thanks to molecular technology improvements, DNA sequencing technology has revealed intratumor genetic heterogeneity, surprisingly. In addition, while the morphological and functional features of each normal cell form its own cellular identity, the observation of deviations in cellular identities in tumor cells without DNA-based mutations helped us to understand that not only gene mutations but also changes in epigenetic regulatory mechanisms are common in the process of carcinogenesis (Liang et al. 2019; Klemm et al. 2019).
It is generally accepted that human cancer cells have epigenetic abnormalities, which is the main topic of this chapter, and that global and/or focal epigenetic alterations may play a key role in the initiation and progression of tumorigenesis (Jones and Baylin 2007; Hassler and Egger 2012; Lafave et al. 2022; Bond et al. 2020). Significant changes in different epigenetic regulatory mechanisms characterize the cancer epigenome. In the process of tumor formation, genetic and epigenetic mechanisms are intertwined and mutually benefit from each other. Genetic mutations in epigenetic regulators can cause alterations in the cancer epigenome, while changes in epigenetic processes can result in genetic mutations (You and Jones 2012).
1.1 History of Epigenetics
The fundamental concepts of genetics and heredity were established by Mendel’s theories in 1865, the isolation of the DNA molecule in 1869, and the discovery of the double helix structure of DNA almost a century later, in 1959. Conrad H. Waddington, a developmental biologist, created the term “epigenetics” to describe a novel biology area focusing on the connections between gene and protein expression (Waddington 2012). In 1957, Waddington put forth the renowned epigenetic landscape, in which a rough surface (which represents extra- and intracellular environmental factors) allows a ball, representing a cell, to travel in various directions (Goldberg et al. 2007). The discovery of the high mobility group (HMG) proteins in the mid-1970s and early 1980s helped us realize that specific proteins, besides the histones, may play an architectural function in chromatin and affect how phenotypes are expressed. Even though the overall structure of DNA was roughly recognized relatively early in the twentieth century, the field of epigenetics could take off until the discovery of specific enzymes acting as writers and erasers of epigenetic marks in the 1990s and 2000s. The well-known markers, including DNA methylation and post-translational histone modifications, were quickly found after understanding the DNA-double helix structure. DNA methylation was first observed in 1965. Histone modifications, such as methylation, acetylation, ubiquitylation, and phosphorylation, were documented from 1962 to 1977 (Peixoto et al. 2020).
Although Waddington’s definition initially concerned the interpretation of the involvement of epigenetics in embryonic development and the link between genotype and phenotype, the definition of “epigenetics” has changed accordingly over the last 80 years and has been redefined multiple times. Understanding how a fertilized egg may develop into an organism made up of hundreds of different types of specialized cells, each of which expresses a specific set of genes with the same genetic material, has long been a goal of researchers. It is now widely acknowledged that specific gene expression patterns determine cellular identity. Establishing and maintaining this expression pattern is necessary. The coordinated action of hundreds of transcription factors, which bind to specific DNA sequences to activate or inhibit the transcription of cell lineage genes, is crucial for maintaining the pluripotency of the initial cell and establishing different cell types. The establishment of this phase concerns the mechanisms by which the genotype produces the phenotype during development, similar to Waddington’s first definition of epigenetics. In the maintenance phase, non-DNA sequence-specific chromatin cofactors are involved in setting up and maintaining the chromatin states throughout cell division and for extended periods, even in the lack of transcription factors. This stage is similar to Nanney’s original definition of epigenetics as the meiotic/mitotic inheritance of alternate chromatin states without changes in DNA sequence. This definition was later expanded upon by Riggs and Holliday and further changed by Bird and others (Felsenfeld 2014; Peixoto et al. 2020; Cavalli and Heard 2019).
1.2 Epigenetics and Epigenome
Although all body cells have essentially the same genetic material and hence the same genes, they are categorized into about 200 cell types depending on morphological and functional features. A highly controlled arrangement of DNA into chromatin is necessary to access the fundamental data of the DNA sequence and establish cell type-specific gene expression profiles that are tightly regulated, both temporally and spatially.
It is well known that chromatin, a macromolecular complex made up of DNA and histone proteins, serves as the scaffold for packing the genome into microscopic nuclei. The ability of genes to be silenced or activated is significantly related to the arrangement of the genome into the compact structure. Although there are various factors affecting both local and global chromatin architecture, the covalent modifications of DNA and histones are mainly involved in the coordination of this process. Since specific combinations of genes are expressed in corresponding cell types, cell type has its own distinctive feature known as cell identity. Cellular identity is formed during embryogenesis by constraining the developmental potential of embryonic cells toward tissue-specific stem cells and specialized cell types with differentiation programs. These dynamic events take place in cells that have the same genetic information. In normal cells, the genes having roles in the function of a particular cell type are maintained in an accessible state, while the genes without functions are silenced through epigenetic mechanisms.
The epigenetic mechanisms restrict each cell type’s potential; thus, the cell’s fate depends on the epigenetic regulation of the genetic code. Therefore, epigenetic mechanisms determine each cell type’s potential and play vital roles in mammalian development, differentiation, and homeostasis. The complex interplay between these systems is stable during cell division to preserve cellular identity. However, they also respond to intrinsic cellular signals during development or extrinsic ones for adapting to environmental cues through epigenomic features.
The epigenome combines cellular information encoded in the genome with molecular/chemical information of extracellular and environmental origin. The epigenome and the genome establish their unique gene expression program to define the functional identity unique to each cell type, developmental, or disease process. At the same time, the epigenome plays a role in the development of the organism’s ability to respond to environmental stimuli in some cases. Therefore, unlike the fixed genome, the epigenome exhibits dynamic and variable behavior in its response to intracellular and extracellular stimuli.
As a result, while epigenetics is concerned with the processes that control when and how specific genes are activated or silenced, epigenomics deals with the analysis of epigenetic alterations across multiple genes in a cell or an entire organism,
2 Epigenetic Machinery
Epigenetic modifications provide chromatin organization by creating inherited transcription conditions responsible for maintaining cellular function, i.e., epigenetic regulation occurs through chromatin modifications, which are formed by the packaging of histone and histone-binding proteins with DNA. Epigenetic machinery is composed of four main groups: DNA methylation, histone post-translational modifications, non-coding RNAs (ncRNAs), and chromatin remodeling (Fig. 3.1). However, many subgroups within each main group, together with chromatin rearrangement complexes, regulate gene transcription by controlling chromatin organization. These are cytosine methylation and, recently detailed, hydroxymethylation-induced DNA modifications, ATP-based chromatin rearrangement, and non-coding RNA-mediated pathways, including microRNA and long non-coding RNA.

The epigenetic machinery. A collection of related components that work in concert to control both transcriptional and post-transcriptional levels of gene expression make up the epigenetic machinery
Previously, these mechanisms have been extensively reviewed elsewhere, we will summarize them in normal cells and then their roles in the carcinogenesis process in detail.
2.1 DNA Methylation
DNA methylation is the most extensively studied chemical modification in mammals and is now well-known to play a significant regulatory role in the regulation of epigenetic gene expression, developmental processes, cellular differentiation, cell identity establishment, and tissue homeostasis. It alters the functional state of the regulatory areas but has no effect on the cytosine Watson-Crick base pairing rule. Therefore, it exhibits the traditional “epigenetic” signature and has fundamental functions in numerous stable epigenetic suppression mechanisms, including genomic imprinting, X-chromosome inactivation, tissue-specific gene expression, chromosome stability, repression of transposable elements, and aging (Turpin and Salbert 2022; Tucci et al. 2019; Anvar et al. 2021; Cavalli and Heard 2019; Neidhart 2015; Eden et al. 2003; Karpf and Matsui 2005; Smith and Meissner 2013).
The chemical mechanism underlying DNA methylation is the covalent transfer of a methyl (CH3) group from S′Adenosyl methionine to the fifth carbon of the pyrimidine ring of the cytosine (C) base (5-methylcytosine, 5mC) in the CpG dinucleotide under the catalytic action of DNA methyltransferases (DNMTs) (Schübeler 2015; Turpin and Salbert 2022; Ross and Bogdanovic 2019).
However, CpG dinucleotide content of the human genome is not equally distributed throughout the genome. CpG dinucleotides are concentrated in areas with large repetitive genomic sequences scattered all over the genome, such as centromeric repeats, intergenic regions, and retrotransposon elements, and they are generally methylated (70–80%) (Deaton and Bird 2011; Turpin and Salbert 2022). The hypermethylation of large repetitive genomic regions such as pericentromeric, centromeric, and telomeric areas is crucial for maintaining chromosome stability and proper chromosome division, as well as the restriction of the production of transposable elements, such as LINE-1 by hypermethylation (Ortiz-Barahona et al. 2020; Sharma et al. 2010; Roberti et al. 2019; Neidhart 2015). In contrast, less than 10% of total CpGs are found at the 5′ ends of many human genes as CpG-rich DNA stretches called “CpG islands” (CGI). While transcription is facilitated by the chromatin structure adjacent to CGI promoters, transcription and, consequently, gene expression is inhibited if CpG islands are methylated. The amount of methylation varies across the genome, and substantially methylated regions typically have lower transcriptional activity (Neidhart 2015). The majority of CGIs usually remain unmethylated during development and in differentiated tissues. Nearly 60% of CGIs in normal somatic cells are mainly localized in gene promoters and the first exon regions, primarily housekeeping genes (Deaton and Bird 2011). However, CGI promoters of some genes that should be transcriptionally silent for a long term during normal development become hypermethylated, such as imprinted genes, the genes located on inactive X-chromosomes, or genes that are exclusively expressed in germ cells but not appropriate to their expressions in somatic cells (Jones and Baylin 2007; Sharma et al. 2010). Besides, CGI hypermethylation in primarily developmentally significant, tissue-specific genes has also been reported (Handy et al. 2011; Roberti et al. 2019).
The genome-wide analyses of the methylome have shown that the methylation position in the transcriptional unit affects gene regulation. Previous studies revealed that although hypermethylation of CGI promoters is blocking the initiation of transcription, gene body methylation may even enhance the elongation of transcription for prevention of the intragenic promoters transcriptions and be involved in alternative splicing regulation (Bond et al. 2020; Neri et al. 2017; Ortiz-Barahona et al. 2020).
On the other hand, DNA methylation alterations occur not only in CGIs and promoters but also in the sequences up to 2 kb from CGIs, which are called CGI “shores.” The methylation of CpG shores is associated with transcriptional repression, and methylation patterns in these zones have been reported as tissue-specific, indicating that they play a role in tissue differentiation. Moreover, CGI “shelves,” which are located 2 kb upstream and downstream of the CGI shores, have also been identified in the DNA methylation studies. The DNA methylations in different regions and the GC content of these regions have different effects on gene expressions (Nishiyama and Nakanishi 2021; Jones and Baylin 2007).
2.1.1 DNA Methyltransferases
During the epigenetic tags incorporation, writers add the marks to chromatin/DNA, whereas readers mediate transcriptional consequences of epigenetic alterations, and finally, erasers remove the added tags.
DNA methyltransferases (DNMTs) are the enzymes responsible for adding the methyl group from S-adenosyl-l-methionine (Ross and Bogdanovic 2019) to cytosine, i.e., DNMTs are DNA methylation “writers.” The family comprises five members: DNMT1, DNMT2, DNMT3a, DNMT3b, and DNMT3L. DNA methylation involves three key stages; establishment (de novo methylation), maintenance of methylation, and demethylation. Of DNMT family members, DNMT3A and DNMT3B in combination with DNMT3L are regarded as de novo methylation enzymes targeting unmethylated CpG dinucleotides and establishing new DNA methylation patterns. DNMT3L serves as an accessory partner to the de novo methylation activity of DNMT3A. DNMT3A and DNMT3B play vital roles during early development, and the inactivation of these enzymes results in early embryonic lethality. DNMT1 enzyme recognizes the hemimethylated DNA strands and is responsible for maintaining the methylation process during replication by binding to hemimethylated parental DNA and copying the methylation pattern to fully methylated daughter strands. In the case of aberrant DNA methylation, DNMTs play critical roles. Overexpression of DNMT1, DNMT3a, and DNMT3b has been reported in various solid tumors, such as glioblastoma, gastric, colorectal, pancreatic, hepatic, and lung cancers. In cervical cancers, higher DNMT1 expression was reported in about 70% of the cells, linking to a worse prognosis (Neidhart 2015; Schübeler 2015; Jones and Baylin 2007; Lafave et al. 2022).
2.1.2 Methyl-CpG Recognition Proteins
Gene transcription may be impacted by DNA methylation in two different ways: First, DNA methylation itself may physically prevent transcriptional proteins from attaching to the gene. Transcription factors, such as AP-2, c-Myc, E2F, and NF-kB, may be prevented from binding to promoter sites by DNA methylation (Kulis and Esteller 2010). Second, and perhaps more crucially, the established methylated DNA sequences can be read by methyl-CpG binding domain protein (MeCP) families, which then enlist histone deacetylases, a family of enzymes responsible for repressive epigenetic alterations that suppress gene expression and preserve genome integrity (Clouaire and Stancheva 2008; Cheng et al. 2021). MBD1, MBD2, MBD4, and MeCP2 are among the proteins with methyl-CpG binding domains (MBD) and are involved in gene transcription regulation through the cooperation of other proteins. Histone deacetylases and other chromatin remodeling proteins that can change histones are subsequently recruited to the locus by MBDs, resulting in the formation of compact, inactive chromatin known as heterochromatin (Jones and Baylin 2007). It is crucial to understand the relationship between DNA methylation and chromatin structure. Methyl-CpG-binding domain protein 2 (MBD2) regulates the transcriptional silence of hypermethylated genes in cancer, and the lack of methyl-CpG-binding protein 2 (MeCP2) has been linked to Rett syndrome. In contrast to the other four family members, MBD3 attaches to hydroxymethylated DNA rather than methylated DNA (Yildirim et al. 2011). The other family which able to bind 5-mC consists of the ubiquitin-like proteins UHRF1 and UHRF2 (containing PHD and RING fingers domains 1 and 2), which are SET- and RING finger-associated (SRA) domain-containing proteins (Vaughan et al. 2018). Many of these proteins are known to insert repressive histone marks (such as lysine deacetylation and histone lysine/arginine methylation) at their binding sites, either directly or by uptake of proteins that catalyze reactions. Thus, the process of nucleosome remodeling, chromatin compaction, and complex chromatin modifications occur, resulting in transcriptional repression due to the limited access of transcription factors to the promoter.
As previously mentioned, DNMT1 recognizes the hemimethylated DNA for copying the methylated parental DNA strand to form a fully methylated DNA double helix. Therefore, it is responsible for maintaining the methylation process during the replication. The versatile protein UHRF1 is a crucial cofactor for DNMT1 in the process of DNA maintenance methylation (Sharif et al. 2007). The multi-domain protein UHRF1 controls epigenetic changes and mediates between DNA methylation and histone modifications. Through its central SET- and RING-associated (SRA) and C-terminal really fascinating new gene domains, UHRF1 preferentially recognizes hemimethylated DNA and exchanges it by methylating cytosines via its SRA domain at the replication fork. DNMT1 is attracted to its target sites on the freshly synthesized DNA strand by this base-flipping mechanism during the S phase, exposing the unaltered cytosine to DNMT1 (Qin et al. 2015; Berkyurek et al. 2014).
The results of MBD2 inhibition on colon and lung cancer carcinogenesis inhibition seem encouraging. MBD3 interacts with other proteins, including MBD2 and HDAC, to control the methylation process even though it does not directly bind to DNA that has been methylated. MBD4 mutations have been reported in colorectal cancer, endometrial carcinoma, and pancreatic cancers. Additionally, this mutation unexpectedly influences not just CpG sites but also the stability of the entire genome. Because of the interaction between MBD4 and MMR, MBD4 can potentially be crucial for DNA damage repair. In contrast, MeCP2 and the UHRF family seem to stimulate tumor growth when expressed (Mudbhary et al. 2014; Cheng et al. 2021; Cheng et al. 2019).
2.1.3 5-Hydroxymethyl Cytosine and TET Enzymes
The enzyme family of 2-oxoglutarate-dependent dioxygenases (2-OGDD) gained a new member in 2009, named Ten-eleven translocation methylcytosine dioxygenases (TET proteins). The ten-eleven translocation (t(10;11)(q22;q23)), which is rarely seen in acute myeloid and lymphocytic leukemia cases, inspired the name of the TET proteins. This structural chromosome aberration caused the fusion of TET1 gene located on chromosome 10q22 with the mixed lineage leukemia 1 (MLL1) gene on chromosome 11q23. TET1 is a Fe(II) and 2-keto glutarate-dependent enzyme involved in the conversion of 5-methyl cytosine dioxygenase to 5-hydroxymethylcytosine (hmC) (Tahiliani et al. 2009). Subsequently, the other members of the TET family, TET2 and TET3, were identified in humans and were shown to possess similar catalytic activity. It is known that the hydroxylation of the 5mC substrate at the CpG dinucleotides to 5hmC can be followed by the sequential oxidation of 5hmC to 5-formyl cytosine (5fC) and to 5-carboxyl cytosine (5caC) by the catalytic activity of the TET enzymes (Ito et al. 2011). For the completion of DNA demethylation, DNA repair enzyme thymine DNA glycosylase (TDG) enzyme recognizes any of these base changes from the genome, which results in the creation of an abasic site. DNA repair mechanisms in the cell (Base excision repair BER) recognize the abasic sites and restore the cytosine in the 5-mC locus (He et al. 2011b).
The TET enzymes are the only recognized “methylation editors” because they catalyze the repetitive oxidation of 5-mC, leading to the demethylation of 5-mC. Because of the demethylation activity of TETs, they can activate transcription and so, they have vital roles in various cellular processes, including embryogenesis, cell differentiation, and tumorigenesis (Ross and Bogdanovic 2019).
2.1.4 TET Proteins and DNA Demethylation
As a 2-oxoglutarate/Fe(II)-dependent oxygenase (2OG oxygenase), TETs are iron/ketoglutarate (Fe(II)/KG) dependent dioxygenases. The double-stranded β-helix (DSBH) and the cysteine-rich domain are at the core catalytic domain at the carboxyl terminus. While the cysteine-rich domain wraps around the DSBH core for stabilizing the overall structure and TET-DNA interaction, the DSBH domain with conserved residues brings Fe(II), KG, and 5mC together for oxidation. Since the methyl group is not involved in the TET–DNA interface, TET can accept various cytosine modifications (Ito et al. 2011; Kao et al. 2016). TET1 and TET3 have a CXXC-type zinc-binding domain, distinguishing methylated and unmethylated DNA at their amino terminus. However, TET2 does not encode a CXXC domain, instead, it is located close to the IDAX gene, directly interacting with TET2 and coding a CXCC domain similar to that of other TETs (Pastor et al. 2013).
There are two mechanisms for 5-mC demethylation: passive and active (Fig. 3.2). These mechanisms differ from each other according to whether they are replication dependent or not. Passive demethylation is a replication-dependent mechanism in which modified 5mC tags dilute through consecutive cell divisions in the lack of DNMT1-mediated methylation maintenance, and consequently gradually declining degree of methylation. In contrast, the active demethylation mechanism corresponds to a replication-independent mechanism in which methylated Cs are eliminated and replaced with unmodified cytosines through enzymatic activities (Wu and Zhang 2017).

DNA methylation and demethylation mechanisms. DNMT proteins carry out the methylation of cytosines in DNA. The most crucial methylation regulators are DNMT3A/B, while DNMT1 is principally responsible for protecting the 5mC mark during DNA replication. Since DNMT1 does not recognize 5-hmC, it may represent an intermediary in passive demethylation by replication. Two active demethylation mechanisms have recently been identified. The majority of the data points to a route in which TET (Ten-eleven translocation) dioxygenases, which use α-ketoglutarate (α-KG) and iron as cofactors, undergo three consecutive oxidation processes to change 5mC into 5hmC, 5fC, and 5caC. TDG (thymine DNA glycosylase) proteins then identify 5fC and 5caC, activating the base excision repair (BER) process. Additionally, there is evidence for a mechanism in which TDG-mediated BER comes after AID/APOBEC proteins, a group of cytidine deaminases, deaminate 5hmC to 5hmU
When 5mC is oxidized to 5fC or 5caC, TDG-mediated excision of 5fC or 5caC and BER-dependent repairment of the abasic site can restore unmodified cytosine through the TDG-BER pathway (He et al. 2011b; Wu and Zhang 2017). This process is defined as active modification–active removal (AM–AR) and is independent of DNA replication (Kohli and Zhang 2013). On the other hand, DNA replication can result in the dilution of the oxidized 5mC in restoring the unmodified cytosine pathway; this time, the mechanism is known as active modification-passive dilution. Hemi-modified CpG dyads are produced during DNA replication when unmodified cytosine is integrated into the freshly generated strand. UHRF1 detects a 5mC: C dyad, which aids in bringing DNMT1 to the hemi-5mC location. A CpG site that has been changed with 5hmC, 5fC, or 5caC may become demethylated during several cycles of DNA replication (Wu and Zhang 2017). In regulating the active TET-mediated DNA demethylation, all genes involved can be regulated at the transcriptional, post-transcriptional, and post-translational levels. Moreover, factors belonging to specific genomic regions at which the demethylation process is targeted may also be effective.
The 2-Oxoglutarate (2-OG), also known as α-ketoglutarate (α-KG) and vitamin C, regulates the activity of TET enzymes. In the TET-mediated oxidation processes, oxygen and α-KG are needed as substrates, while Fe(II) is necessary as a cofactor to produce CO2 and succinate (Kohli and Zhang 2013). Isocitrate dehydrogenase 1 (IDH1), IDH2, and IDH3 are the enzymes responsible for producing α-KG from isocitrate in the Krebs cycle (Losman and Kaelin 2013; Shekhawat et al. 2021). IDH1 or IDH2 overexpression promotes 5hmC synthesis in cells (Waitkus et al. 2015). However, as seen in melanoma and glial tumors, the decreased 5hmC level is linked to IDH2 downregulation (Fig. 3.3). In addition, cancer-related IDH mutations cause inhibition of TET activity through the production of 2hydroxyglutarate (2HG) instead of α-KG. The mutant product 2HG is an oncometabolite that challenges α-KG for binding to TET (Xu et al. 2011).

Schematic overview of a cell related to the involvement of IDH1 and IDH2 mutations and the resulting loss of TET2 protein demethylation ability in the DNA demethylation process
Preimplantation and primordial germ cell development, stem cell differentiation and maintenance, and neuronal functions are biological processes with a global hypomethylation condition that is maintained by 5-hmC through active DNA demethylation. Abnormal DNA demethylation is one of the primary cancer epigenetics subjects and the relation between TET and 5-hmC levels with clinical outcomes in different cancers will be discussed later.
2.2 Abnormal Epigenomic Reprogramming in Cancer
Tumor biology is a complex process involving many different mechanisms. Genomic and epigenetic anomalies play a role in the initiation and development of cancer. The genetic and epigenetic basis of cancer has been studied over the past 10 years, and the presence of high-frequency changes in numerous epigenetic regulators has clearly demonstrated the crucial role of epigenetic dysregulation in carcinogenesis. During tumorigenesis, the epigenome undergoes many changes, including genome-wide loss of DNA methylation, especially along the repetitive sequences of the genome, regional hypermethylation, mainly in CpG promoter islands of tumor suppressor genes, global changes in histone modification marks, and alterations in networks involving ncRNAs.
Comprehensive investigations of the human cancer genomes have shown that various cancer types have mutations in many key players in the epigenetic control of gene expression, DNA repair, and DNA replication. Cancer initiation and progression frequently result from mutations in epigenetic writers, readers, and editors, as well as components involving chromatin remodeling complex.
2.2.1 Cancer-Specific DNA Methylation Alterations
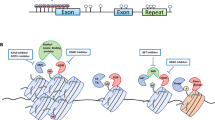
A diagram summarizing the most significant DNA methylation alterations seen in human malignancies is given in Fig. 3.4. These occurrences include DNA hypermethylation at gene promoters, frequently occurring on CpG islands and rendering the afflicted gene silencing. Hypomethylation, or loss of DNA methylation, affects the entire genome and is frequently found in repeated areas of the genome.

A schematic diagram representing the most significant DNA methylation alterations seen in normal and tumor genomes and genome-scale consequences of methylation alterations. Unmethylated CpG sites are shown by white circles, while methylated CpG sites are shown by red circles. The transcription start location and ongoing loss of transcription following DNA methylation are indicated by the arrows. Exons are demonstrated with green boxes, while the location of repetitive sequences and regulated regions is indicated by the blue rectangle
2.2.2 Global DNA Hypomethylation
The genome-wide DNA hypomethylation is one of the epigenetics-related hallmarks of cancer and occurs in various genomic regions, including repetitive sequences and regulatory regions. It results in abnormal gene expression, derepression of imprinted genes and retrotransposons, and chromosomal instability (Berdasco and Esteller 2010; Li et al. 2023; Mazloumi et al. 2022; Lozano-Ureña et al. 2021). As already mentioned, hypermethylated pericentromeric, centromeric, and telomeric sequences, preserving chromosomal stability and proper cell division in normal cells, are hypomethylated in tumor cells. Although the majority of CpGs in the genome are known to be 80% methylated, CpG methylation levels in cancer are typically between 40% and 60% (Baylin and Jones 2016). Loss of hypermethylation leads to cell division errors, disrupted chromosome stability, and increased mutation events during multistage carcinogenesis, all classical hallmarks of cancer. The presence of a high frequency of numerical and complex structural chromosome abnormalities are examples seen in tumors (Mazloumi et al. 2022; Pappalardo and Barra 2021). Retrotransposons that are repressed in healthy cells, such as LINEs (long interspaced nuclear elements) and Alu sequences, can be reactivated in cancer cells due to global hypomethylation (Ortiz-Barahona et al. 2020). Studies indicate that up to 50% of cancerous tumors may exhibit retrotransposition activation, which frequently results in structural and copy number changes as well as the induction of oncogene activity. Since the silencing of repetitive genomic regions is through the DNA methylation and repressive chromatin mark, histone H3 lysine 9 (H3K9) methylation, the hypomethylation probably allows the gene expression activation at these repetitive regions (Pfeifer 2018). In most cancer types, including bladder cancer, hepatocellular carcinoma, gastrointestinal stromal tumor, colon cancer, extra-hepatic cholangiocarcinoma, chronic lymphocytic leukemia, ovarian carcinoma, and lung carcinoma, LINE-1 hypomethylation is highly recurrent and tightly correlated with global hypomethylation. It is interesting to note that LINE-1 hypomethylation frequently increases along with the tumor’s histological grade and a poor prognosis, particularly in gastrointestinal malignancies (Zheng et al. 2019; Igarashi et al. 2010; Ikeda et al. 2013; Zhang et al. 2020; Baba et al. 2018).
Furthermore, abnormal hypomethylation is also seen in regulatory DNA regions that are normally methylated and repressed. These sequences become hypomethylated in cancer, which can interfere with the repression of normally silenced genes and cellular functions, leading to active transcription of proto-oncogenes, genomic instability, tumorigenesis, and metastasis (Mazloumi et al. 2022).
Through genome-wide sequencing studies, it has been revealed that DNA hypomethylation occurs specifically in DNA blocks called partially methylated domains (PMDs) (Nishiyama and Nakanishi 2021; Hansen et al. 2014). PMDs comprise about half of the genome, usually located in gene-sparse genomic locations, and coincide with nuclear lamina-associated domains and late replication sites (Berman et al. 2012; Hon et al. 2012). They represent a repressive chromatin structure associated with a high somatic mutation rate (Brinkman et al. 2019). Despite this general trend, their location shows some degree of cell type specificity (Schroeder et al. 2011). The enriched genomic regulatory features, which often include promoters and insulators, containing or defined by CTCF regions, are in the boundaries of PMDs (Salhab et al. 2018; Decato et al. 2020).
The gene-specific promoter DNA hypomethylation can also be involved in carcinogenesis. A subset of genes that fall into the germline-specific genes category is activated in cancers as a result of loss of DNA methylation at their promoter regions. Although the information related to the oncogenic potential remains limited, the group of genes, so-called cancer-germline genes, whose expressions are only active during spermatogenesis, can become activated in tumors through promoter hypomethylation. These genes were first identified in melanoma tumors as cytotoxic T lymphocyte antigens, and some of them are known as MAGE (melanoma antigen gene). These genes have an appropriate biomarker potential for malignancy diagnosis and prospective therapeutic targets since they are not expressed in normal somatic tissues but show unique cancer-specific expression patterns. About 250 cancer-germline genes have been identified and although the localizations are dispersed on different chromosomes, X-chromosome hosts many of these genes. The MAGE family, which has more than 50 family members and is evolutionary conserved, is a significant group of these genes. These genes produce ubiquitin ligases, which play a role in reproductive organ germ cell development. Several MAGE proteins can bind to and inhibit well-known tumor suppressor proteins such as TP53 and Retinoblastoma (De Souza et al. 2013) (Ladelfa et al. 2012). Activation of MAGEA11 is frequently observed in prostate cancer and has been associated with increased tumor cell growth. Besides activation of MAGEB2, another MAGE family member, has been reported in various tumors, such as lung carcinoma, and head and neck carcinoma (Van Tongelen et al. 2017). The BORIS/CTCFL gene family, which codes for a homolog of the insulator protein CCCTC binding factor (CTCF), is one intriguing member of the cancer-testis gene family. The encoded protein BORIS/CTCFL causes an increase in telomerase reverse transcriptase (hTERT) gene expression, encouraging cell immortalization and elevated expression revealed in testicular and ovarian cancers (Renaud et al. 2011).
The overexpression of c-MYC has been determined in various cancer types. The hypomethylated condition of the c-MYC promoter is correlated with its oncogenic potential and resulted from the hypomethylation-related reactivation of the transcriptionally silent retrotransposons (Fatma et al. 2020). The c-MYC promoter hypomethylation and aggressive cancer development correlation has been revealed in about 86,4% of gastric adenocarcinoma samples (De Souza et al. 2013)
Genomic imprinting is an epigenetic marking process that causes the monoallelic gene expression depending on parental origin. As is well known, imprinting patterns vary between tissues. They are regulated by imprinting control regions (ICRs), which are differentially methylated regions (DMRs), to form the parental-specific methylation pattern (Ferguson-Smith 2011). DNA methylation is the most crucial mechanism to govern imprinted gene expression in coordination with other epigenetic mechanisms, including H3K27me3 modification. They play crucial roles in various biological processes, including embryonic and placental growth, fetal development, and adult metabolism. Deletion of these sequences results in loss of imprinting (LOI), which leads to changes in the expression of imprinted genes in the cluster. LOI affects physiological functions and is the cause of the development of imprinting syndromes, including Angelman, Prader-Willi, and Beckwith-Wiedemann syndromes. Furthermore, the dysregulation of the imprinting pattern or the LOI has been described as the most common and early event in different tumors such as esophageal or colorectal cancer, or gliomas, meningiomas, and chronic myeloid leukemia (Jelinic and Shaw 2007). H19, the first reported imprinted gene in humans, and the other IGF2 imprinted gene are both growth regulatory genes that frequently regulate reciprocally. Zhang et al. (2018) and Yang et al. (2021) have reported the role of H19 overexpression in the promotion of leukemogenesis of AML (Zhang et al. 2018; Yang et al. 2021). The loss of the IGF2 imprint gene, related to the Beckwith–Wiedemann syndrome, is also a risk factor for cancer, e.g., colorectal cancer or development of Wilms tumor. The dysregulated expressions of maternally expressed CDKN1C (p57KIP2), H19, MEG3 or paternally expressed IGF2, PEG3, Contactin 3 (CNTN3), and DLK1 imprinted genes have been reported as biomarkers associated with the development of high-grade glial tumors and/or prediction of overall survival of patients (Lozano-Urena et al. 2021). Recent studies highlight the potential roles of epigenetic instability of imprinted domains in human cancers and suggest further studies necessary to determine potential use as cancer biomarkers (Bildik et al. 2022; Kim et al. 2015).
2.2.2.1 DNA Methyltransferases (DNMTs) and DNA Methylation
The hypomethylation of CpG sites of the genome typically results in the activation of gene expression, whereas the hypermethylation of the sites in enhancers or promoters results in transcriptional silencing (Morgan et al. 2018). DNA methyltransferases (DNMTs), as was previously discussed, are crucial for DNA methylation in the genome. DNMTs regulate the dynamic DNA methylation patterns of embryonic and adult cells in mammals in conjunction with other factors. On the other hand, cancer is typically identified by the abnormal function of DNMTs. As can be expected, there is a close relationship between the aberrant functions of DNMTs and cancer, as well. Common somatic mutations across tumors have been reported by recent large-scale cancer genomics consortia, including The Cancer Genome Atlas (TCGA) and the Genomics Evidence Neoplasia Information Exchange (GENIE). Although many somatic mutations exist in epigenetic regulators, relatively few mutations have been detected in DNMT enzymes (Han et al. 2019). A limited percentage of colon cancer patients have DNMT1 mutations; contrarily, a significant incidence of DNMT3A somatic mutations is seen in patients with acute myeloid leukemia (AML) (Hájková et al. 2012; Lee and Kim 2021).
Focal increases in DNA methylation associated with extensive hypomethylation are hallmarks of cancer genomes. A recent study by Lopez-Mayodo et al. showed a tight correlation between loss of TET function and cancer, as well as the interaction between DNMT3A and TET2 mutations in hematological malignancies. They emphasized that the distinctive pattern of global hypomethylation paired with localized hypermethylation reported in various cancer genomes may be primarily due to loss of TET function (López-Moyado et al. 2019).
2.2.2.2 Focal DNA Hypermethylation and Tumor Suppressor Genes
The aberrant hypermethylation of CpG islands (CGI) in the 5′ regions of cancer-related genes is a well-documented DNA methylation alteration in cancer. An alternate pathway to mutation for the deactivation of genes with tumor suppressor activity is this alteration, which can be intimately linked to transcriptional silencing. Accordingly, 60% of all gene promoters contain CpG islands, most of which are unmethylated throughout healthy development or adult cell renewal processes. Therefore, the more open chromatin states and active or ready to be activated, the expression status of these genes is fundamentally dependent on this unmethylated status. Contrarily, methylated CpG island promoters are so common in malignancies (5–10% of CGI genes) and are known to contribute to carcinogenesis directly. These cancer-specific features of the genes have opened up new options for epigenetic therapy, which targets epigenetic modifications for therapeutic reversal (Baylin and Jones 2016).
In order for malignant cells to maintain their uncontrolled development, cancer-related hypermethylation of CpG islands at promoter regions affects genes implicated in all regulatory circuits that control cell proliferation and homeostasis. At every stage of cancer development, hypermethylation events can occur and interact with both other epigenetic mechanisms and genomic abnormalities. Tumor-associated epigenetic lesions are far more common than genetic mutations, according to studies of DNA sequencing and genome-wide methylation data (Vogelstein et al. 2013). Between 5 and 10% of CpG island-containing promoters may be hypermethylated due to cancer.
Genome-wide CGI hypermethylation is evident not only in the majority of primary and metastatic tumors (Costello et al. 2000). However, it is also present in premalignant lesions, such as actinic keratosis lesions of the skin (Rodríguez-Paredes et al. 2018) and early stages of lung cancer (Vrba and Futscher 2019). It makes the most sense to explain a tumor-causing role for a hypermethylated gene in cancer when the methylation event impacts regulatory gene sequences like enhancers or promoter regions. The role of DNA methylation in these situations is typically blocking the related gene expression.
It should be emphasized that 5mC frequently exists in the gene body of active genes, and its effects here may frequently be the opposite of those they have in promoters. At least on a global scale, gene body or transcribed region hypermethylation is linked to increased gene expression levels, and it may encourage carcinogenesis by activating oncogenes if this condition occurs in genes with oncogenic characteristics (Liang and Weisenberger 2017). Nevertheless, CpG island hypermethylation more frequently will result in gene silencing when it affects promoters. If the impacted genes are involved in functional pathways, including cell proliferation control, genomic stability, activation of apoptosis or senescence, DNA repairing, and invasion and metastasis, then methylation-induced silencing events may have a tumor-promoting effect (Pfeifer 2018).
The role of promoter hypermethylation in the repression of gene expression was initially discovered in the retinoblastoma tumor suppressor gene (RB1) promoter region in patients with retinoblastoma (Greger et al. 1989), and then several tumor suppressor genes whose gene expression is repressed by DNA hypermethylation have been found in tumor tissues. Similar to germline mutation in familial malignancies, DNA hypermethylation in these genes is in a tissue-specific manner (Li et al. 2021a).
2.2.2.3 Roles of DNA Methylation Aberrations in Cell Proliferation
Cells need external stimuli such as growth factors, mitogens, and hormones for proliferation. Compared to normal cells, tumor cells use different ways to maintain these proliferative signals. They can activate proliferative pathways by deregulating downstream mediators, stimulating cells from the tumor microenvironment to provide them with mitogens (paracrine signaling), or producing their own mitogens (autocrine signaling). An essential component of growth control systems is the restriction of signaling pathways that promote proliferative processes. An important family of protein kinases called cyclin-dependent kinases (CDKs) controls the cell cycle. For CDKs to engage in their kinase activity, they need to be bound to the cyclins. In addition to cyclins, CDK inhibitors (CDKi) also control CDK activity. Cyclins and CDKi, together, are responsive to the stimuli through signal transduction pathways for dividing or staying quiescent of cells. Evading antiproliferative signaling at the different cell cycle checkpoints through epigenetic mechanisms is a characteristic feature of cancer cells. For instance, CDK inhibitor protein-coding genes, including cyclin-dependent kinase inhibitor 2A (CDKN2A), also known as p16INK4a, and a related gene CDKN2B (p15INK4a), located next to the CDKN2A locus, are involved in the regulation of cell cycle progression. The suppression of these genes by promoter hypermethylation has been reported in various cancer types. An essential mechanism for controlling cell proliferation is cell cycle-promoting kinase inhibition, and it is predicted that inactivating this mechanism may enhance cell growth. Breast, lung, head and neck cancers, gliomas, and melanomas are tumors associated with the inactivation of CDKN2A through promoter hypermethylation. Importantly, base substitution mutations, loss of homozygosity, promoter methylation, and other mutually exclusive events can all inactivate CDKN2A (Ortiz-Barahona et al. 2020; Pfeifer 2018).
In the mitogen-activated protein kinase (MAPK) pathway, a serial set of protein kinase cascades are involved, which is activated through the binding of mitogen to membrane receptors. The protein kinase cascades involved in the mitogen-activated protein kinase (MAPK) pathway are triggered by mitogen binding to membrane receptors, which then activate transcription factors to promote gene expression. Both activating mutations in signaling molecules and modifications to membrane receptors have the ability to constitutively activate the MAPK pathway. For example, a valine to glutamic acid alteration (V600E) in the B-RAF (B-Raf serine/threonine) gene gives rise to constitutive kinase activation, and this substitution is primarily seen in melanomas. Additionally, promoter hypermethylation-related inactivation of the PTPRR (ERK phosphatases protein tyrosine phosphatase receptor type R) and DUSP1 (dual specificity phosphatase 1 gene) genes have been reported in colon cancer (Laczmanska et al. 2013) and oral cavity carcinomas, respectively, meaning leading to MAPK cascade activation (Khor et al. 2013).
In the recent study by Xiang et al., they suggested that the tumor-specific reduced protein expression of PLCD1 (phospholipase C delta1) resulting from promoter hypermethylation could be used as a novel biomarker for early detection and prognostic prediction in colorectal cancers. They also reported that the gene plays important roles in proliferation, migration, invasion, cell cycle progression, and epithelial-mesenchymal transition. The PLCD1 is a negative regulator of the phosphatidylinositol 3-kinase (PI3K)-AKT pathway, another example of a dysregulated proliferative pathway in cancer (Xiang et al. 2019).
The familial cancer syndrome adenomatous polyposis coli is linked to germline mutations of the tumor suppressor gene adenomatous polyposis coli (APC), which predisposes its carriers to early-onset colorectal cancer. APC is a negative regulator of the Wingless/Int (WNT) signaling pathway. The other growth-promoting module, the WNT pathway, is especially relevant for intestinal stem cells and their malignancies. Epigenetic alterations in this pathway often result in higher β-catenin expression. Not only in colon cancer, but also APC promoter hypermethylation has been reported in breast, pancreatic, lung, and gastric cancers (Liu et al. 2021a; Zhou et al. 2020; Liang et al. 2017).
2.2.2.4 Role of DNA Methylation Changes in Evasion of Apoptosis
Success in tumor development depends not only on maintaining active cell proliferation but also on preventing the programmed cell death that would occur if the pathways were to become dysregulated. A high number of proliferative signals, significant DNA damage caused by the proliferation itself, hypoxia, or externally harmful substances can all cause apoptosis. The primary DNA damage sensor, p53 (TP53), directly controls the transcription of growth arrest genes when it activates in response to significant DNA damage. By epigenetically suppressing p53 targets like stratifin (SFN), tumoral cells can continue the cell cycle despite p53 activity. Stratifin is an important G2/M cell cycle checkpoint regulator and is expressed in response to DNA damage stress via a p53-dependent mechanism. SFN promoter hypermethylation is seen in various tumor types, including small-cell lung cancer (SCLC), prostate, endometrial, and breast cancers (Chauhan et al. 2021).
In normal tissues, if cells are unable to repair DNA damage, p53 activates the intrinsic apoptotic pathway, in which the pro- and anti-apoptotic members of the Bcl-2 family of regulatory proteins take roles in regulation. This route results in the release of cytochrome C and the creation of apoptosomes. The suppression of proapoptotic Bcl-2 family members (BCL2-Associated X Protein (BAX)), BIM (BCL2L11), BCL2 Binding Component 3 or PUMA (BBC3) or silencing of apoptotic peptidase activating factor 1 (APAF1) are examples of cancer-associated epigenetic dysregulation that prevents the development of this cascade (Ortiz-Barahona et al. 2020; Neophytou et al. 2021).
One of the hallmarks of cancer is the evasion of apoptosis. Many pro-apoptotic genes have been discovered to be silenced by methylation in malignant tumors. Death-associated protein kinase (DAPK), an example of hypermethylation-related silenced pro-apoptotic genes, has been revealed in many cancer types as well as in B-cell malignancies. Similarly, neuroblastomas and other malignancies have been shown to have methylation of the caspase 8 gene (CASP8), which encodes a cysteine protease controlled in a death-receptor-dependent and independent way. The paralogue of the well-known tumor suppressor TP53, TP73, has the ability to induce apoptosis. The TP73 promoter is methylated in some malignancies, including neuroblastomas and melanomas (Pfeifer 2018; Ortiz-Barahona et al. 2020).
The hippo signaling pathway is a route that manages cell proliferation and death to govern organ growth. The Hippo signaling pathway is important in inducing apoptosis and limiting cell proliferation. This signaling pathway has grown in importance in human cancer research, as unregulated cell division is a hallmark of many malignancies. MST1 and MST2 (Mammalian sterile 20-like kinases 1 and 2) are present in the pathway’s core kinase cassette. Soft tissue sarcomas have been shown to have methylated MST1 and MST2 promoters (Pfeifer 2018). The Ras association domain family (RASSF) of proteins is one of the few positive regulators of MST kinases discovered. The hypermethylation of the RASSF family member, RASSF1A, is practically seen in all human cancers and is mostly already methylated in early preneoplastic lesions. Through the MST1/2 kinases, RASSF1A positively regulates the Hippo growth control system, including its pro-apoptotic output (Motavalli et al. 2021; Malpeli et al. 2019).
2.2.2.5 Promotion of Genome Instability by DNA Methylation Alterations
As aforementioned, in addition to a global loss of DNA methylation at repeated sequences in the genome resulting in chromosomal instability, impaired genomic maintenance machinery results in the greater mutability of malignant cells. Changes to this machinery could occur at the DNA damage detection level or at the repairing mechanism itself. Any of these inactive levels make identifying and repairing genetic mistakes more difficult, which may speed up cell division and prevent apoptosis. Either inactivating mutations or promoter hypermethylation-related silencing can result in the loss of these functionalities. Consequently, both levels of DNA methylation can exhibit abnormalities. The hypermethylated Ataxia telangiectasia mutated promoter has been discovered in glioma, breast, and colorectal cancers (Begam et al. 2017). The DNA double-strand break (DSB) sensor ATM phosphorylates multiple important proteins in response to damage, which can result in cell cycle arrest, DNA repair, or apoptosis. The checkpoint kinase 2 (CHK2), a serine-threonine kinase, is also hypermethylated and silent in gliomas (Wang et al. 2010). The DNA repair apparatus is extensive and tailored to diverse forms of damage, from recombination mechanisms for double-strand breaks (DSBs) to mechanisms for single base or nucleotide damage
Depending on which repair mechanisms have been impaired, the inactivation of DNA repair function will probably lead to an increase in the frequency of mutations, either at the single base level or the chromosomal level. Tumors have impaired DNA repair mechanisms, most notably because of mutations in the germline. Xeroderma pigmentosum gene variants, for instance, can induce errors in nucleotide excision repair (e.g., XPA, XPC, and XPF). The mutations in DNA mismatch repair genes cause a hypermutator phenotype that frequently shows up as microsatellite instability. Base excision repair impairment is less frequently linked to cancer. Mutations in BRCA1, BRCA2, and RAD51 genes impair DNA double-strand break repair and recombination repair processes. Both sporadic cancers and familial cancer predisposition syndromes, particularly colorectal malignancies with microsatellite instability, have been linked to mutations in DNA mismatch repair genes. Although Lynch syndrome is due to inherited mutations in DNA mismatch repair genes, including MSH2, MLH1, MSH6, or PMS2, a majority of mismatch repair deficient sporadic colorectal tumors do not contain mutations; instead, the promoter of the MLH1 gene is frequently hypermethylated, and biallelic methylation-mediated inactivation causes the loss of protein production. The inactivation of MLH1 is a convincing illustration of a driver methylation event in carcinogenesis because of causes the loss of function similar to gene mutation (Keum and Giovannucci 2019).
The MGMT (O6-methylguanine methyltransferase) is a DNA repair gene, encoding a DNA repair protein that removes mutagenic and cytotoxic alkyl groups from the O6 position of guanine and restores the guanine to its original state, i.e., repairs O6-alkylated guanine residues in genomic DNA. By pairing thymine instead of cytosine during DNA replication, guanine-O6 methylation creates a methylated nucleotide with impaired base pairing potential, which encourages G:C to A:T mutations. The promoter of the gene is CpG rich and is epigenetically inactivated through DNA methylation, and consequently, methylation silencing of MGMT diminishes its O6-alkylguanine repairing efficiency. The epigenetically inactivated MGMT is seen in colorectal, gastric, non-small-cell lung cancers, head, and neck squamous cell carcinomas, and significantly in gliomas (Uddin et al. 2020). However, alkylating agents such as Temozolomide (TMZ) are among the most used chemotherapeutic drugs in cancer treatment and are known to cause cell cycle arrest at G2/M, which ultimately leads to apoptosis. Adding methyl groups at the N7 and O6 sites on guanines and the O3 site on adenines in genomic DNA is the mechanism through which TMZ causes cytotoxicity. When the O6 site on guanine is alkylated, a thymine rather than a cytosine match opposite the methylguanine during the following DNA replication, and DNA mismatch errors occur. The mismatches of methylated DNA can be repaired by base excision or DNA mismatch repair pathways through the involvement of a DNA glycosylase like alkylpurine-DNA-N-glycosylase (APNG) or a demethylating enzyme like MGMT. Thus, DNA mismatch repair by active MGMT causes the development of a resistance mechanism against TMZ. In contrast, epigenetically silenced MGMT sensitizes the tumor to TMZ. Glioma patients with a methylated MGMT gene have been shown to have a higher survival rate when treated with the alkylating agent TMZ compared to patients with an unmethylated promoter, possibly due to increased cell killing by the chemotherapy agent (Kukreja et al. 2021; Śledzińska et al. 2021).
2.3 Histon Modifications in Cancer
Histone proteins are essential for nucleosome components. In eukaryotes, chromatin is organized into nucleosomes, each formed of a histone octamer and a fragment of surrounding DNA. There are six histones: H1, H2A, H2B, H3, H4, and H5, highly rich in lysine and arginine, two positively charged amino acids (Neganova et al. 2022; Zhao et al. 2021). Since Vincent Allfrey’s pioneering work in 1964, it has been known that histones are post-translationally modified (PMTs) (Allfrey et al. 1964). Histon proteins’ amino and carboxy termini can undergo transcription-regulating changes, including methylation, acetylation, phosphorylation, sumoylation, ubiquitination, and ADP-ribosylation. They may also act as recognition modules for specific binding proteins (Audia and Campbell 2016).
Histone alterations are classified as active or repressive based on their effects on gene expression. The steady-state cell maintains a balance between particular modifications and modifiers to preserve chromatin structure, execute the correct gene expression program, and regulate the biological outcome. Disruption of this balance in the cell may change the phenotype, leading to the disease’s formation and progression (Zhao and Shilatifard 2019; Markouli et al. 2021). Deregulation of these mechanisms results in the development and progression of cancer due to the increased activation of oncogenes or the inhibition of tumor suppressor activity.
2.3.1 Histone Acetylation
Histone acetyltransferases (HATs) and histone deacetylases (HDACs) regulate acetylation, a reversible modification of the ε-amino group on lysine residues. HATs transfer the acetyl group of acetyl coenzyme A to the terminal of histone amino acid. Acetylation of the histone tails neutralizes the positively charged lysines, disrupting the connection between the tail and the negatively charged nucleosomal DNA to facilitate chromatin opening and enhance active transcription by making DNA accessible to transcription factors. The lysine residues of non-histone proteins are known to be acetylated such as p53, Rb, and MYC. Therefore, these enzymes are also called lysine acetyltransferases (KATs). In contrast, HDACs remove the terminal acetyl group of histone lysine, resulting in a compact chromatin structure that inhibits transcription (Neganova et al. 2022; Audia and Campbell 2016) (Fig. 3.5).

Schematic mechanism of histone acetylation and deacetylation
Acetylated lysines might provide a unique signal for regulatory factors or chromatin remodeling complexes to target specific domains. Bromodomains were discovered to function as acetyl-lysine recognition modules, guiding enzymes with these domains to specific locations on chromosomes. In addition to transcriptional regulation, new functions for histone acetylation have been identified, including nucleosome assembly, chromatin folding, heterochromatic silencing, DNA damage repair, and replication (Cohen et al. 2011; Zhang et al. 2015).
Numerous studies have shown that aberrant expression or activity of HATs and HDACs significantly affects the cancer acetylome (Li et al. 2019). Depending on the target genes (e.g., tumor suppressor and proto-oncogenes), hyperacetylation and hypoacetylation may disrupt the normal cell cycle, prevent or reverse differentiation, block apoptosis, and enhance cell proliferation, contributing to the formation and metastasis of a cancer phenotype (Di Cerbo and Schneider 2013). Alterations in global histone acetylation, specifically acetylation of H4 at lysine (K)16, have been associated with various cancers and may have predictive significance in some cases (Seligson et al. 2009; Fraga et al. 2005).
Several studies have suggested the dual roles of HATs as oncogenes and tumor suppressors. HAT mutations and altered expression without DNA mutation have been detected in multiple cancers (Chen et al. 2013; Di Cerbo and Schneider 2013).
Well-studied human HAT families are GNAT (HAT1, GCN5, PCAF), MYST (Tip60, MOF, MOZ, MORF, HBO1), and p300/CBP. p300/CBP includes the HAT domain, the bromodomain (BRD), and three cysteine and histidine-rich domains. Germline mutation of CBP causes Rubinstein-Taybi syndrome and increased susceptibility to childhood cancers, probably due to loss of the second allele. p300 has also been linked to hematological malignancies (Cheng et al. 2019; Di Cerbo and Schneider 2013). CBP- and p300-null chimeric mice developed hematological malignancies (Rebel et al. 2002). Several p300 missense mutations have been detected in colorectal adenocarcinoma, gastric adenocarcinoma, and breast cancer (Gayther et al. 2000; Cheng et al. 2019). Small-cell lung cancers and non-Hodgkin B-cell lymphomas have been shown to have mutations close to the HAT catalytic domain that lead to a loss of enzymatic activity (Peifer et al. 2012; Pasqualucci et al. 2011). However, impaired activation of HATs, which are also responsible for the acetylation of tumor suppressor genes such as p53 and Rb, can induce tumorigenesis.
On the other hand, oncogenic effects may result from abnormal activation or localization of p300/CBP. MLL-CBP t(11;16)(q23;p13), MLL-p300 t(11;22)(q23;q13), MOZ-CBP t(8;16)(p11;p13), and MOZ-p300 t(8;22)(p11;q13) have been identified in acute myeloid leukemia (AML), myeloid/lymphoid, or mixed lineage leukemia (MLL) (Cohen et al. 2011). In addition, it has been shown that p300 can modulate some fusion protein activity by acetylation, such as AML1-ETO t(8;21)(q22;q22), which is the most common fusion protein in AMLs. Depletion of p300 impaired its ability to promote leukemic transformation by inhibiting acetylation of AML1-ETO (Wang et al. 2011). The relationship between histone alterations and malignancy in hematological cancers has been broadly studied compared to solid tumors. High p300 expression has been related to poor prognosis in laryngeal squamous cell carcinoma and small-cell lung cancer (Chen et al. 2013; Gao et al. 2014).
Histone acetyltransferase TIP60 regulates apoptosis and DNA damage repair by acetylation of some tumor suppressor genes in addition to histones. Mutations of the human TIP60 gene have been identified in head and neck squamous carcinomas, ductal breast carcinomas, and low-grade B-cell lymphomas (Di Cerbo and Schneider 2013). Low TIP60 mRNA expression was associated with poor overall survival and recurrence-free survival in breast cancer (McGuire et al. 2019). It has also been found that TIP60 can inhibit viability and invasion of lung cancer cells through downregulation of the AKT signaling pathway (Yang et al. 2017). Another acetyltransferase, GCN5, has been shown to regulate gene transcription by catalyzing the acetylation of lysine residues on multiple histones, including H2b, H3, and H4, in addition to transcription factors such as FBP1 and N-Myc. GCN5 mRNA is upregulated in some cancers (Yin et al. 2015).
HDACs are divided into four groups classes I, II, III, and IV. HDAC overexpression has been reported in solid and hematological cancers and is associated with advanced disease and poor patient outcomes. Therefore, HDACs have become promising therapeutic targets (Hosseini and Minucci 2018).
High expression of HDAC1 and 2 is associated with reduced patient survival in colorectal carcinomas. The overexpression of HDAC1, 2, and 6 and HDAC1, 2, and 3 have been described in diffuse large B-cell lymphomas (DLBCL)/peripheral T-cell lymphomas and classical Hodgkin lymphomas, respectively (Dell’Aversana et al. 2012). HDAC6 and HDAC10 have been downregulated in human hepatocellular carcinoma (HCC) tissues and in patients with lung and stomach cancer, respectively, and associated with poor prognosis (Li and Seto 2016). It has been observed that HDAC4 is critical for regulating chromosome structure, while low HDAC4 expression is associated with chromosomal instabilities in high-grade glioma (Cheng et al. 2015). Class III HDACs, known as sirtuins, which play essential roles in regulating gene expression, apoptosis, autophagy, DNA damage repair and, genome stability, have been studied broadly. Increased or decreased class III HDAC expression levels have been detected in myeloid leukemia, prostate and ovarian carcinoma, gliomas, gastric carcinomas, non-melanoma, and melanoma skin cancers (Benedetti et al. 2015).
In addition to alterations in the expression level of HDACs, their enzymatic activity also contributes to cancer development. Some HDACs have been reported to be attracted to target genes by oncogenic proteins such as aberrant HDAC1, 2, or 3 recruitment by AML1-ETO fusion protein. Recruitment of HDACs prevents myeloid differentiation and results in cellular transformation by suppressing AML1 target genes (Falkenberg and Johnstone 2014). Somatic HDAC1 mutations and homozygous HDAC4 deletions have been detected in liposarcomas and melanomas. Also, HDAC2 loss-of-function mutations have been observed in sporadic carcinomas with microsatellite instability and hereditary non-polyposis colorectal cancer syndrome (Hosseini and Minucci 2018; Ropero et al. 2006).
HDACs affect the expression of many cell cycle regulators and also may directly interact with proteins implicated in tumor development, migration, and metastasis. HDAC1 and 2 suppress the expression of the cell cycle inhibitors p21 and p27. HDAC2 knocked down cells have shown an increase in p21Cip1/WAF1 expression independent of p53 in colorectal cancer cells (Huang et al. 2005).
Protein readers play an important role in histone post-translational modifications as well as HATs and HDACs. Readers identify particular locations, attract transcription factors or chromatin-associated protein complexes, and bind to histones to facilitate the localization of enzymes to specific targets (Liu et al. 2021b). The functional protein domains known as bromodomains (BRDS) can identify acetylated lysine residues in histones and other non-histone proteins. Additionally, they can serve as transcription factors and transcriptional coregulators. Another important family, Bromodomain and the extra-terminal domain-containing proteins (BET) include four family members: BRD2, BRD3, BRD4, and BRDT. These proteins play crucial functions as gene transcription activity mediators.
Genetic rearrangements of BRD-containing proteins have been associated with some aggressive tumor types. Nuclear protein midline carcinoma (NMC) of the testis is a highly aggressive tumor associated with translocations involving the NUT protein. BRD4–NUT rearrangements are observed in two-thirds of cases. BRD–NUT blocks cellular differentiation. BRD4–NUT stimulates CBP/p300 HAT activity and inactivation of p53. With recent studies, BET proteins have become potential therapeutic targets against testicular carcinoma, multiple myeloma, lymphoma, lung cancer, and neuroblastoma (Muller et al. 2011; Neganova et al. 2022; Cheng et al. 2019).
The reversible nature of epigenetic modifications has provided the basis for the development of anti-cancer strategies for the regulation of cancer epigenetics. HDAC inhibitors (HDACi) continue to be explored as promising anti-cancer drugs by modulating histone and non-histone proteins, regulating processes such as inhibiting cancer cell invasion, inducing apoptosis, and immunogenicity. Vorinostat, belinostat, Panobinostat, and romidepsin are FDA-approved HDAC inhibitors (Roberti et al. 2019; Karagiannis and Rampias 2021). BET inhibitors (iBETs) that bind reversibly to the bromodomain of BET proteins continue to be studied to suppress oncogenic networks.
2.3.2 Histone Methylation
The methylation of histones is a process that occurs mainly at lysines (K) and arginines (R) and plays essential functions in differentiation and development. Dynamic methylation processes require methyl transferases as “writers,” demethylases as “erasers,” and effector proteins as “readers.” Lysine methyltransferases (KMTs) and arginine methyltransferases (PRMTs) are enzymes that transfer methyl groups from S-adenosyl methionine (SAM). Lysine demethylases (KDMs) remove methyl groups from histone lysine residues (Fig. 3.6).

Methylation sites in histone 3 and the enzymes (KMTs and KDMs) involved in process
The effects of methylation on histones can be correlated with various gene expression statuses. For instance, methylation of H3K9, H3K27, and H4K20 inhibits gene expression, whereas methylation of H3K4, H3K36, and H3K79 stimulates gene expression but the final effect on chromatin is affected by the interaction of several histone modifications known as histone crosstalk. The same modification may have distinct functional effects depending on the methylation status (e.g., H3K4me2 and H3K4me3) and chromosomal position (Izzo and Schneider 2010). The involvement of histone methylation in transcriptional regulation is associated with chromatin structure, recruitment of transcriptional factors, interactions with initiation and elongation factors, and effects on RNA processing (Zhao and Shilatifard 2019).
Although methylation and demethylation processes’ role in cancer development/progression remains unclear, it is known that abnormalities in the methylation of various lysine residues by histone lysine methyl transferases can alter gene expression specific to certain neoplastic and normal cell types (Neganova et al. 2022). As expected, misregulation of KMTs has been associated with numerous cancers, such as EZH2 overexpression has been detected in breast, bladder, and prostate malignancies, and NSD2 has been associated with tumor aggressiveness and poor prognosis in various types of cancer (Albert and Helin 2010).
All KMTs have SET (Suppressor of variegation, Enhancer of Zeste, Trithorax) domain for their catalytic activity, except disruptor of telomeric silencing 1-like (DOT1L) methyltransferase. The human genome encodes 48 proteins containing SET domains. KMTs also methylate lysines in non-histone proteins. SET7/9, for instance, can stabilize the tumor suppressor p53 by methylating K372 (Chuikov et al. 2004; Cheng et al. 2019; Albert and Helin 2010).
MLL1 (KMT2A), which specifically methylates histone H3 lysine 4, is implicated in various forms of cancer with loss of function and rearrangement. Leukemogenesis can be induced by MLL fusion proteins that alter the proliferation and differentiation of hematopoietic cells. HOXA9 transcriptional regulation is disrupted due to an increase in H3K4me3 elicited by MLL1 translocation in myeloid and lymphoid leukemias. More than 50 MLL fusion proteins have been identified in AML, ALL, and MLLs (Audia and Campbell 2016; Neganova et al. 2022).
Methyltransferase DOT1L catalyzes H3K79 methylation, which occurs in the core of histone H3 rather than on its N-terminal tail and is thought to increase gene expression. H3K79 methylation regulates chromatin structure, transcription, DNA damage response, and cell cycle processes. Misregulation of these mechanisms via aberrant DOT1L function and defects in H3K79 methylation can lead to aneuploidy, telomere elongation, and disturbances in cell proliferation (Ljungman et al. 2019; Guppy et al. 2017). The identification of abnormal upregulation of H3K79 methylation in leukemia led to the development of the DOT1L inhibitor (Zhao and Shilatifard 2019). DOT1L is recruited by MLL fusion partners, resulting in aberrant H3K79 methylation that leads to increased transcription of MLL fusion target genes. DOT1L also has an effect on the development and progression of some solid tumors such as breast, lung, and ovarian cancers (Neganova et al. 2022; Song et al. 2020).
Enhancer of zeste homolog 2 (EZH2), one of the best-studied HMT enzymes involved in oncogenesis, is responsible for the di- and trimethylation of H3K27 (H3K27me2 and -me3). The members of the enhancer of zeste homolog family are the catalytic components of polycomb repressor complexes (PRCs) responsible for gene silencing (Cohen et al. 2011). EZH2 has the potential to function as an oncogene by playing a role in the H3K27me3-mediated aberrant silencing of the promoters of some tumor suppressor genes. EZH2 overexpression and gain-of-function mutations have been associated with many types of cancer. Overexpression of EZH2 has been linked to some solid tumors such as prostate, bladder, colon, and breast cancers and is also associated with aggressive and metastatic disease in prostate cancer (Chase and Cross 2011). B-cell lymphoma cell lines and lymphoma samples with heterozygous EZH2Y641 mutations have exhibited elevated H3K27me3 (Yap et al. 2011). Dysregulation of EZH2 in cancer may occur with the effect of multiple microRNAs. For example, targeting EZH2, miR-101 also regulates cell proliferation, invasion, and tumor growth. Loss of miR-101 has been shown in prostate cancer to lead to overexpression of EZH2 (Varambally et al. 2008). EZH2 loss-of-function mutations have also demonstrated a potential tumor suppressor role in hematologic malignancies (Khan et al. 2013).
H3K9 mono-, di-, or trimethylation is associated with different chromatin states, aberrantly regulated in multiple cancers. For example, H3K9me3 correlates with transcriptionally inactive chromatin and acts as a specific binding platform for heterochromatin protein 1 (HP1). The SUV39H1 and SUV39H2 enzymes preferentially trimethylate H3K9 and are crucial in forming constitutive heterochromatin, primarily pericentric heterochromatin (Lachner et al. 2001; Cohen et al. 2011). Dysregulation of members of the H3K9 methyltransferase family has been demonstrated in numerous cancers. KMT1A/SUV39H1 has been overexpressed in breast cancer but has not been correlated with disease progression (Patani et al. 2011).
Histone demethylases can be classified into two groups: The lysine-specific demethylases (LSDs) and Jumonji C (JmjC) domain-containing histone demethylases (KDM2–8) (Cheng et al. 2019). The first reported lysine demethylase specific for residues H3K4 and H3K9 is LSD1 (KDM1A), which has been identified as overexpressed in several cancer types. Non-histone proteins such as p53, E2F1, and HIF-1 are also demethylated by KDM1A (Sterling et al. 2021). For example, LSD1 has been shown to suppress p53 function by inhibiting the interaction of p53 with p53-binding protein 1 (53BP1) (Huang et al. 2007).
KDM2A promotes tumor growth and invasion in lung cancer by increasing ERK1/2 and JNK1/2 activities through H3K36 demethylation at the DUSP3 promoter (Wagner et al. 2013). KDM2B is thought to function as an oncogene and plays a critical role in the development and maintenance of leukemia cells (He et al. 2011a). Similarly, KDM3 enzymes are overexpressed in various tumors and implicated in oncogenic processes. KDM3A has been demonstrated to control the invasion and apoptosis of breast cancer cells and maintain myeloma cells’ survival (D’oto et al. 2016). KDM4B and KDM4C catalyze the demethylation of H3K9me3/me2 mark and have been shown that amplified in medulloblastoma, malignant peripheral nerve sheath tumors, and squamous cell carcinoma. KDM4B also plays an important role in the regulation of the N-Myc pathway in neuroblastoma. Glioblastoma stem cells exhibit lower levels of H3K9me3/me2 and H3K27me3/me2 than differentiated cells (Mallm et al. 2020; Yang et al. 2015).
KDM5 subfamily catalyzes only H3K4me3/me2, gene activating marks. KDM5 family members may be involved in the downregulation of tumor suppressors and oncogenes (Sterling et al. 2021). KDM5A is overexpressed in several cancer types. For instance, KDM5A-mediated-H3K4me3 demethylation results in downregulation of the expression of genes encoding the tumor suppressor proteins p16 and p27 in breast cancer (Yang et al. 2019). Furthermore, KDM5B inhibits their oncogenic potential by reducing H3K4me3/me2 on oncogenes such as Hox/Meis in leukemia stem cells (Wong et al. 2015). It has been reported that low KDM5C levels in renal cancer cells trigger genomic instability and are associated with poor prognosis in patients (Rondinelli et al. 2015). Disruption of the histone demethylase KDM6A, the first reported mutation in cancers, leads to cell cycle dysregulation. The roles of KDM6 enzymes appear context-dependent in cancer. Tumor suppressor and oncogenic effects have been observed in different studies (D’oto et al. 2016).
2.3.3 Histone Ubiquitination
Ubiquitin (Ub) is a highly conserved, 76-amino acid regulatory protein. In 1977, Gold Knopf et al. identified histone ubiquitination that is involved in many cellular processes, including transcription, DNA repair, and genome stability. Ubiquitination is a modification that tags substrate proteins with Ub and involves a multi-step enzymatic process, including ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin-protein ligase to attach to the substrate. In this enzymatic process, Ub is adenosine triphosphate-dependently activated and transferred to E2. Finally, a ubiquitin ligase binds ubiquitin to the specific lysine residue. Deubiquitinating enzymes (DUB, also known as ubiquitin-specific peptidases (USPs)) remove ubiquitin (Ub) from target proteins (Fig. 3.7). Considering its cellular functions, it is not surprising that aberrant ubiquitination induces oncogenesis by altering the expression of oncogenes and promoting cancer cell proliferation as with other PMTs (Jeusset and McManus 2019; Deng et al. 2020).

Schematic illustration of the ubiquitination process
Although polyubiquitination of canonical protein is a mark for proteasome-mediated degradation, histone ubiquitination has been associated with controlling various pathways and activities rather than degradation. H2A and H2B are the most abundant ubiquitinated proteins in the nucleus. Although H1, H3, and H4 ubiquitination have been reported, the biological function of these modifications has yet to be fully elucidated (Thompson et al. 2013).
H2A may be either mono- or polyubiquitinated, but H2B is often monoubiquitinated. H2B monoubiquitination (H2Bub1) is a crucial modification for transcriptional activation and tumor suppression. Loss of global H2Bub1 has been reported in breast, lung, and parathyroid cancers and has also been correlated with poor survival in colorectal cancer patients (Cole et al. 2015; Melling et al. 2016). It has been shown that a reduction in H2Bub1 affects the transcriptional mechanism of the ER and may also potentially play a role in estrogen-independent proliferation. H2Bub1 levels have been reported to be decreased in both primary and metastatic breast cancers, although they remain unchanged in benign breast tissue (Prenzel et al. 2011; Wu et al. 2015; Dwane et al. 2017) Depletion E3 ligase RNF20, which is responsible for H2B ubiquitination, has increased cell migration, eliciting transformation and tumorigenesis. RNF20 promoter hypermethylation in primary breast cancer cells and mutation at low frequency in colorectal cancer have been reported (Shema et al. 2008; Marsh and Dickson 2019). Rearrangements of the mixed lineage leukemia proto-oncogene MLL1 initiate aggressive forms of acute leukemia and are associated with poor prognosis. It has been shown that suppression of RNF20, which is required for MLL fusion-mediated leukemogenesis, leads to inhibition of cell proliferation (Wang et al. 2013). H2Bub1 is also required to recruit players in the DNA repair pathways (Moyal et al. 2011). Failure to repair DNA can cause chromosomal instability and contribute to the tumorigenic process (Thompson et al. 2013). On the other hand, some studies have shown that high levels and/or activity of H2Bub1 and its E3 ligases may have an oncogenic effect (reviewed in (Wright et al. 2011)). USP22 is the best-characterized DUB of H2BK120ub1. USP22 overexpression was reported to be associated with more aggressive tumors and poor prognosis in breast cancer (Zhang et al. 2011).
Lysine 119 in H2As is the most frequently observed ubiquitination site. Really interesting new gene 1A (RING1A) and RING1B, and B-lymphoma Moloney murine leukemia virus insertion region 1 homolog (BMI1) are ubiquitin ligases responsible for the monoubiquitination of H2AK119 that plays a central role in transcriptional repression by coordinating with H3K27 trimethylation. USP16 and breast cancer type 1 susceptibility protein (BRCA1)-associated protein 1 (BAP1) are DUBs for H2AK119ub1. Mammals have two primary Polycomb group complexes, PRC1 and PRC2. H2A monoubiquitination is also involved in X inactivation. RING1B and H2Aub affect the initiation of imprinted and random X-chromosome inactivation. Loss of ubiquitylation of histone H2A in BRCA1-deficient mice resulted in disrupting structural heterochromatin and gene silencing integrity in the repeat regions (Zhu et al. 2011).
2.3.4 Histone Phosphorylation
Histone phosphorylation is a reversible PMT that usually occurs at serine (S), threonine(T), and tyrosine (Y) residues of histone tails and is controlled by various kinases and phosphatases. Histones H1, H2A, H2B, H3, and H4 are phosphorylated at multiple sites. It has been implicated in DNA repair, regulation of transcription, apoptosis, and chromatin remodeling (Shanmugam et al. 2018).
Phosphorylation of the histone H2A subtype, H2AX, at the Ser139 position occurs in response to DNA damage and is mediated by ATM and ATR (Podhorecka et al. 2010). Histone phosphorylation has been found to be associated with transcriptional regulation and gene expression, particularly genes that regulate cell cycle and proliferation. For example, H3S10 and 28, H2BS32 phosphorylations have been related to activation of epidermal growth factor (EGF)-mediated gene transcription. Aurora B, responsible for H3S10 phosphorylation, has been identified as being overexpressed in various solid tumors, including breast and colorectal cancers (Hosseini and Minucci 2018). An increase in H3S10 phosphorylation has been observed in breast cancer, esophageal squamous cell carcinoma, gastric cancer, glioblastoma, melanoma, and nasopharyngeal carcinoma (Komar and Juszczynski 2020). Small-cell lung cancers (SCLC) with c-MYC amplification/high expression have been shown to respond to Aurora B inhibitors (Helfrich et al. 2016).
H3Y41 phosphorylation and displacement of HP1α can lead to oncogene activation, inducing tumorigenesis. H3Y41 phosphorylation of Janus kinase 2 (JAK2) has been observed to cause disrupting chromatin binding by heterochromatin protein 1α (HP1α). Inhibition of JAK2 activity reduces the phosphorylation of H3Y41 in the promoter of the hematopoietic oncogene Imo2 and expression and also increases HP1 α binding at the same site in human leukemic cells (Dawson et al. 2009). Gene amplification, mutation, and/or rearrangement of JAK2 have been shown in several hematological malignancies.
2.3.5 Other Modifications
SUMOylation is a negative regulator and is known to reduce transcriptional activity. Small ubiquitin-like modifier (SUMO) pathway is involved in carcinogenesis, the regulation of DNA damage repair, immune responses, carcinogenesis, cell cycle progression, and apoptosis. Blocking sumoylation results in decreased proliferative capacity and induction of antitumor immune response in cancer cells. Key pathways related to cancer, such as PI3K/AKT/mTOR, JAK-STAT, MAPK/ERK cascade, TGF signaling, and EMT pathway, are subjected to SUMO control. Some tumor suppressor genes and proto-oncogenes are also SUMO targets (Shanmugam et al. 2018; Lara-Ureña et al. 2022).
O-GlcNAcylation is catalyzed by O-Linked N-acetylglucosamine (O-GlcNAc) transferase (OGT) and O-GlcNAcase (OGA). Alteration of these processes may lead to tumorigenesis (Forma et al. 2014). Low expression of OGA in hepatocellular carcinoma tissues has been suggested to be a prognostic marker for tumor recurrence (Zhu et al. 2012). It has been shown that global GlcNAcylation levels are significantly elevated in tumor tissues, and there is a significant increase in metastatic lymph nodes compared to the corresponding primary tumor tissues (Gu et al. 2010). Overexpression of OGT has been reported to alter mitotic histone post-translational modifications of histone H3 in Lys-9, Ser-10, Arg-17, and Lys-27 (Sakabe and Hart 2010).
2.4 Chromatin Remodelers
Chromatin remodeling complexes are regulators that remodel nucleosomes in an ATP-dependent manner and have essential roles in DNA damage repair, recombination, replication, and transcriptional control, and aberrations in this process can induce carcinogenesis. SWI/SNF, ISWI, INO80, and NuRD/Mi-2 are the best-characterized remodelers (Nair and Kumar 2012).
SWItch/Sucrose Non-Fermentable (SWI/SNF) chromatin remodeling complex uses energy from ATP dephosphorylation to alter chromatin accessibility by chromatin repositioning, exchanging specific or all nucleosome cores, and histone dimer eviction (Tsuda et al. 2021). The SWI/SNF complex is known to control transcription by regulating acetylated histone H3K27. Alterations in genes encoding SWI/SNF remodeling factors such as ARID1A have been identified in about 8% of human cancers. ARID1A has a role in the ability of the SWI/SNF complex to inhibit cell growth and prevent genomic instability (Krishnamurthy et al. 2022; Tsuda et al. 2021). ARID1A mutations were observed in 13% of hepatocellular carcinoma, 9.6% of gastrointestinal adenocarcinoma, 2.5% of malignant melanoma, and 57% of ovarian clear cell carcinoma (Okawa et al. 2017).
Imitation switch (ISWI) family, which is included in the ATPase family, is involved in many cellular processes, such as transcriptional regulation, DNA damage response, repair, and recombination. ISWI subunits are thought to be involved in tumorigenesis by regulating oncogenic gene transcription. Somatic mutations, copy number variations, and gene fusions have been identified in various tumor types for ISWI subunits (Li et al. 2021b).
Ino80 ATPase is a member of the SNF2 family of ATPases and a component of the INO80 ATP-dependent chromatin remodeling complex (INO80). Ino80 overexpression has been shown to promote proliferation in the immortalized cervical epithelial cell line and non-small-cell lung cancer cells. It is thought that INO80 binds to enhancer regions near cancer-associated genes, promoting their expression (Hu et al. 2016; Zhang et al. 2017). Ino80 silencing also inhibited melanoma cell proliferation, anchorage-independent growth, and tumorigenesis (Zhou et al. 2016).
Nucleosome remodeling and deacetylase complex (Mi-2/NuRD) that function in gene repression contain histone deacetylases (HDAC1/2), metastasis-associated (MTA1/2) proteins, and methyl CpG-binding domain (MBD) proteins. Overexpression of MTA1 has been observed in gastrointestinal and esophageal carcinomas and breast adenocarcinomas (Fu et al. 2011; Toh and Nicolson 2009). It was shown that PML-RARa binds and recruits NuRD to target genes, including the tumor suppressor gene RARβ2. Knockdown of the NuRD complex in leukemic cells prevented histone deacetylation and chromatin compaction and promoted cellular differentiation by disrupting stable silencing and DNA and histone methylation (Morey et al. 2008).
2.5 miRNAs in Cancer
Understanding how cancer begins and progresses is essential for cancer prevention, early detection, and treatment. Since changes in gene expression also have important effects on cancer, microRNA (miRNA) research has also been a focus in recent years.
microRNAs are a type of non-coding RNA, 19–25 nucleotides in length, that regulate gene expression post-transcriptionally. A microRNA can target hundreds of genes and affect their expression (Lu and Rothenberg 2018). miRNA sequences can be located within introns, exons of non-coding RNAs and a intron of pre-mRNA (pre-messenger RNA). Most miRNAs are expressed by RNA polymerase II (RNA pol II), but some are transcribed by RNA polymerase III (Borchert et al. 2006; Lee et al. 2004).
2.5.1 miRNA Biogenesis and Functions
miRNA biogenesis occurs by two different pathways; canonical and non-canonical pathways.
2.5.1.1 Canonical miRNA Biogenesis
Most intergenic miRNAs use their own promoter region. miRNA sequences are located in exons or introns of non-coding RNAs. Polymerase II synthesizes pi-miRNAs containing at least 1 hairpin structure. pi-miRNAs are divided into structures called precursor miRNAs (pre-miRNAs). Each pre-miRNA is about 70 nt long, and this process takes place in the nucleus. Then, pre-miRNAs are exported to the cytoplasm. Drosha, DiGeorge syndrome critical region gene 8 (DGCR8), XPO5, and Ras-related nuclear protein (RAN) are involved in this process (Saliminejad et al. 2019). The microprocessor complex, consisting of Nuclear RNAase III DROSHA and its cofactor DGCR8, serves in the cleavage of pi-miRNA to form pre-miRNA (Nguyen et al. 2018). The Ran/GTP/Exportin 5 complex is involved in the transport of pre-miRNA into the cytoplasm. In the cytoplasm, the pre-miRNA is cleaved into a double strand, one of which is the passenger strand and the other is the guide strand. This process is catalyzed by Dicer, an RNAase III enzyme (Peng and Croce 2016). The mature miRNA gets its name from the 5′ or 3′ directionality of the strands. Both strands can be loaded into the Argonaute (AGO) protein family. Which strand will bind to AGO depends on the cell type and cell environment. The unloaded strand is identified as the passenger strand and is degraded by AGO2 (O’Brien et al. 2018).
Repression of transcription by miRNA is classically mediated by miRNA-induced silencing complex (miRISC). The miRISC allows to recognize 3’UTR region of the target mRNA. However, it is stated that mRNA can be recognized in the 5’-UTR and even in protein-coding sequences. The target mRNA is recognized by the sequences on it called miRNA response elements (Saliminejad et al. 2019). The degree of complementarity of miRNA with mRNA determines whether it is repressed by AGO2 or miRISC. Full complementarity between miRNA and mRNA activates AGO2 endonuclease activity and mRNA is cleaved (Fig. 3.8) (O’Brien et al. 2018).

Biogenesis and functions of miRNA
It has been stated that miRNA can suppress translation in three different ways: (i) Ago2 interacts with TNRC6, which recruits the CCR4-NOT deadenylase complex. So, the mRNA is deadenylated and degraded. (ii) TNRC6 interacts with the Dcp 1/2 cap complex, which cleaves the 5′ capped mRNA and destabilizes the mRNA. (iii) With the binding of Ago 2, mRNA is rendered inaccessible for ribosome attachment and function, which inhibits the translation process. When Ago 2 binds, the mRNA cannot interact with the ribosome and the translation process is suppressed. Transfer of Ago 2 with mature miRNA to the nucleus is via Importin 8, while TNRC6 is transported via Importin β. In the nucleus, RISC is assembled and RISC can be transported via Exportin1 (Fig. 3.8) (Liu et al. 2018).
It has long been known that miRNAs play a role in gene regulation post-transcriptionally. However, it has also been discovered to interact with long non-coding RNA (lncRNA), circular RNA (circRNA), and pseudogenes. This information indicates that while investigating the effects of miRNAs on diseases, the processes in question are much more complex. These functions of miRNAs will be explained in the sections on the effects of miRNAs in cancer.
2.5.1.2 Non-canonical miRNA Biogenesis
Although many different pathways have been described for non-canonical miRNA biogenesis, the well-recognized are Drosha- and Dicer-independent pathways. As an example of the Drosha/DGCR8-independent pathway, mirtrons produced from mRNA introns, have the property of being dicer substrates. Then it is included in the canonical pathway (O’Brien et al. 2018; Saliminejad et al. 2019). In the Dicer-independent pathway, the endogenous hairpin transcripts are short to become Dicer substrates and therefore require AGO2 (Fig. 3.8) (O’Brien et al. 2018).
2.6 The Role of miRNA in Cancer
Among the non-coding RNAs that play critical roles in gene regulation, microRNAs (miRNAs) are the most studied type of non-coding RNA in different types of cancer. The association between miRNAs and cancer was first discovered in CLL patients with 13q deletion. Two microRNAs (miR-15a and miR-16-1), deleted or downregulated, were discovered in the majority of CLL patients (Calin et al. 2002). After a while, it was determined that these microRNAs role as tumor suppressors by suppressing the BCL2 (B-cell lymphoma 2) gene (Cimmino et al. 2005).
miRNAs act as tumor suppressors (oncosupressor-miR) or oncogenes (onko-miR) depending on the functions of the genes they target. One of the well-known oncosuppressor-miRs is let-7. Expression of let-7 has been shown to be decreased in various cancers and associated with poor prognosis (Boyerinas et al. 2010). OncomiRs generally contribute to tumor development by targeting genes that control cell division, differentiation, and apoptosis (Lujambio and Lowe 2012). miR-21 is the first miRNA discovered in humans. As a result of transcript profiling studies conducted after many miRNA discoveries, miR-21 was shown to be upregulated in various cancers such as breast cancer, chronic lymphoblastic leukemia, lung cancer, prostate cancer, colon cancer, and glioblastoma. Subsequent function studies have shown that miR-21 has oncogenic activity (Selcuklu et al. 2009).
2.6.1 Proliferation and miRNAs in Cancer
Suppression of cell differentiation and maintenance of proliferation is one of the very important mechanisms in tumorigenesis. The role of miRNAs in cell cycle progression was first proven by Hatfield et al. They showed that G1/S transition was suppressed when DICER-1 knockout in Drosophila germline stem cells. This proved that miRNAs have a role in the normal G1/S transition (Hatfield et al. 2005).
The E2F family of transcription factors controls cell proliferation. E2F1 acts as a tumor suppressor and induces transcription of the target gene in the transition from G1 to S stage. After c-MYC is activated, miR-17-92 inhibits the translation of E2F1. Since C-MYC also directly induces mir-17-92, this mechanism is evidence of a normal cell cycle process under normal conditions (Coller et al. 2007). The overexpression of miR-17-92 cluster has been demonstrated to have oncogenic functions in many cancer types (Kalkan and Atli 2016; Fang et al. 2017; Gruszka and Zakrzewska 2018).
2.6.2 Apoptosis and miRNAs in Cancer
Evasion of apoptosis is an important mechanism for tumor cells, and the cells can choose many different pathways for this. Although the most common mechanism is the loss of TP53 function, upregulation of anti-apoptotic regulators and suppression of pro-apoptotic regulators can also occur (Peng and Croce 2016).
Activation of miR-192, miR-194, and miR-215 by TP53 and suppression of MDM2 by targeting mRNA transcribed from the MDM2 gene has been demonstrated in multiple myeloma. Because the MDM2 gene is the negative regulator of TP53 (Nag et al. 2013), downregulation of these miRNAs is an important mechanism in the development of multiple myelomas (Pichiorri et al. 2010). In a recent study, it was shown that the expression of miRNA-331-3p is downregulated in nasopharyngeal carcinoma patients and that overexpression of this miRNA leads to inhibition of phosphorylation of Phosphoinositide 3-kinase (PI3K) and Serine/threonine kinase (AKT). miRNA-331-3p has been shown to suppress proliferation and induce apoptosis (Xuefang et al. 2020).
2.6.3 Invasion, Metastasis, and miRNAs in Cancer
Epithelial-mesenchymal transition (EMT) is a very important mechanism for invasion and metastasis. Activation of EMT is required for cell migration and invasion, while mesenchymal-to-epithelial transition (MET) is required for metastasis outgrowth (Tan et al. 2018). EMT is characterized by loss of adhesion, decreased expression of E-cadherin, acquisition of mesenchymal markers, and mobilization of the cell.
Many transcription factors such as Snail, Slug, Twist, ZEB1, and ZEB2 are involved in the EMT process. The miR-205 and miR-200 family have been shown to be epithelial markers and suppressors. The miR-200 family target ZEB1/2 and act to suppress EMT. In contrast, ZEB1 directly binds to the promoter regions of miR-200 genes and represses its transcription. That is, there is a double negative feedback loop. While expression of the MiR-200 family is absent in metaplastic breast cancer cells, ZEB1 and ZEB2 are highly present in invasive mesenchymal cells (Zhang and Ma 2012). miR-99a inhibits the expression of E2F and adhesion G protein-coupled receptor E2 (ADGRE2), thereby suppressing the EMT process. miR-5188 targets the Fork-headed Box Protein O1 (FOXO1) gene and can activate the Wnt signaling pathway via β-catenin, thereby EMT is induced (Pan et al. 2021).
Thanks to the studies on the effects of miRNA on EMT, a lot of information has been obtained about cancer development and metastasis, and it is even among the subjects of drug resistance studies.
2.6.4 Angiogenesis and miRNAs in Cancer
One of the necessary mechanisms for tumor growth and metastasis is angiogenesis. It has been determined that miRNAs are effective in the mechanism of angiogenesis.
miR-34a is one of the most studied miRNAs in cancer and is known to have a suppressive effect on angiogenesis. miR-34a achieves this effect through the interactions of Silent Information Regulator 1 (Sirt1), Foxo1, Notch1 and Tp53. The mir-29 family also inhibits angiogenesis and tumorigenesis and has been shown to be downregulated in many varieties of cancers. miR-29b targets AKT3 and inhibits Akt3-mediated vascular endothelial growth factor (VEGF) and C-myc activations (Lahooti et al. 2021).
Considering that miRNAs are highly effective in angiogenesis and tumorigenesis, their potential to be a treatment target is quite high.
2.6.5 Non-canonical Function of miRNA in Cancer
For a long time, miRNAs were considered to suppress expression by targeting only mRNAs. however, in recent years, evidence has been presented that it both suppresses and increases expression. In recent studies, it has been shown that miRNAs also target the 5’UTR regions of mRNAs and have an effect on increasing transcription (Semina et al. 2021). It has been found that miR-1254 together with Ago/2 and iRISC, interacts with the 5’UTR region of mRNA of cell cycle and apoptosis regulator (CCAR1) and causes its upregulation, thus re-sensitizing mammary cancer cells resistant to tamoxifen (Li et al. 2016). Human miR-369-3 can activate the translation of tumor necrosis factor-α (TNF-α) mRNA when the cell cycle is stopped but suppresses it when cell division occurs. These data support that miRNAs have many functions in the cytoplasm, apart from targeting and suppressing mRNAs (Semina et al. 2021).
Evidence that miRNAs regulate expression in the nucleus has recently been found. It also performs the function of repressing transcription through traditional RISC in the nucleus. They also bind to promoter regions, alter the epigenetic profile, and regulate gene expression (Liu et al. 2018).
In the nucleus, the RNA-Ago complex can directly target non-coding transcripts and modify epigenetic modifications to serve as a scaffold on which epigenetic factors will be recruited. In a study, it was shown that three signaling molecules were activated in response to endoplasmic reticulum stress, and PERK, which is among these molecules, induced miR-211. It was determined that miR-211 increased methylation in the promoter of the proapoptotic transcription factor C/EBP homologous protein (CHOP), which resulted in decreased CHOP expression (Chitnis et al. 2012).
In addition to all these, it has been observed that miRNAs also connect with non-AGO proteins in tumor cells. Downregulation of miR-328 expression has been observed in the blast crisis of chronic myeloid leukemia (CML). It was found that miR-128 directly binds to hnRNP E2 and rescues the translation of the differentiation-inducing transcription factor CEBPA mRNA (Dragomir et al. 2022; Eiring et al. 2010).
The encoding of mRNA-encoded peptides (miPEP) by pri-miRNAs is one of the non-canonical actions of miRNAs. It has been determined that pri-miRNAs transcribed from MIR200A and MIR200B in prostate cancer encode miPEP200a and miPEP200b and these miPEPs show antioncogenic effect by inhibiting migration (Dragomir et al. 2022).
2.6.6 Deregulation of miRNA Expression in Cancer
After realizing that the expression of miRNAs was deregulated in tumor cells, many studies were conducted. Understanding the mechanisms that cause the dysregulation of cancer miRNA expression is very important for tumorigenesis, development, metastasis, and treatment.
One of the most common causes of miRNA expression changes in cancer cells is numerical and structural anomalies in the genome (such as amplification, deletion, and translocation). 13q deletions in CLL, which led to the establishment of the first association between miRNAs and cancer, are an example of decreased expression of miR-16-1 and miR-15a due to copy number loss (Calin et al. 2002). The miR-17-92 cluster has been amplified in lung and B-cell lymphoma, and it has been found to undergo a translocation that will lead to overexpression in T-cell acute lymphoblastic leukemia (Peng and Croce 2016). The relationship between chromosome breaks and miRNA localization was first discovered in the sample with t(8;17) anomaly. The miR-142 gene was determined to be located at a distance of 50 nt from the break point of chromosome 17, where it was included in t(8;17), and it was likely that the regulatory elements of miR-142 increased the expression of C-MYC (Calin and Croce 2006).
The expression of miRNAs is controlled by many different transcription factors. Two of these transcription factors are Tp53 and C-Myc, which are known to have important effects on tumorigenesis. C-Myc binds to the promoter of miR-17-92, which has oncogenic properties and activates its transcription. In addition, it suppresses the transcriptional activity of tumor suppressor miRNAs such as mir-15a, miR-26, miR-29, mir-30, and let-7 families (Chang et al. 2008). Expression of the miR-34 family is controlled by Tp53. When cell stress increases, miR-34 activates TP53. Expression of miR-145 is also induced by upregulated TP53. However, the miR-143/145 cluster is suppressed by the RAS signal. RAS-responsive element-binding protein 1 (RREB1) transcriptionally represses the miR-143/145 cluster, and then miR-143/145 represses the expression of RREB1 (Ali Syeda et al. 2020).
One of the factors affecting miRNA expression is epigenetic changes. It has been determined that, like the hypermethylation of CpG islands in the promoters of tumor suppressor genes, the expression of miR-124 is also suppressed due to hypermethylation in their promoters in leukemia, lymphoma, breast, colon, and liver cancers (Lujambio et al. 2007; Ali Syeda et al. 2020).
Another mechanism that causes miRNA deregulation is mutations. The first discovered germline mutation in miRNA was detected in miR-16-1 (Calin et al. 2005). The most mutated miRNAs in the analysis of all cancers were MIR1324, MIR1303, and MIR4686, whereas MIR142, which has driver mutations in DLBCL, CLL, acute myeloid leukemia (ALL), and other kinds of lymphoma, was the most mutated miRNA in a particular cancer (Dragomir et al. 2022). Mutations or expression changes can be observed in DNA sequences encoding all proteins involved in miRNA biogenesis as well as in miRNA genes. Various mutations or change of expression have been detected in DROSHA, DICER, DGCR8, AGO, and EXPORTIN 5 genes, which are involved in miRNA biogenesis, in different cancer types (Ali Syeda et al. 2020; Peng and Croce 2016).
2.7 Circulating miRNA in Cancer
Extracellular miRNAs are highly durable and stable. Extracellular miRNAs exist as part of vesicles or as a soluble form of protein-containing complexes. HnRNPA2B1 and HnRNPA1 proteins regulate the loading of miRNAs into exosomes by identifying particular sequence patterns. As the suppression of neutral sphingomyelinase 2 (nSMase2), an enzyme involved in ceramide production, downregulates exosome secretion and releases exosomal miRNAs into the extracellular environment, exosomal miRNAs can be exported outside the cells through a ceramide-dependent mechanism. Although various distinct routes for miRNA entry into cells have been postulated, the mechanisms for exosomal miRNAs uptake by cells are currently poorly understood. Exosomes can enter cells through a variety of methods, including endocytosis, phagocytosis, and micropinocytosis. Another is a direct fusion of exosomes with the plasma membrane. Exosome-free miRNAs can also enter cells by way of certain receptors. Exosomes that contain miRNAs that are produced by tumor cells can be taken up by the recipient cells. MiRNAs can affect the development of tumors by promoting or inhibiting cell invasion, metastasis, and tumor neoangiogenesis. Exosomal miRNAs can potentially modify the extracellular matrix or attract and activate immune cells, which can both have an impact on the tumor microenvironment (Semina et al. 2021).
The first circulating miRNAs were discovered in patients with diffuse large B-cell lymphoma. As a result of subsequent studies, it was shown that miRNAs could be used to determine tumor grades or to evaluate treatment responses. Unlike mRNAs, their ability to stay for a long time without degradation also provides an advantage in using miRNAs as biomarkers (Smolarz et al. 2022).
2.8 miRNA-based Biomarkers in Cancer
After the discovery of the roles of miRNAs in cancer, it was inevitable to investigate the relationships between miRNAs and cancer types and disease prognosis. There is a large amount of data proving that many miRNAs can be diagnostic and prognostic markers. In addition to all these, miRNAs have become a treatment target in cancer.
There are many studies proving that miRNAs will show clinical benefits as diagnostic and prognostic markers (He et al. 2020). In a study investigating the role of miRNAs in triple-negative breast cancer (TNBC), databases such as PubMed, ScienceDirect, Springer, Web of Science, and Scopus were searched and 197/1233 articles were extensively reviewed. Many miRNAs have been reported that have the potential to be of prognostic and diagnostic importance, e.g., miR-9, miR-21, miR-93, miR-181a/b, miR-182, miR-221, miR-321, miR-155, miR-10b, miR-29, miR-222, miR-373, miR-145, miR-199a-5p, miR-200 family, miR-203, and miR-205 (Sabit et al. 2021).
MiR-155-5p, an oncogenic miRNA, regulates important transcription factors such as E2F2, hypoxia-inducible factor 1 (HIF1), and FOXO3. One study showed that the upregulation of miR-155-5p is associated with short overall survival in cases of chronic lymphocytic leukemia (CLL) (Papageorgiou et al. 2017).
Although hematuria is the most common symptom of bladder cancer (BC), hematuria is not a definitive diagnostic marker. In a study conducted, urinary cell-free microRNA expression differences were investigated to distinguish patients with BC from patients with hematuria, and the ratio of miR-612–miR-4511 was found to be significantly higher in BC (Piao et al. 2019).
One of the biggest problems in cancer treatment is the late detection of cancer. Plasma/serum circular miRNA can be used in the diagnosis of breast, colorectal, stomach, lung, pancreatic, and hepatocellular cancer. Circular miRNAs may contribute to the discovery of the primary origin of metastatic tumors of unknown primary tissue. In addition, circular miRNAs can be used as a marker in disease follow-up (Cui et al. 2019).
For example, it has been shown that miR-125b suppresses cell proliferation in ovarian, thyroid, and oral cancers, but induces proliferation in prostate cancers (Cui et al. 2019). Although hematuria is the most common symptom of bladder cancer (BC), hematuria is not a definitive diagnostic marker. In a study conducted, urinary cell-free microRNA expression differences were investigated to distinguish patients with BC from patients with hematuria, and the ratio of miR-6124 to miR-4511 was found to be significantly higher in BC (Piao et al. 2019). As another example, elevated levels of circulating miR-122 were found to correlate with metastatic recurrence in stage II-III breast cancer patients (Wu et al. 2012). In another study, it was determined that miR-375 and miR-200b in serum were expressed higher in patients with metastatic prostate cancer than in patients with localized cancer (Bryant et al. 2012).
2.9 miRNA-Based Therapies in Cancer
The regulatory role of miRNAs in many cancer types has made them a therapeutic target. The miRNA-based therapy methods in cancer have two approaches: increasing the activities of miRNAs that act as tumor suppressors and suppressing the functions of oncoMIRs.
Tumor suppressor miRNAs are downregulated in tumor cells and miRNA mimics are used to function as before. miRNA mimics are chemically modified (2’-O’methoxy) double-stranded RNA molecules (Menon et al. 2022). The size of miRNA is smaller than the protein, which gives it an advantage in terms of penetration into the cell. The first study to show the tumor suppressor function of Let-7 and its potential for treatment was conducted in 2008. In mouse models, it has been demonstrated that tumor growth can be inhibited by restoring let-7 (Esquela-Kerscher et al. 2008). Another study with mouse models of lung cancer demonstrated that metastasis and tumor growth could be suppressed through chemically synthesized miR-34a and a lipid-based delivery vehicle (Wiggins et al. 2010).
For the suppression of oncomiRs, small molecule inhibitors and complementary oligonucleotides such as anti-miRNA oligonucleotide (AMOs) (Amodeo et al. 2013), locked-nucleic acid antisense oligonucleotides (LNAs), antagomirs, and miRNA sponges have been developed. AMO is a DNA sequence complementary to the target miRNA and prevents the miRNA from binding to the target mRNA. LNA-AMOs are more stable and more sensitive than just AMOs. It was created as a result of the modification of AMOs. Antagomirs and miRNA sponges are longer nucleic acids that prevent miRNAs from binding to their targets (Mollaei et al. 2019; Fu et al. 2021). For example, in a study by Chen et al. (2014), it was shown that miRNA sponges successfully suppressed miR-23b expression both in vitro and in vivo, and reduced glioma angiogenesis, invasion, and migration (Chen et al. 2014).
2.9.1 Approaches for miRNA Therapeutic Delivery
Although the direct injection of miRNA mimics or inhibitors into tumor tissue is limited due to their application to localized and easily accessible solid tumors, it is an advantage that the probability of rejection by healthy organs is minimal. The development of a systemic delivery approach is needed to treat other types of cancer and metastatic tumors. For this, miRNAs must not deteriorate in the bloodstream in a short time, be able to be transported to target cells, and not cause an immunological response. Some chemical modifications are performed on miRNA oligonucleotides to increase miRNA stability and protect it from nucleases. LNAs are examples of modified nucleotides. LNA-anti-mir-122 has been shown to regulate the expression of mRNA in the liver of mice, depending on the level of miR-122 (Forterre et al. 2020).
Although viral and non-viral vectors are generally used for miRNA delivery, adverse immune responses occur against viral vectors. Tumor suppressor pri-miRNAs are inserted into a plasmid. A viral promoter, a restriction enzyme gene and an antibiotic resistance gene are contained in this plasmid. The plasmid is delivered to tumor cells in a viral vector and the mature miRNA suppresses translation or induces degradation of the target mRNA. The low cost of DNA plasmids is an advantage. Furthermore, the untranslated miRNA is transferred to the nucleus and its continuous expression is ensured. In addition, because it is translated in tumor cells, less off-target effects occur compared to synthetic miRNA sequences (Hosseinahli et al. 2018).
For the non-viral delivery system to be successful, it must prevent nuclease-mediated degradation and carry endogenous miRNA or miRNA-expressing vectors. Delivery can be accomplished using techniques such as gene gun, electroporation, or ultrasound, or using organic-based, inorganic-based, or polymer-based carriers. Although non-viral systems have less toxicity and immunological effects, low transfection efficiency is considered a disadvantage of this method (Menon et al. 2022). Radiotherapy is used in the treatment of head and neck cancer, but its clinical effect is inhibited by both the side effects of radiation and radioresistance. RNA therapeutics therefore have great potential as radiosensitizers as they can target radioresistance-specific pathways. High-density lipoprotein nanoparticle (HDL NPs) was used in a head and neck cancer cell line in a 2022 study to deliver miR-34a. As a result of the study, it was observed that proliferation decreased and apoptosis increased (Dehghankelishadi et al. 2022). Besides biomaterials, polymeric vectors (PEIs, polylactic-co-glycolic acid/PLGA, chitosans, and dendrimers) and inorganic materials (gold, diamond, silica, and iron oxide) are also used in the non-viral vectors delivery system. Among these polymers, PLGA is an FDA-approved biodegradable polymer (Forterre et al. 2020; Menon et al. 2022).
Another miRNA delivery system is the use of outer membrane vesicles (OMVs) of Escherichia coli as nanoscale spherical vesicles (Menon et al. 2022). In a 2022 study, An inexpensive and potentially mass-produced method was found for the preparation of engineered OMV with overexpressed pre-miRNA. In this study, it was discovered that OMV can be discharged from parent E. coli and inherit an overexpressed tRNALys-pre-miRNA that is used directly for the treatment of tumors. It was suggested that the OMV-based platform is a flexible and effective method to directly and specifically target individualized tumor therapy (Cui et al. 2022).
Many studies have shown that the use of miRNA-based therapies together with other treatment options such as chemotherapy and radiotherapy induces the therapeutic effect and prevents drug resistance (He et al. 2020; Menon et al. 2022).
Understanding the molecular mechanism of cancer increases the chances of treatment success. Understanding the role of miRNAs in cancer also shows the potential to be used as a treatment target or tool in the future. However, one of the most important problems is that a miRNA has more than one target. Another problem is choosing the right miRNA delivery system. Today, pre-clinical and clinical studies continue. In the future, personalized treatment options based on miRNA are expected to be developed.
3 Conclusion
In this chapter, we have reviewed the known epigenetic mechanisms in normal cells and their roles in the carcinogenesis process. The molecular processes that lead to promoter hypermethylation, genome-wide DNA demethylation, histone modifications, and non-coding RNAs were highlighted in cancer cells. The long-held conventional belief that the genetic code is the primary determinant of cellular gene function and that its change is the primary cause of human diseases has been called into question by the epigenetic revolution that has occurred in the area of biology during the last decades. The packaging of the genome may be just as important as the genome itself in regulating the vital cellular activities necessary for maintaining a cellular identity as well as in the development of disease states like cancer, according to recent developments in the field of cancer epigenetics. All cells of an individual have the same genome, but they might have different epigenotypes depending on their epigenetic markings, which are suitable for different tissues, stages of development, or environmental conditions. The partially improved treatment approaches have been made possible by a deeper understanding of the worldwide patterns of these epigenetic modifications and their related changes in cancer. Several genetic and epigenetic abnormalities, including structural variants, copy number variations, single nucleotide polymorphisms, mutations, and epigenetic dysregulations, are addressed to cancer hallmarks. To advance personalized and precision medicine and improve cancer treatment, it is crucial to comprehend the intricate interplay of genetic and epigenetic modifications. Combinatorial promising approaches that combine several epigenetic therapeutic modalities with conventional chemotherapy have a strong chance of treating cancer successfully in the future. These methods may also enable cancer cells, particularly cancer stem cells, which are resistant to conventional chemotherapy, to become more sensitive. We may be able to successfully reset the altered cancer epigenome with increased knowledge of cancer stem cells and the development of more targeted epigenetic medicine.
Compliance with Ethical Standards
Funding: The authors declare no competing financial interest.
Conflict of Interest: The authors declare no conflicts of interest.
Ethical Approval: This chapter does not contain any studies with human participants performed by the authors.
Abbreviations
- 2HG:
-
2hydroxyglutarate
- 2-OGDD:
-
2-Oxoglutarate-dependent dioxygenases
- 5caC:
-
5-Carboxylcytosine
- 5fC:
-
5-Formylcytosine
- 5mC:
-
5-Methylcytosine
- ADGRE2:
-
Adhesion G protein-coupled receptor E2
- AML:
-
Acute myeloid leukemia
- APAF1:
-
Peptidase activating factor 1
- BC:
-
Bladder cancer
- BER:
-
Base excision repair
- CGI:
-
CpG islands
- CHK2:
-
Checkpoint kinase 2
- CLL:
-
Chronic lymphocytic leukemia
- CTCF:
-
CCCTC binding factor
- DGCR8:
-
DiGeorge syndrome critical region gene 8
- DMRs:
-
Differentially methylated regions
- DNMTs:
-
DNA methyltransferases
- DSB:
-
Double-strand break
- DUSP1:
-
Dual specificity phosphatase 1 gene
- E1:
-
Ubiquitin-activating enzyme 1
- E2:
-
Ubiquitin-conjugating enzyme 2
- EMT:
-
Epithelial-mesenchymal transition
- FOXO1:
-
Fork-headed Box Protein O1
- GENIE:
-
Genomics Evidence Neoplasia Information Exchange
- H2Bub1:
-
H2B monoubiquitination
- HATs:
-
Histone acetyltransferases
- HDACs:
-
Histone deacetylases
- HIF1:
-
Hypoxia-inducible factor 1
- HMG:
-
High mobility group
- HP1α:
-
Heterochromatin protein 1α
- hTERT:
-
Telomerase reverse transcriptase
- ICRs:
-
Imprinting control regions
- ISWI:
-
Imitation switch
- JAK2:
-
Janus kinase 2
- KATs:
-
Lysine acetyltransferases
- KDMs:
-
Lysine demethylases
- KMTs:
-
Lysine methyltransferases
- LINEs:
-
Long interspaced nuclear elements
- LOI:
-
Loss of imprinting
- MAGE:
-
Melanoma antigen gene
- MBD:
-
Methyl-CpG binding domains
- MBD2:
-
Methyl-CpG-binding domain protein 2
- MeCP:
-
Methyl-CpG binding domain protein
- MeCP2:
-
Methyl-CpG-binding protein 2
- MET:
-
Mesenchymal-to-epithelial transition
- MGMT:
-
O6-methylguanine methyltransferase
- miRISC:
-
miRNA-induced silencing complex
- miRNA:
-
microRNA
- MLL1:
-
Mixed lineage leukemia 1
- ncRNAs:
-
Non-coding RNAs
- PLCD1:
-
Phospholipase C delta1
- PMDs:
-
Partially methylated domains
- PRMTs:
-
Arginine methyltransferases
- PTPRR:
-
ERK phosphatases protein tyrosine phosphatase receptor type R
- RAN:
-
Ras-related nuclear protein
- S:
-
Serine
- SAM:
-
S-adenosyl methionine
- SCLC:
-
Small-cell lung cancers
- SRA:
-
SET- and RING-associated
- SUMO:
-
Small ubiquitin-like modifier
- SWI/SNF:
-
Switching defective/sucrosenon-fermenting complex
- T:
-
Threonine
- TCGA:
-
The Cancer Genome Atlas
- TDG:
-
Thymine DNA glycosylase
- TET proteins:
-
Ten-eleven translocation methylcytosine dioxygenases
- TMZ:
-
Temozolomide
- TNBC:
-
Triple-negative breast cancer
- Ub:
-
Ubiquitin
- USPs:
-
Ubiquitin-specific peptidases
- Y:
-
Tyrosine
References
Albert M, Helin K (2010) Histone methyltransferases in cancer. Seminars in cell & developmental biology. Elsevier, pp 209–220
Ali Syeda Z, Langden SSS, Munkhzul C, Lee M, Song SJ (2020) Regulatory mechanism of microRNA expression in cancer. Int J Mol Sci 21
Allfrey VG, Faulkner R, Mirsky A (1964) Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci 51:786–794
Amodeo V, Bazan V, Fanale D, Insalaco L, Caruso S, Cicero G, Bronte G, Rolfo C, Santini S, Russo A (2013) Effects of anti-miR-182 on TSP-1 expression in human colon cancer cells: there is a sense in antisense? Expert Opin Ther Targets 17(11):1249–1261. https://doi.org/10.1517/14728222.2013.832206
Anvar Z, Chakchouk I, Demond H, Sharif M, Kelsey G, Van Den Veyver IB (2021) DNA methylation dynamics in the female germline and maternal-effect mutations that disrupt genomic imprinting. Genes 12:1214
Audia JE, Campbell RM (2016) Histone modifications and cancer. Cold Spring Harb Perspect Biol 8:A019521
Baba Y, Yagi T, Sawayama H, Hiyoshi Y, Ishimoto T, Iwatsuki M, Miyamoto Y, Yoshida N, Baba H (2018) Long interspersed Element-1 methylation level as A prognostic biomarker in gastrointestinal cancers. Digestion 97:26–30
Basu S, Dong Y, Kumar R, Jeter C, Tang DG (2021) Slow-cycling (dormant) cancer cells in therapy resistance, cancer relapse and metastasis. Seminars in cancer biology. Elsevier
Baylin SB, Jones PA (2016) Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol 8:A019505
Begam N, Jamil K, Raju SG (2017) Promoter Hypermethylation of the ATM gene as A novel biomarker for breast cancer. Asian Pacific J Cancer Prevent 18:3003
Benedetti R, Conte M, Altucci L (2015) Targeting histone deacetylases in diseases: where are we? Antioxid Redox Signal 23:99–126
Berdasco M, Esteller M (2010) Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev Cell 19:698–711
Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y, Noushmehr H, Lange CPE, van Dijk JM, Tollenaar RAEM, Van Den Berg D, Laird PW (2012) Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina–associated domains. Nat Genet 44(1):40–46. https://doi.org/10.1038/ng.969
Berkyurek AC, Suetake I, Arita K, Takeshita K, Nakagawa A, Shirakawa M, Tajima S (2014) The DNA methyltransferase Dnmt1 directly interacts with the SET and RING finger-associated (SRA) domain of the multifunctional protein Uhrf1 to facilitate accession of the catalytic center to hemi-methylated DNA. J Biol Chem 289:379–386
Bildik G, Liang X, Sutton MN, Bast RC Jr, Lu Z (2022) DIRAS3: an imprinted tumor suppressor gene that regulates RAS and PI3K-driven cancer growth motility autophagy and tumor dormancy. Mol Cancer Ther 21(1):25–37. https://doi.org/10.1158/1535-7163.MCT-21-0331
Bond DR, Uddipto K, Enjeti AK, Lee HJ (2020) Single-cell epigenomics in cancer: charting a course to clinical impact. Epigenomics 12:1139–1151
Borchert GM, Lanier W, Davidson BL (2006) RNA polymerase III transcribes human MicroRNAs. Nat Struct Mol Biol 13:1097–1101
Boyerinas B, Park SM, Hau A, Murmann AE, Peter ME (2010) The role of Let-7 in cell differentiation and cancer. Endocr Relat Cancer 17:F19–F36
Brinkman AB, Nik-Zainal S, Simmer F, Rodriguez-Gonzalez F, Smid M, Alexandrov LB, Butler A, Martin S, Davies H, Glodzik D (2019) Partially methylated domains are hypervariable in breast cancer and fuel widespread CpG island hypermethylation. Nat Commun 10:1–10
Bryant RJ, Pawlowski T, Catto JW, Marsden G, Vessella RL, Rhees B, Kuslich C, Visakorpi T, Hamdy FC (2012) Changes in circulating microRNA levels associated with prostate cancer. Br J Cancer 106:768–774
Calin GA, Croce CM (2006) MicroRNAs and chromosomal abnormalities in cancer cells. Oncogene 25:6202–6210
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L, Kipps T, Negrini M, Bullrich F, Croce CM (2002) Frequent deletions and down-regulation of micro-RNA genes Mir15 and Mir16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 99:15524–15529
Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE, Iorio MV, Visone R, Sever NI, Fabbri M, Iuliano R, Palumbo T, Pichiorri F, Roldo C, Garzon R, Sevignani C, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM (2005) A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med 353:1793–1801
Cavalli G, Heard E (2019) Advances in epigenetics link genetics to the environment and disease. Nature 571:489–499
Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT (2008) Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet 40:43–50
Chase A, Cross NC (2011) Aberrations of EZH2 in cancer aberrations of EZH2 in cancer. Clin Cancer Res 17:2613–2618
Chauhan S, Sen S, Chauhan SS, Pushker N, Tandon R, Kashyap S, Vanathi M, Bajaj MS (2021) Stratifin in ocular surface squamous neoplasia and its association with P53. Acta Ophthalmol 99:E1483–E1491
Chen Y-F, Luo R-Z, Li Y, Cui B-K, Song M, Yang A-K, Chen W-K (2013) High expression levels of COX-2 and P300 are associated with unfavorable survival in laryngeal squamous cell carcinoma. Eur Arch Otorhinolaryngol 270:1009–1017
Chen L, Zhang K, Shi Z, Zhang A, Jia Z, Wang G, Pu P, Kang C, Han L (2014) A lentivirus-mediated Mir-23b sponge diminishes the malignant phenotype of glioma cells in vitro and in vivo. Oncol Rep 31:1573–1580
Cheng W, Li M, Cai J, Wang K, Zhang C, Bao Z, Liu Y, Wu A (2015) HDAC4, A prognostic and chromosomal instability marker, refines the predictive value of MGMT promoter methylation. J Neuro-Oncol 122:303–312
Cheng Y, He C, Wang M, Ma X, Mo F, Yang S, Han J, Wei X (2019) Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther 4:1–39
Cheng W-L, Feng P-H, Lee K-Y, Chen K-Y, Sun W-L, Van Hiep N, Luo C-S, Wu S-M (2021) The role of EREG/EGFR pathway in tumor progression. Int J Mol Sci 22:12828
Chitnis NS, Pytel D, Bobrovnikova-Marjon E, Pant D, Zheng H, Maas NL, Frederick B, Kushner JA, Chodosh LA, Koumenis C, Fuchs SY, Diehl JA (2012) Mir-211 is a prosurvival MicroRNA that regulates chop expression in a PERK-dependent manner. Mol Cell 48:353–364
Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, McKinney K, Tempst P, Prives C, Gamblin SJ (2004) Regulation of P53 activity through lysine methylation. Nature 432:353–360
Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM (2005) Mir-15 and Mir-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A 102:13944–13949
Clouaire T, Stancheva I (2008) Methyl-Cpg binding proteins: specialized transcriptional repressors or structural components of chromatin? Cell Mol Life Sci 65:1509–1522
Cohen I, Poręba E, Kamieniarz K, Schneider R (2011) Histone modifiers in cancer: friends or foes? Genes Cancer 2:631–647
Cole AJ, Clifton-Bligh R, Marsh DJ (2015) Histone H2B monoubiquitination: roles to play in human malignancy. Endocr Relat Cancer 22:T19–T33
Coller HA, Forman JJ, Legesse-Miller A (2007) “Myc’ed messages”: Myc induces transcription of E2F1 while inhibiting its translation via a microRNA polycistron. PLoS Genet 3:E146
Costello JF, Frühwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomäki P, Lang JC (2000) Aberrant Cpg-island methylation has non-random and tumour-type–specific patterns. Nat Genet 24:132–138
Cui M, Wang H, Yao X, Zhang D, Xie Y, Cui R, Zhang X (2019) Circulating microRNAs in cancer: potential and challenge. Front Genet 10:626
Cui C, Guo T, Zhang S, Yang M, Cheng J, Wang J, Kang J, Ma W, Nian Y, Sun Z, Weng H (2022) Bacteria-derived outer membrane vesicles engineered with over-expressed pre-Mirna as delivery nanocarriers for cancer therapy. Nanomedicine 45:102585
D’oto A, Tian Q-W, Davidoff AM, Yang J (2016) Histone demethylases and their roles in cancer epigenetics. J Med Oncol Ther 1:34
Dawson MA, Bannister AJ, Göttgens B, Foster SD, Bartke T, Green AR, Kouzarides T (2009) JAK2 phosphorylates histone H3Y41 and excludes HP1α from chromatin. Nature 461:819–822
De Souza CRT, Leal MF, Calcagno DQ, Costa Sozinho EK, Borges BDN, Montenegro RC, Dos Santos AKCR, Dos Santos SEB, Ribeiro HF, Assumpção PP (2013) MYC deregulation in gastric cancer and its clinicopathological implications. PLoS One 8:E64420
Deaton AM, Bird A (2011) Cpg Islands and the regulation of transcription. Genes Dev 25:1010–1022
Decato BE, Qu J, Ji X, Wagenblast E, Knott SR, Hannon GJ, Smith AD (2020) Characterization of universal features of partially methylated domains across tissues and species. Epigenetics Chromatin 13:1–14
Dehghankelishadi P, Maritz MF, Badiee P, Thierry B (2022) High density lipoprotein nanoparticle as delivery system for radio-sensitising Mirna: an investigation in 2D/3D head and neck cancer models. Int J Pharm 617:121585
Dell’Aversana C, Lepore I, Altucci L (2012) HDAC modulation and cell death in the clinic. Exp Cell Res 318:1229–1244
Deng L, Meng T, Chen L, Wei W, Wang P (2020) The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct Target Ther 5:1–28
Di Cerbo V, Schneider R (2013) Cancers with wrong Hats: the impact of acetylation. Brief Funct Genomics 12:231–243
Dragomir MP, Knutsen E, Calin GA (2022) Classical and noncanonical functions of MiRNAs in cancers. Trends Genet 38:379–394
Dwane L, Gallagher WM, Chonghaile TN, O’connor DP (2017) The emerging role of non-traditional ubiquitination in oncogenic pathways. J Biol Chem 292:3543–3551
Eden A, Gaudet F, Waghmare A, Jaenisch R (2003) Chromosomal instability and tumors promoted by DNA hypomethylation. Science 300:455–455
Eiring AM, Harb JG, Neviani P, Garton C, Oaks JJ, Spizzo R, Liu S, Schwind S, Santhanam R, Hickey CJ, Becker H, Chandler JC, Andino R, Cortes J, Hokland P, Huettner CS, Bhatia R, Roy DC, Liebhaber SA, Caligiuri MA, Marcucci G, Garzon R, Croce CM, Calin GA, Perrotti D (2010) Mir-328 functions as an RNA decoy to modulate Hnrnp E2 regulation of Mrna translation in leukemic blasts. Cell 140:652–665
Esquela-Kerscher A, Trang P, Wiggins JF, Patrawala L, Cheng A, Ford L, Weidhaas JB, Brown D, Bader AG, Slack FJ (2008) The Let-7 Microrna reduces tumor growth in mouse models of lung cancer. Cell Cycle 7:759–764
Falkenberg KJ, Johnstone RW (2014) Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov 13:673–691
Fang LL, Wang XH, Sun BF, Zhang XD, Zhu XH, Yu ZJ, Luo H (2017) Expression, regulation and mechanism of action of the Mir-17-92 cluster in tumor cells (review). Int J Mol Med 40:1624–1630
Fatma H, Maurya SK, Siddique HR (2020) Epigenetic modifications of C-MYC: role in cancer cell reprogramming, progression and chemoresistance. Seminars in cancer biology. Elsevier
Felsenfeld G (2014) A brief history of epigenetics. Cold Spring Harb Perspect Biol 6:A018200
Ferguson-Smith AC (2011) Genomic imprinting: the emergence of an epigenetic paradigm. Nat Rev Genet 12:565–575
Forma E, Jóźwiak P, Bryś M, Krześlak A (2014) The potential role of O-Glcnac modification in cancer epigenetics. Cell Mol Biol Lett 19:438–460
Forterre A, Komuro H, Aminova S, Harada M (2020) A comprehensive review of cancer microrna therapeutic delivery strategies. Cancers (Basel) 12
Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K (2005) Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is A common hallmark of human cancer. Nat Genet 37:391–400
Fu J, Qin L, He T, Qin J, Hong J, Wong J, Liao L, Xu J (2011) The TWIST/Mi2/Nurd protein complex and its essential role in cancer metastasis. Cell Res 21:275–289
Fu Z, Wang L, Li S, Chen F, Au-Yeung KK, Shi C (2021) Microrna as an important target for anticancer drug development. Front Pharmacol 12:736323
Gao Y, Geng J, Hong X, Qi J, Teng Y, Yang Y, Qu D, Chen G (2014) Expression of P300 and CBP is associated with poor prognosis in small cell lung cancer. Int J Clin Exp Pathol 7:760
Garraway LA, Lander ES (2013) Lessons from the cancer genome. Cell 153:17–37
Gayther SA, Batley SJ, Linger L, Bannister A, Thorpe K, Chin S-F, Daigo Y, Russell P, Wilson A, Sowter HM (2000) Mutations truncating the EP300 acetylase in human cancers. Nat Genet 24:300–303
Goldberg AD, Allis CD, Bernstein E (2007) Epigenetics: a landscape takes shape. Cell 128:635–638
Greger V, Passarge E, Höpping W, Messmer E, Horsthemke B (1989) Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum Genet 83:155–158
Gruszka R, Zakrzewska M (2018) The oncogenic relevance of Mir-17-92 cluster and its paralogous Mir-106b-25 and Mir-106a-363 clusters in brain tumors. Int J Mol Sci 19
Gu Y, Mi W, Ge Y, Liu H, Fan Q, Han C, Yang J, Han F, Lu X, Yu W (2010) Glcnacylation plays an essential role in breast cancer metastasis. Cancer Res 70:6344–6351
Guppy BJ, Jeusset LM, McManus KJ (2017) The relationship between DOT1L, histone H3 methylation, and genome stability in cancer. Curr Mol Biol Reports 3:18–27
Hájková H, Marková J, Haškovec C, Šárová I, Fuchs O, Kostečka A, Cetkovský P, Michalová K, Schwarz J (2012) Decreased DNA methylation in acute myeloid leukemia patients with DNMT3A mutations and prognostic implications of DNA methylation. Leuk Res 36:1128–1133
Han M, Jia L, Lv W, Wang L, Cui W (2019) Epigenetic enzyme mutations: role in tumorigenesis and molecular inhibitors. Front Oncol 9:194
Hanahan D (2022) Hallmarks of cancer: new dimensions. Cancer Discov 12:31–46
Handy DE, Castro R, Loscalzo J (2011) Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation 123:2145–2156
Hansen KD, Sabunciyan S, Langmead B, Nagy N, Curley R, Klein G, Klein E, Salamon D, Feinberg AP (2014) Large-scale hypomethylated blocks associated with Epstein-Barr virus–induced B-cell immortalization. Genome Res 24:177–184
Hassler MR, Egger G (2012) Epigenomics of cancer–emerging new concepts. Biochimie 94:2219–2230
Hatfield SD, Shcherbata HR, Fischer KA, Nakahara K, Carthew RW, Ruohola-Baker H (2005) Stem cell division is regulated by the microRNA pathway. Nature 435:974–978
He J, Nguyen AT, Zhang Y (2011a) KDM2b/JHDM1b, an H3k36me2-specific demethylase, is required for initiation and maintenance of acute myeloid Leukemia. Blood J Am Soc Hematol 117:3869–3880
He Y-F, Li B-Z, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L (2011b) Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333:1303–1307
He B, Zhao Z, Cai Q, Zhang Y, Zhang P, Shi S, Xie H, Peng X, Yin W, Tao Y, Wang X (2020) Mirna-based biomarkers, therapies, and resistance in cancer. Int J Biol Sci 16:2628–2647
Helfrich BA, Kim J, Gao D, Chan DC, Zhang Z, Tan A-C, Bunn PA (2016) Barasertib (AZD1152), A small molecule Aurora B inhibitor, inhibits the growth of SCLC cell lines in vitro and in Vivobarasertib inhibits small-cell lung cancer cell line growth. Mol Cancer Ther 15:2314–2322
Hon GC, Hawkins RD, Caballero OL, Lo C, Lister L, Pelizzola M, Valsesia A, Ye Z, Kuan S, Edsall LE, Camargo AA, Stevenson BJ, Ecker JR, Bafna V, Strausberg RL, Simpson AJ, Ren B (2012) Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res 22(2):246–258. https://doi.org/10.1101/gr.125872.111
Hosseinahli N, Aghapour M, Duijf PHG, Baradaran B (2018) Treating cancer with Microrna replacement therapy: a literature review. J Cell Physiol 233:5574–5588
Hosseini A, Minucci S (2018) Alterations of histone modifications in cancer. Epigenetics in human disease. Elsevier
Hu J, Liu J, Chen A, Lyu J, Ai G, Zeng Q, Sun Y, Chen C, Wang J, Qiu J (2016) Ino80 promotes cervical cancer tumorigenesis by activating Nanog expression. Oncotarget 7:72250
Huang B, Laban M, Leung CH, Lee L, Lee C, Salto-Tellez M, Raju G, Hooi S (2005) Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ 12:395–404
Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, Opravil S, Shiekhattar R, Bedford MT, Jenuwein T (2007) P53 is regulated by the lysine demethylase LSD1. Nature 449:105–108
Igarashi S, Suzuki H, Niinuma T, Shimizu H, Nojima M, Iwaki H, Nobuoka T, Nishida T, Miyazaki Y, Takamaru H (2010) A novel correlation between LINE-1 hypomethylation and the malignancy of gastrointestinal stromal tumorsline-1 hypomethylation and malignancy of gists. Clin Cancer Res 16:5114–5123
Ikeda K, Shiraishi K, Eguchi A, Shibata H, Yoshimoto K, Mori T, Baba Y, Baba H, Suzuki M (2013) Long interspersed nucleotide element 1 hypomethylation is associated with poor prognosis of lung adenocarcinoma. Ann Thorac Surg 96:1790–1794
Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333:1300–1303
Izzo A, Schneider R (2010) Chatting histone modifications in mammals. Brief Funct Genomics 9:429–443
Jelinic P, Shaw P (2007) Loss of imprinting and cancer. J Pathol 211(3):261–268. 10.1002/(ISSN)1096-9896. https://doi.org/10.1002/path.v211:3 10.1002/path.2116
Jeusset LM, McManus KJ (2019) Developing targeted therapies that exploit aberrant histone ubiquitination in cancer. Cell 8:165
Jones PA, Baylin SB (2007) The epigenomics of cancer. Cell 128:683–692
Kalkan R, Atli EI (2016) The impacts of miRNAs in glioblastoma progression. Crit Rev Eukaryot Gene Expr 26(2):137–142. 10.1615/CritRevEukaryotGeneExpr.v26.i2. https://doi.org/10.1615/CritRevEukaryotGeneExpr.2016015964
Kao S-H, Wu K-J, Lee W-H (2016) Hypoxia, epithelial-mesenchymal transition, and TET-mediated epigenetic changes. J Clin Med 5:24
Karagiannis D, Rampias T (2021) HDAC inhibitors: dissecting mechanisms of action to counter tumor heterogeneity. Cancers 13:3575
Karpf AR, Matsui S-I (2005) Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res 65:8635–8639
Keum N, Giovannucci E (2019) Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol 16:713–732
Khan S, Jankowska A, Mahfouz R, Dunbar A, Sugimoto Y, Hosono N, Hu Z, Cheriyath V, Vatolin S, Przychodzen B (2013) Multiple mechanisms deregulate EZH2 and histone H3 lysine 27 epigenetic changes in myeloid malignancies. Leukemia 27:1301–1309
Khor GH, Froemming GRA, Zain RB, Abraham MT, Omar E, Tan SK, Tan AC, Vincent-Chong VK, Thong KL (2013) DNA methylation profiling revealed promoter hypermethylation-induced silencing of P16, DDAH2 and DUSP1 in primary oral squamous cell carcinoma. Int J Med Sci 10:1727
Kim J, Bretz CL, Lee S (2015) Epigenetic instability of imprinted genes in human cancers. Nucleic Acids Res 43(22):10689–10699. https://doi.org/10.1093/nar/gkv867
Klemm SL, Shipony Z, Greenleaf WJ (2019) Chromatin accessibility and the regulatory epigenome. Nat Rev Genet 20:207–220
Kohli RM, Zhang Y (2013) TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502:472–479
Komar D, Juszczynski P (2020) Rebelled epigenome: histone H3S10 phosphorylation and H3S10 kinases in cancer biology and therapy. Clin Epigenetics 12:1–14
Krishnamurthy N, Kato S, Lippman S, Kurzrock R (2022) Chromatin remodeling (SWI/SNF) complexes, cancer, and response to immunotherapy. J Immunother Cancer 10:E004669
Kukreja L, Li CJ, Ezhilan S, Iyer VR, Kuo JS (2021) Emerging epigenetic therapies for brain tumors. NeuroMolecular Med:1–9
Kulis M, Esteller M (2010) DNA methylation and cancer. Adv Genet 70:27–56
Lachner M, O’carroll D, Rea S, Mechtler K, Jenuwein T (2001) Methylation of histone H3 lysine 9 creates A binding site for HP1 proteins. Nature 410:116–120
Laczmanska I, Karpinski P, Bebenek M, Sedziak T, Ramsey D, Szmida E, Sasiadek MM (2013) Protein tyrosine phosphatase receptor-like genes are frequently hypermethylated in sporadic colorectal cancer. J Hum Genet 58:11–15
Ladelfa MF, Peche LY, Toledo MF, Laiseca JE, Schneider C, Monte M (2012) Tumor-specific MAGE proteins as regulators of P53 function. Cancer Lett 325:11–17
Lafave LM, Savage RE, Buenrostro JD (2022) Single-cell epigenomics reveals mechanisms of cancer progression. Ann Rev Cancer Biol 6:167–185
Lahooti B, Poudel S, Mikelis CM, Mattheolabakis G (2021) Mirnas as anti-angiogenic adjuvant therapy in cancer: synopsis and potential. Front Oncol 11:705634
Lara-Ureña N, Jafari V, García-Domínguez M (2022) Cancer-associated dysregulation of Sumo regulators: proteases and ligases. Int J Mol Sci 23:8012
Lee JE, Kim M-Y (2021) Cancer epigenetics: past, present and future. Seminars in cancer biology. Elsevier
Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN (2004) Microrna genes are transcribed by RNA polymerase II. EMBO J 23:4051–4060
Li Y, Seto E (2016) Hdacs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med 6:A026831
Li G, Wu X, Qian W, Cai H, Sun X, Zhang W, Tan S, Wu Z, Qian P, Ding K, Lu X, Zhang X, Yan H, Song H, Guang S, Wu Q, Lobie PE, Shan G, Zhu T (2016) CCAR1 5’ UTR as a natural mirancer of Mir-1254 overrides tamoxifen resistance. Cell Res 26:655–673
Li Y, Li Z, Zhu W-G (2019) Molecular mechanisms of epigenetic regulators as activatable targets in cancer theranostics. Curr Med Chem 26(8):1328–1350. https://doi.org/10.2174/0929867324666170921101947
Li Y, Chen X, Lu C (2021a) The interplay between DNA and histone methylation: molecular mechanisms and disease implications. EMBO Rep 22:E51803
Li Y, Gong H, Wang P, Zhu Y, Peng H, Cui Y, Li H, Liu J, Wang Z (2021b) The emerging role of ISWI chromatin remodeling complexes in cancer. J Exp Clin Cancer Res 40:1–27
Li Y, Fan Z, Meng Y, Liu S, Zhan H (2023) Blood-based DNA methylation signatures in cancer: a systematic review. Biochim Biophys Acta (BBA) - Mol Basis Dis 1869(1):166583. https://doi.org/10.1016/j.bbadis.2022.166583
Liang G, Weisenberger DJ (2017) DNA methylation aberrancies as a guide for surveillance and treatment of human cancers. Epigenetics 12:416–432
Liang T-J, Wang H-X, Zheng Y-Y, Cao Y-Q, Wu X, Zhou X, Dong S-X (2017) APC hypermethylation for early diagnosis of colorectal cancer: a meta-analysis and literature review. Oncotarget 8:46468
Liang Y, Xu P, Zou Q, Luo H, Yu W (2019) An epigenetic perspective on tumorigenesis: loss of cell identity, enhancer switching, and namirna network. seminars in cancer biology. Elsevier, pp 1–9
Liu H, Lei C, He Q, Pan Z, Xiao D, Tao Y (2018) Nuclear functions of mammalian micrornas in gene regulation, immunity and cancer. Mol Cancer 17:64
Liu F, Lu X, Zhou X, Huang H (2021a) APC gene promoter methylation as a potential biomarker for lung cancer diagnosis: a meta-analysis. Thoracic Cancer 12:2907–2913
Liu X-Y, Guo C-H, Xi Z-Y, Xu X-Q, Zhao Q-Y, Li L-S, Wang Y (2021b) Histone methylation in pancreatic cancer and its clinical implications. World J Gastroenterol 27:6004
Ljungman M, Parks L, Hulbatte R, Bedi K (2019) The role of H3K79 methylation in transcription and the DNA damage response. Mutat Res/Rev Mutat Res 780:48–54
López-Moyado IF, Tsagaratou A, Yuita H, Seo H, Delatte B, Heinz S, Benner C, Rao A (2019) Paradoxical association of TET loss of function with genome-wide DNA hypomethylation. Proc Natl Acad Sci 116:16933–16942
Losman J-A, Kaelin WG (2013) What a difference a hydroxyl makes: mutant IDH,(R)-2-hydroxyglutarate, and cancer. Genes Dev 27:836–852
Lozano-Urena A, Jimenez-Villalba E, Pinedo-Serrano A, Jordan-Pla A, Kirstein M, Ferron SR (2021) Aberrations of genomic imprinting in glioblastoma formation. Front Oncol 11:630482
Lu TX, Rothenberg ME (2018) Microrna. J Allergy Clin Immunol 141:1202–1207
Lujambio A, Lowe SW (2012) The microcosmos of cancer. Nature 482:347–355
Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setién F, Casado S, Suarez-Gauthier A, Sanchez-Cespedes M, Git A, Spiteri I, Das PP, Caldas C, Miska E, Esteller M (2007) Genetic unmasking of an epigenetically silenced microrna in human cancer cells. Cancer Res 67:1424–1429
Mallm JP, Windisch P, Biran A, Gal Z, Schumacher S, Glass R, Herold-Mende C, Meshorer E, Barbus M, Rippe K (2020) Glioblastoma initiating cells are sensitive to histone demethylase inhibition due to epigenetic deregulation. Int J Cancer 146:1281–1292
Malpeli G, Innamorati G, Decimo I, Bencivenga M, Nwabo Kamdje AH, Perris R, Bassi C (2019) Methylation dynamics of RASSF1A and its impact on cancer. Cancers 11:959
Marei HE, Althani A, Afifi N, Hasan A, Caceci T, Pozzoli G, Morrione A, Giordano A, Cenciarelli C (2021) P53 Signaling in cancer progression and therapy. Cancer Cell Int 21:1–15
Markouli M, Strepkos D, Basdra EK, Papavassiliou AG, Piperi C (2021) Prominent role of histone modifications in the regulation of tumor metastasis. Int J Mol Sci 22:2778
Marsh DJ, Dickson K-A (2019) Writing histone Monoubiquitination in human malignancy—the role of RING finger E3 ubiquitin ligases. Genes 10:67
Marusyk A, Janiszewska M, Polyak K (2020) Intratumor heterogeneity: the Rosetta stone of therapy resistance. Cancer Cell 37:471–484
Mazloumi Z, Farahzadi R, Rafat A, Asl KD, Karimipour M, Montazer M, Movassaghpour AA, Dehnad A, Charoudeh HN (2022) Effect of aberrant DNA methylation on cancer stem cell properties. Exp Mol Pathol 104757
McGranahan N, Swanton C (2017) Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 168:613–628
McGuire A, Casey M, Shalaby A, Kalinina O, Curran C, Webber M, Callagy G, Holian E, Bourke E, Kerin M (2019) Quantifying Tip60 (Kat5) stratifies breast cancer. Sci Rep 9:1–14
Melling N, Grimm N, Simon R, Stahl P, Bokemeyer C, Terracciano L, Sauter G, Izbicki JR, Marx AH (2016) Loss of H2Bub1 expression is linked to poor prognosis in nodal negative colorectal cancers. Pathol Oncol Res 22:95–102
Menon A, Abd-Aziz N, Khalid K, Poh CL, Naidu R (2022) Mirna: a promising therapeutic target in cancer. Int J Mol Sci 23
Mollaei H, Safaralizadeh R, Rostami Z (2019) MicroRNA replacement therapy in cancer. J Cell Physiol 234:12369–12384
Morey L, Brenner C, Fazi F, Villa R, Gutierrez A, Buschbeck M, Nervi C, Minucci S, Fuks F, Di Croce L (2008) MBD3, a component of the Nurd complex, facilitates chromatin alteration and deposition of epigenetic marks. Mol Cell Biol 28:5912–5923
Morgan A, Davies TJ, McAuley MT (2018) The role of DNA methylation in ageing and cancer. Proc Nutr Soc 77:412–422
Motavalli R, Marofi F, Nasimi M, Yousefi M, Khiavi FM (2021) Association of hippo signalling pathway with epigenetic changes in cancer cells and therapeutic approaches: a review. Anti-Cancer Agents Med Chem 21:1520–1528
Moyal L, Lerenthal Y, Gana-Weisz M, Mass G, So S, Wang S-Y, Eppink B, Chung YM, Shalev G, Shema E (2011) Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol Cell 41:529–542
Mudbhary R, Hoshida Y, Chernyavskaya Y, Jacob V, Villanueva A, Fiel MI, Chen X, Kojima K, Thung S, Bronson RT (2014) UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell 25:196–209
Muller S, Filippakopoulos P, Knapp S (2011) Bromodomains as therapeutic targets. Expert Rev Mol Med 13
Nag S, Qin J, Srivenugopal KS, Wang M, Zhang R (2013) The MDM2-P53 pathway revisited. J Biomed Res 27:254–271
Nair SS, Kumar R (2012) Chromatin remodeling in cancer: a gateway to regulate gene transcription. Mol Oncol 6:611–619
Neganova ME, Klochkov SG, Aleksandrova YR, Aliev G (2022) Histone modifications in epigenetic regulation of cancer: perspectives and achieved progress. Seminars in cancer biology. Elsevier, pp 452–471
Neidhart M (2015) DNA methylation and complex human disease. Academic Press
Neophytou CM, Trougakos IP, Erin N, Papageorgis P (2021) Apoptosis deregulation and the development of cancer multi-drug resistance. Cancers 13:4363
Neri F, Rapelli S, Krepelova A, Incarnato D, Parlato C, Basile G, Maldotti M, Anselmi F, Oliviero S (2017) Intragenic DNA methylation prevents spurious transcription initiation. Nature 543:72–77
Nguyen TA, Park J, Dang TL, Choi YG, Kim VN (2018) Microprocessor depends on hemin to recognize the apical loop of primary microrna. Nucleic Acids Res 46:5726–5736
Nishiyama A, Nakanishi M (2021) Navigating the DNA methylation landscape of cancer. Trends Genet 37:1012–1027
O’Brien J, Hayder H, Zayed Y, Peng C (2018) Overview of microrna biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne) 9:402
Okawa R, Banno K, Iida M, Yanokura M, Takeda T, Iijima M, Kunitomi-Irie H, Nakamura K, Adachi M, Umene K (2017) Aberrant chromatin remodeling in gynecological cancer. Oncol Lett 14:5107–5113
Ortiz-Barahona, V., Joshi, R. S. & Esteller, M. Use of DNA methylation profiling in translational oncology. Seminars in cancer biology. 2020 Elsevier
Pan G, Liu Y, Shang L, Zhou F, Yang S (2021) EMT-associated micrornas and their roles in cancer stemness and drug resistance. Cancer Commun (Lond) 41:199–217
Papageorgiou SG, Kontos CK, Diamantopoulos MA, Bouchla A, Glezou E, Bazani E, Pappa V, Scorilas A (2017) Microrna-155-5p overexpression in peripheral blood mononuclear cells of chronic lymphocytic leukemia patients is a novel, independent molecular biomarker of poor prognosis. Dis Markers 2017:2046545
Pappalardo XG, Barra V (2021) Losing DNA methylation at repetitive elements and breaking bad. Epigenetics Chromatin 14:1–21
Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J (2011) Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 471:189–195
Pastor WA, Aravind L, Rao A (2013) Tetonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol 14:341–356
Patani N, Jiang WG, Newbold RF, Mokbel K (2011) Histone-modifier gene expression profiles are associated with pathological and clinical outcomes in human breast cancer. Anticancer Res 31:4115–4125
Peifer M, Fernández-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T (2012) Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 44:1104–1110
Peixoto P, Cartron P-F, Serandour AA, Hervouet E (2020) From 1957 to nowadays: a brief history of epigenetics. Int J Mol Sci 21:7571
Peng Y, Croce CM (2016) The role of micrornas in human cancer. Signal Transduct Target Ther 1:15004
Pfeifer GP (2018) Defining driver DNA methylation changes in human cancer. Int J Mol Sci 19:1166
Piao XM, Jeong P, Kim YH, Byun YJ, Xu Y, Kang HW, Ha YS, Kim WT, Lee JY, Woo SH, Kwon TG, Kim IY, Moon SK, Choi YH, Cha EJ, Yun SJ, Kim WJ (2019) Urinary cell-free microrna biomarker could discriminate bladder cancer from benign hematuria. Int J Cancer 144:380–388
Pichiorri F, Suh SS, Rocci A, De Luca L, Taccioli C, Santhanam R, Zhou W, Benson DM Jr, Hofmainster C, Alder H, Garofalo M, Di Leva G, Volinia S, Lin HJ, Perrotti D, Kuehl M, Aqeilan RI, Palumbo A, Croce CM (2010) Downregulation of P53-inducible micrornas 192, 194, and 215 impairs the P53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell 18:367–381
Podhorecka M, Skladanowski A, Bozko P (2010) H2AX phosphorylation: its role in DNA damage response and cancer therapy. J Nucl Acids 2010
Prenzel T, Begus-Nahrmann Y, Kramer F, Hennion M, Hsu C, Gorsler T, Hintermair C, Eick D, Kremmer E, Simons M (2011) Estrogen-dependent gene transcription in human breast cancer cells relies upon proteasome-dependent monoubiquitination of histone H2B. Cancer Res 71:5739–5753
Qin W, Wolf P, Liu N, Link S, Smets M, Mastra FL, Forné I, Pichler G, Hörl D, Fellinger K (2015) DNA methylation requires a DNMT1 ubiquitin interacting motif (UIM) and histone ubiquitination. Cell Res 25:911–929
Rebel VI, Kung AL, Tanner EA, Yang H, Bronson RT, Livingston DM (2002) Distinct roles for CREB-binding protein and P300 in hematopoietic stem cell self-renewal. Proc Natl Acad Sci 99:14789–14794
Recasens A, Munoz L (2019) Targeting cancer cell dormancy. Trends Pharmacol Sci 40:128–141
Renaud S, Loukinov D, Alberti L, Vostrov A, Kwon Y-W, Bosman FT, Lobanenkov V, Benhattar J (2011) BORIS/CTCFL-mediated transcriptional regulation of the Htert telomerase gene in testicular and ovarian tumor cells. Nucleic Acids Res 39:862–873
Roberti A, Valdes AF, Torrecillas R, Fraga MF, Fernandez AF (2019) Epigenetics in cancer therapy and nanomedicine. Clin Epigenetics 11:1–18
Robinson NJ, Parker KA, Schiemann WP (2020) Epigenetic plasticity in metastatic dormancy: mechanisms and therapeutic implications. Ann Transl Med 8
Rodríguez-Paredes M, Bormann F, Raddatz G, Gutekunst J, Lucena-Porcel C, Köhler F, Wurzer E, Schmidt K, Gallinat S, Wenck H (2018) Methylation profiling identifies two subclasses of squamous cell carcinoma related to distinct cells of origin. Nat Commun 9:1–9
Rondinelli B, Rosano D, Antonini E, Frenquelli M, Montanini L, Huang D, Segalla S, Yoshihara K, Amin SB, Lazarevic D (2015) Histone demethylase JARID1C inactivation triggers genomic instability in sporadic renal cancer. J Clin Invest 125:4625–4637
Ropero S, Fraga MF, Ballestar E, Hamelin R, Yamamoto H, Boix-Chornet M, Caballero R, Alaminos M, Setien F, Paz MF (2006) A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat Genet 38:566–569
Ross SE and Bogdanovic O (2019) TET enzymes, DNA demethylation and pluripotency. Biochem Soc Trans 47:875–885
Sabit H, Cevik E, Tombuloglu H, Abdel-Ghany S, Tombuloglu G, Esteller M (2021) Triple negative breast cancer in the era of Mirna. Crit Rev Oncol Hematol 157:103196
Sakabe K, Hart GW (2010) O-Glcnac transferase regulates mitotic chromatin dynamics. J Biol Chem 285:34460–34468
Salhab A, Nordström K, Gasparoni G, Kattler K, Ebert P, Ramirez F, Arrigoni L, Müller F, Polansky JK, Cadenas C, Hengstler JG, Lengauer T, Manke T, DEEP Consortium, Walter J (2018) A comprehensive analysis of 195 DNA methylomes reveals shared and cell-specific features of partially methylated domains. Genome Biol 19(1). https://doi.org/10.1186/s13059-018-1510-5
Saliminejad K, Khorram Khorshid HR, Soleymani Fard S, Ghaffari SH (2019) An overview of micrornas: biology, functions, therapeutics, and analysis methods. J Cell Physiol 234:5451–5465
Schroeder DI, Lott P, Korf I, LaSalle JM (2011) Large-scale methylation domains mark a functional subset of neuronally expressed genes. Genome Res 21(10):1583–1591
Schübeler D (2015) Function and information content of DNA methylation. Nature 517:321–326
Selcuklu SD, Donoghue MT, Spillane C (2009) Mir-21 as a key regulator of oncogenic processes. Biochem Soc Trans 37:918–925
Seligson DB, Horvath S, McBrian MA, Mah V, Yu H, Tze S, Wang Q, Chia D, Goodglick L, Kurdistani SK (2009) Global levels of histone modifications predict prognosis in different cancers. Am J Pathol 174:1619–1628
Semina EV, Rysenkova KD, Troyanovskiy KE, Shmakova AA, Rubina KA (2021) Micrornas in cancer: from gene expression regulation to the metastatic niche reprogramming. Biochemistry (Mosc) 86:785–799
Shanmugam MK, Arfuso F, Arumugam S, Chinnathambi A, Jinsong B, Warrier S, Wang LZ, Kumar AP, Ahn KS, Sethi G (2018) Role of novel histone modifications in cancer. Oncotarget 9:11414
Sharif J, Muto M, Takebayashi S-I, Suetake I, Iwamatsu A, Endo TA, Shinga J, Mizutani-Koseki Y, Toyoda T, Okamura K (2007) The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450:908–912
Sharma S, Kelly TK, Jones PA (2010) Epigenetics in cancer. Carcinogenesis 31:27–36
Shekhawat J, Gauba K, Gupta S, Choudhury B, Purohit P, Sharma P, Banerjee M (2021) Ten–eleven translocase: key regulator of the methylation landscape in cancer. J Cancer Res Clin Oncol 147:1869–1879
Shema E, Tirosh I, Aylon Y, Huang J, Ye C, Moskovits N, Raver-Shapira N, Minsky N, Pirngruber J, Tarcic G (2008) The histone H2B-specific ubiquitin ligase RNF20/Hbre1 acts as a putative tumor suppressor through selective regulation of gene expression. Genes Dev 22:2664–2676
Śledzińska P, Bebyn MG, Furtak J, Kowalewski J, Lewandowska MA (2021) Prognostic and predictive biomarkers in gliomas. Int J Mol Sci 22:10373
Smiley JA, Kundracik M, Landfried DA, Barnes SR, Axhemi AA (2005) Genes of the thymidine salvage pathway: thymine-7-hydroxylase from a rhodotorula glutinis cDNA library and iso-orotate decarboxylase from Neurospora crassa. Biochimica Et Biophysica Acta (BBA)-General Subjects 1723:256–264
Smith ZD, Meissner A (2013) DNA methylation: roles in mammalian development. Nat Rev Genet 14:204–220
Smolarz B, Durczyński A, Romanowicz H, Szyłło K, Hogendorf P (2022) Mirnas in cancer (review of literature). Int J Mol Sci 23
Song Z, Wei Z, Wang Q, Zhang X, Tao X, Wu N, Liu X, Qian J (2020) The role of DOT1L in the proliferation and prognosis of gastric cancer. Biosci Reports 40
Sterling J, Menezes SV, Abbassi RH, Munoz L (2021) Histone lysine demethylases and their functions in cancer. Int J Cancer 148:2375–2388
Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324:930–935
Tan W, Liu B, Qu S, Liang G, Luo W, Gong C (2018) Micrornas and cancer: key paradigms in molecular therapy. Oncol Lett 15:2735–2742
Thompson LL, Guppy BJ, Sawchuk L, Davie JR, McManus KJ (2013) Regulation of chromatin structure via histone post-translational modification and the link to carcinogenesis. Cancer Metastasis Rev 32:363–376
Toh Y, Nicolson GL (2009) The role of the MTA family and their encoded proteins in human cancers: molecular functions and clinical implications. Clin Exp Metastasis 26:215–227
Tsuda M, Fukuda A, Kawai M, Araki O, Seno H (2021) The role of the SWI/SNF chromatin remodeling complex in pancreatic ductal adenocarcinoma. Cancer Sci 112:490–497
Tucci V, Isles AR, Kelsey G, Ferguson-Smith AC, Bartolomei MS, Benvenisty N, Bourc’his D, Charalambous M, Dulac C, Feil R (2019) Genomic imprinting and physiological processes in mammals. Cell 176:952–965
Turpin M, Salbert G (2022) 5-Methylcytosine turnover: mechanisms and therapeutic implications in cancer. Front Mol Biosci 9
Uddin MS, Al Mamun A, Alghamdi BS, Tewari D, Jeandet P, Sarwar MS, Ashraf GM (2020) Epigenetics of glioblastoma multiforme: from molecular mechanisms to therapeutic approaches. Seminars in cancer biology. Elsevier
Van Tongelen A, Loriot A, De Smet C (2017) Oncogenic roles of DNA hypomethylation through the activation of cancer-germline genes. Cancer Lett 396:130–137
Varambally S, Cao Q, Mani R-S, Shankar S, Wang X, Ateeq B, Laxman B, Cao X, Jing X, Ramnarayanan K (2008) Genomic loss of Microrna-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 322:1695–1699
Vaughan RM, Dickson BM, Cornett EM, Harrison JS, Kuhlman B, Rothbart SB (2018) Comparative biochemical analysis of UHRF proteins reveals molecular mechanisms that uncouple UHRF2 from DNA methylation maintenance. Nucleic Acids Res 46:4405–4416
Vogelstein B, Kinzler KW (2004) Cancer genes and the pathways they control. Nat Med 10:789–799
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW (2013) Cancer genome landscapes. Science 339:1546–1558
Vrba L, Futscher BW (2019) DNA methylation changes in biomarker loci occur early in cancer progression. F1000Research:8
Waddington CH (2012) The epigenotype. Int J Epidemiol 41:10–13
Wagner KW, Alam H, Dhar SS, Giri U, Li N, Wei Y, Giri D, Cascone T, Kim J-H, Ye Y (2013) KDM2A promotes lung tumorigenesis by epigenetically enhancing ERK1/2 signaling. J Clin Invest 123:5231–5246
Waitkus MS, Diplas BH, Yan H (2015) Isocitrate dehydrogenase mutations in gliomas. Neuro-Oncology 18:16–26
Wang H, Wang S, Shen L, Chen Y, Zhang X, Zhou J, Wang Z, Hu C, Yue W, Wang H (2010) Chk2 down-regulation by promoter hypermethylation in human bulk gliomas. Life Sci 86:185–191
Wang L, Gural A, Sun X-J, Zhao X, Perna F, Huang G, Hatlen MA, Vu L, Liu F, Xu H (2011) The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science 333:765–769
Wang E, Kawaoka S, Yu M, Shi J, Ni T, Yang W, Zhu J, Roeder RG, Vakoc CR (2013) Histone H2B ubiquitin ligase RNF20 is required for MLL-rearranged leukemia. Proc Natl Acad Sci 110:3901–3906
Wiggins JF, Ruffino L, Kelnar K, Omotola M, Patrawala L, Brown D, Bader AG (2010) Development of a lung cancer therapeutic based on the tumor suppressor microrna-34. Cancer Res 70:5923–5930
Wong SH, Goode DL, Iwasaki M, Wei MC, Kuo H-P, Zhu L, Schneidawind D, Duque-Afonso J, Weng Z, Cleary ML (2015) The H3K4-methyl epigenome regulates leukemia stem cell oncogenic potential. Cancer Cell 28:198–209
Wright DE, Wang C-Y, Kao C-F (2011) Flickin’the ubiquitin switch: the role of H2B ubiquitylation in development. Epigenetics 6:1165–1175
Wu X, Zhang Y (2017) TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet 18:517–534
Wu X, Somlo G, Yu Y, Palomares MR, Li AX, Zhou W, Chow A, Yen Y, Rossi JJ, Gao H, Wang J, Yuan YC, Frankel P, Li S, Ashing-Giwa KT, Sun G, Wang Y, Smith R, Robinson K, Ren X, Wang SE (2012) De novo sequencing of circulating mirnas identifies novel markers predicting clinical outcome of locally advanced breast cancer. J Transl Med 10:42
Wu Y, Chen P, Jing Y, Wang C, Men Y-L, Zhan W, Wang Q, Gan Z, Huang J, Xie K (2015) Microarray analysis reveals potential biological functions of histone H2B monoubiquitination. PLoS One 10:E0133444
Xiang Q, He X, Mu J, Mu H, Zhou D, Tang J, Xiao Q, Jiang Y, Ren G, Xiang T (2019) The phosphoinositide hydrolase phospholipase C delta1 inhibits epithelial-mesenchymal transition and is silenced in colorectal cancer. J Cell Physiol 234:13906–13916
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim S-H, Ito S, Yang C, Wang P, Xiao M-T (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19:17–30
Xuefang Z, Ruinian Z, Liji J, Chun Z, Qiaolan Z, Jun J, Yuming C, Junrong H (2020) Mir-331-3p inhibits proliferation and promotes apoptosis of nasopharyngeal carcinoma cells by targeting Elf4b-PI3K-AKT pathway. Technol Cancer Res Treat 19:1533033819892251
Yang J, Altahan AM, Hu D, Wang Y, Cheng P-H, Morton CL, Qu C, Nathwani AC, Shohet JM, Fotsis T (2015) The role of histone demethylase KDM4B in Myc signaling in neuroblastoma. JNCI:107
Yang Y, Sun J, Chen T, Tao Z, Zhang X, Tian F, Zhou X, Lu D (2017) Tat-interactive protein-60KDA (TIP60) regulates the tumorigenesis of lung cancer in vitro. J Cancer 8:2277
Yang G-J, Ko C-N, Zhong H-J, Leung C-H, Ma D-L (2019) Structure-based discovery of a selective KDM5A inhibitor that exhibits anti-cancer activity via inducing cell cycle arrest and senescence in breast cancer cell lines. Cancers 11:92
Yang M-Y, Lin P-M, Yang C-H, Hu M-L, Chen I-Y, Lin S-F, Hsu C-M (2021) Loss of ZNF215 imprinting is associated with poor five-year survival in patients with cytogenetically abnormal-acute myeloid leukemia. Blood Cells Mol Dis 90:102577. https://doi.org/10.1016/j.bcmd.2021.102577
Yap DB, Chu J, Berg T, Schapira M, Cheng S-WG, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M (2011) Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood J Am Soc Hematol 117:2451–2459
Yildirim O, Li R, Hung J-H, Chen PB, Dong X, Ee L-S, Weng Z, Rando OJ, Fazzio TG (2011) Mbd3/NURD complex regulates expression of 5-Hydroxymethylcytosine marked genes in embryonic stem cells. Cell 147:1498–1510
Yin Y-W, Jin H-J, Zhao W, Gao B, Fang J, Wei J, Zhang DD, Zhang J, Fang D (2015) The histone acetyltransferase GCN5 expression is elevated and regulated by C-Myc and E2F1 transcription factors in human colon cancer. Gene Expr 16:187
You JS, Jones PA (2012) Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell 22:9–20
Zhang J, Ma L (2012) Microrna control of epithelial-mesenchymal transition and metastasis. Cancer Metastasis Rev 31:653–662
Zhang Y, Yao L, Zhang X, Ji H, Wang L, Sun S, Pang D (2011) Elevated expression of USP22 in correlation with poor prognosis in patients with invasive breast cancer. J Cancer Res Clin Oncol 137:1245–1253
Zhang C, Zhong JF, Stucky A, Chen X-L, Press MF, Zhang X (2015) Histone acetylation: novel target for the treatment of acute lymphoblastic Leukemia. Clin Epigenetics 7:1–10
Zhang S, Zhou B, Wang L, Li P, Bennett B, Snyder R, Garantziotis S, Fargo D, Cox AD, Chen L (2017) INO80 is required for oncogenic transcription and tumor growth in non-small cell lung cancer. Oncogene 36:1430–1439
Zhang T-J, Zhou J-D, Zhang W, Lin J, Ma J-C, Wen X-M, Yuan Q, Li X-X, Xu Z-J, Qian J (2018) H19 overexpression promotes leukemogenesis and predicts unfavorable prognosis in acute myeloid leukemia. Clin Epigenetics 10(1):1–12. https://doi.org/10.1186/s13148-018-0486-z
Zhang W, Klinkebiel D, Barger CJ, Pandey S, Guda C, Miller A, Akers SN, Odunsi K, Karpf AR (2020) Global DNA Hypomethylation in epithelial ovarian cancer: passive demethylation and association with genomic instability. Cancers 12:764
Zhao Z, Shilatifard A (2019) Epigenetic modifications of histones in cancer. Genome Biol 20:1–16
Zhao S, Allis CD, Wang GG (2021) The language of chromatin modification in human cancers. Nat Rev Cancer 21:413–430
Zheng Y, Hlady RA, Joyce BT, Robertson KD, He C, Nannini DR, Kibbe WA, Achenbach CJ, Murphy RL, Roberts LR (2019) DNA methylation of individual repetitive elements in hepatitis C virus infection-induced hepatocellular carcinoma. Clin Epigenetics 11:1–13
Zhou B, Wang L, Zhang S, Bennett BD, He F, Zhang Y, Xiong C, Han L, Diao L, Li P (2016) INO80 governs superenhancer-mediated oncogenic transcription and tumor growth in melanoma. Genes Dev 30:1440–1453
Zhou X, Jiao D, Dou M, Zhang W, Hua H, Chen J, Li Z, Li L, Han X (2020) Association of APC gene promoter methylation and the risk of gastric cancer: a meta-analysis and bioinformatics study. Medicine 99
Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, Nederlof PM, Gage FH, Verma IM (2011) BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 477:179–184
Zhu Q, Zhou L, Yang Z, Lai M, Xie H, Wu L, Xing C, Zhang F, Zheng S (2012) O-Glcnacylation plays a role in tumor recurrence of hepatocellular carcinoma following liver transplantation. Med Oncol 29:985–993
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Gökalp, E.E., Işık, S., Artan, S. (2023). Introduction to Cancer Epigenetics. In: Kalkan, R. (eds) Cancer Epigenetics. Epigenetics and Human Health, vol 11. Springer, Cham. https://doi.org/10.1007/978-3-031-42365-9_3
Download citation
DOI: https://doi.org/10.1007/978-3-031-42365-9_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-42364-2
Online ISBN: 978-3-031-42365-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)