
Abstract
The interaction of platelets with endothelial and inflammatory cells might trigger atherogenesis. Different pathways are responsible for this contribution of platelets to atherogenesis. A significant association has been described between increased platelet activation and the extent of atherosclerosis. Platelet reactivity also plays a key role in determining outcomes of patients undergoing percutaneous coronary intervention (PCI). Despite dual antiplatelet therapy, platelet reactivity increases early after the procedure proportionally to the degree of vascular damage and endothelial dysfunction induced by coronary interventions, and large increases in platelet reactivity are also associated with an increased risk of periprocedural myonecrosis. The interaction between platelets and vessel wall has important clinical implications, especially in patients treated with PCI. These include the appropriate selection of antiplatelet drugs when more aggressive procedures are needed, the prognostic significance of periprocedural variations of platelet reactivity, and the correct timing for platelet function testing.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute coronary syndromes and cerebrovascular accidents are often the first clinical presentation of cardiovascular disease and share common pathophysiological mechanisms, which involve vascular atherosclerosis and thrombosis. Platelets play a key role in hemostasis and thrombosis, but they are also actively involved in atherogenesis and growth of atherosclerotic plaque. The interaction with endothelial and inflammatory cells is the main pathway through which platelets participate in the development of cardiovascular disease, while thrombosis is the final step, leading to the most detrimental clinical manifestation of this process. As a consequence, pharmacological platelet inhibition has become a cornerstone of treatment of patients with cardiovascular disease and especially of those with coronary artery disease (CAD) treated with percutaneous coronary intervention (PCI). Antiplatelet therapy has been shown not only to effectively reduce platelet aggregation but also to inhibit the interaction of platelets with vessel wall and inflammatory components, leading to an improvement in endothelial function and a reduction in inflammation.
In this review article, we examine the available evidence on the interaction between platelets and vascular wall, the clinical implications, and the effects of pharmacological treatment targeting this interaction.
Platelets and Endothelial Function
Platelets are actively involved in the onset of endothelial dysfunction and development of vascular atherosclerosis [1, 2]. Activated platelets (Fig. 1) can in fact interact with inflammatory cells, such as neutrophils and monocytes, endothelial cells, and with endothelial progenitor cells. As long as endothelium is intact, platelets will not interact with vessel wall. However, in the presence of endothelial damage and subsequent inflammation, platelets adhere to activated endothelial cells and this interaction is mediated by platelet receptor glycoprotein IIb/IIIa, involving platelet-bound fibrinogen, fibronectin, and vWF, as well as endothelial receptors, such as intercellular adhesion molecule-1, alphavbeta3 integrin, and glycoprotein Ib [3–6]. Platelet activation itself also induces a local release of granules containing inflammatory substances that further increase the inflammatory response of endothelial cells [7, 8]. The interaction between platelets and the vascular wall involves different steps. Platelet–leukocyte aggregates are first formed, activating leucocyte adhesion receptors and serving as a bridge between leucocytes and the endothelium. After their initial interaction, both platelets and endothelial cells release chemoattractants, such as P-selectin, and provide an adhesive surface for leucocytes [9, 10]. This cascade of interactions leads to an exponential increase in inflammation and cell damage at the level of the vessel wall, creating the molecular and cellular substrate of endothelial dysfunction and vascular atherosclerosis [11].

Pathways of platelet activation (reproduced with permission of Lars Faxalv, Linkoping University, Linkoping, Sweden)
The importance of platelets in the pathogenesis of endothelial dysfunction is also demonstrated by the fact that the antiplatelet drugs improve endothelial function. Platelet inhibition with aspirin has been shown to modulate acetylcholine-induced peripheral vasodilation in patients with atherosclerosis, via inhibition of cyclooxygenase-dependent vasoconstrictors [11]. In addition, glycoprotein IIb/IIIa blockade can improve endothelium-dependent vasodilation in patients with symptomatic coronary artery disease, mainly enhancing nitric oxide bioactivity [12]. The thienopyridine, clopidogrel, also increases nitric oxide production in cultured endothelial cells and improves nitric oxide mediated-vasodilatation in animal models [13, 14]. Furthermore, in patients with stable CAD, clopidogrel administration, both as loading-dose or chronic therapy, improves forearm blood flow and reduces biomarkers of oxidative stress and inflammation [15, 16]. Overall, these findings suggest that clopidogrel could directly improve endothelial function, irrespective of its antiplatelet effect. However, a correlation between the degree of platelet inhibition and the magnitude of the beneficial effect on endothelium has also been demonstrated. In a study of patients with stable angina undergoing elective PCI, Muller et al. [17] investigated the correlation between endothelial function and platelet reactivity after a 600-mg loading dose of clopidogrel. Endothelial function was as assessed with peripheral arterial tonometry (measuring the “Endoscore”), with von Willebrand factor antigen level, and with ristocetin cofactor activity. Platelet reactivity was assessed with soluble P-selectin levels and P2Y12 reaction units (PRU) measured with the VerifyNow assay. Overall, the findings suggested that an impaired endothelial response before clopidogrel is associated with greater platelet reactivity after clopidogrel. As increased platelet reactivity predisposes to higher risk of myonecrosis after PCI, this link might explain the unfavorable PCI outcomes in patients with more severe endothelial impairment.
Consistently, Hamilos et al. [18] appraised whether the improvement in endothelial function induced by clopidogrel was associated with the degree of platelet inhibition provided by the drug. Endothelial function was evaluated before and at least 12 h after 600 mg clopidogrel in 43 patients with stable angina undergoing elective PCI. An improvement in endothelial function after clopidogrel was observed in 20 patients, and this improvement significantly correlated with platelet reactivity. In particular, the endothelial function significantly improved in patients with optimal platelet inhibition (defined as a reduction of platelet reaction unit to below 204 U) after clopidogrel, while it remained unchanged in patients with suboptimal platelet inhibition. Therefore, in addition to a direct effect, the degree of platelet inhibition represents an important additional mechanism by which clopidogrel improves endothelial function. As a considerable interindividual variability in the degree of platelet inhibition after clopidogrel has been demonstrated, and a substantial proportion of patients present suboptimal platelet inhibition and high platelet reactivity even after a 600-mg loading dose of clopidogrel, these findings might have immediate clinical implications. In fact, a more effective antiplatelet therapy might be useful not only to prevent thrombotic events but also to revert endothelial dysfunction, with potential positive impact on clinical outcome. Moving from this assumption, Patti et al. conducted a study comparing the effects of high (150 mg) versus standard clopidogrel (75 mg) maintenance doses on platelet inhibition, inflammation, and endothelial function in patients undergoing PCI [19]. In this randomized crossover study, patients in the 150-mg/day arm had higher platelet inhibition, better flow-mediated vasodilation, and lower high-sensitivity C-reactive protein levels. Based on the evidence of such beneficial effects on endothelium and inflammation, the use of higher doses of clopidogrel might therefore be a useful therapeutic option in selected patients with evident endothelial dysfunction.
Platelets in Atherogenesis
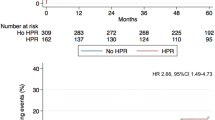
After adhesion to the vessel wall at sites of endothelium damage and activation, platelets contribute to the development of atherosclerotic lesions [1, 2]. Different pathways are responsible for this contribution of platelets to atherogenesis [20–27]. von Willebrand factor mediates the recruitment of platelets at the site of vascular injury and is also a determinant of atherosclerotic plaque development [22]. COX-1-dependent thromboxane has been demonstrated to accelerate atherogenesis in animal models, suggesting that platelet activation increases the rate of plaque formation [23, 24]. P-selectin also stimulates monocytes and macrophages to release chemokines, and it promotes the formation of platelet–monocyte aggregates [25]. From a clinical standpoint, a significant association has been described between increased platelet activation and carotid artery wall thickness [28] and progressive thickening of the artery in patients with type 2 diabetes mellitus and hypertension [29]. Patients with stable CAD present high platelet reactivity and increased levels of circulating monocyte–platelet aggregates [30], which are also early markers of acute myocardial infarction [31]. In addition, platelet reactivity is progressively increased as a function of the number of vascular districts involved by atherosclerosis (i.e., cerebral, cardiac, peripheral) [32]. We have recently investigated the correlation between platelet reactivity and the extension of coronary atherosclerosis in a study of 338 patients undergoing PCI for stable angina loaded with 600 mg clopidogrel [33]. Platelet reactivity was measured with the VerifyNow P2Y12 assay, defining high platelet reactivity (HPR) as PRU value ≥240. The presence of multivessel disease (MVD) and total stent length (TSL) were used as markers of atherosclerosis severity and extension. The results suggested that patients with more extensive coronary atherosclerosis have higher platelet reactivity and higher rates of HPR after clopidogrel, which might partly account for higher risk of periprocedural MI. On this basis, more aggressive platelet inhibition might be particularly beneficial in patients with MVD and extensive coronary atherosclerosis. This hypothesis is corroborated by the results of a subanalysis of the “Enhanced Suppression of the Platelet IIb/IIIa Receptor with Integrilin Therapy” trial [34] that the increased risk of periprocedural myocardial infarction observed in the highest TSL quartile could be largely mitigated by the use of glycoprotein IIb/IIIa inhibitor eptifibatide. This more aggressive antiplatelet strategy resulted into similar rates of periprocedural myocardial infarction in patients with extensive stent use to those observed in patients in the lowest TSL quartile. The potential benefit of antiplatelet drugs in patients with preclinical atherosclerosis is however yet to be demonstrated, and when administered for primary prevention, dual antiplatelet treatment with aspirin and clopidogrel has failed to improve cardiovascular outcomes [35].
Platelet Reactivity in PCI
Platelet reactivity plays a key role in determining outcomes of patients undergoing PCI, both in the periprocedural setting [36, 37] and during follow-up [38, 39], and optimal antiplatelet therapy is crucial for preventing procedural thrombotic complications and recurrent ischemic events [40, 41]. However, due to a wide interindividual variability in baseline platelet reactivity and in response to antiplatelet drugs, a significant proportion of patients still exhibit high platelet reactivity despite treatment and therefore higher thrombogenic risk. Coronary intervention procedures represent a vulnerable time frame during which thrombotic events may occur triggered by coronary manipulation and the use of thrombogenic materials (i.e., coronary wires, balloon catheters, stents) [42, 43]. Moreover, PCI by itself can induce changes in hemostatic markers [42], inflammatory response [43], and platelet activation [44]. Despite ongoing dual antiplatelet therapy, platelet reactivity increases early after the procedure as compared to 24 h later in patients undergoing elective PCI with stent implantation [45–47]. We have recently investigated the impact of different types of coronary interventions on platelet reactivity in patients with stable CAD [48]. We found that an increased platelet reactivity is triggered by more extensive and aggressive coronary interventions (i.e., with multiple stenting or rotational atherectomy), which therefore expose to the risk of suboptimal platelet inhibition despite recommended loading dose of clopidogrel; that periprocedural variations of platelet reactivity are specifically linked to the degree of vascular damage and endothelial dysfunction induced by coronary interventions; and that procedure-related platelet activation is associated with an increased risk of myonecrosis. The evidence of transient platelet activation immediately after PCI has important clinical implications. First, in patients with extensive CAD in whom complex revascularization procedures are planned, a 600-mg loading dose of clopidogrel might not be sufficient to inhibit platelet reactivity in the vulnerable time frame around PCI. In this setting, a high thrombotic milieu is to be expected and therefore an increased risk of ischemic complications might occur. Second, besides the prognostic significance of preprocedural levels, a role for periprocedural variations emerges as a marker of risk for recurrent ischemic complications. In fact, PCI-induced platelet activation might be even more important in determining myocardial damage. Whether more aggressive antiplatelet strategies, such as with glycoprotein IIb/IIIa inhibitors or newer P2Y12 receptor blockers (i.e., prasugrel, ticagrelor), might be useful in improving periprocedural outcomes of patients undergoing extensive coronary revascularization procedures is to be investigated in specifically designed studies. Third, the timing of assessment might significantly influence the results of platelet function tests. For instance, if measured in the first hours following PCI, platelet reactivity might be overestimated as still influenced by the procedure. This might be of particular importance when trying to link residual platelet reactivity assessed post-PCI with future clinical outcomes. The “Gauging Responsiveness with A VerifyNow assay—Impact on Thrombosis And Safety” (GRAVITAS) trial aimed at demonstrating that in PCI patients with high platelet reactivity, the use of high-dose clopidogrel compared with standard dose could improve clinical outcomes at follow-up [49]. Disappointingly, this study failed to demonstrate any benefit from a tailored antiplatelet strategy. However, the fact that platelet reactivity was measured 12 to 24 h post-PCI might have accounted for an improper patient selection. In fact, some of the patients identified as having high platelet reactivity could have been differently classified if platelet function had been assessed before PCI or after a sufficient interval postprocedure. Nevertheless, the timing of platelet function testing is not the only determinant of the negative results of the GRAVITAS trial, where only low-to-intermediate risk patients were enrolled, and recording therefore a number of adverse events is probably not sufficient to show any meaningful differences between different antiplatelet strategies. Similar negative results have been recently achieved by the Assessment by a Double Randomization of a Conventional Antiplatelet Strategy versus a Monitoring-Guided Strategy for Drug-Eluting Stent Implantation and of Treatment Interruption versus Continuation One Year after Stenting study [50], confirming the lack of benefit from platelet function monitoring and treatment adjustment after coronary stenting, as compared with standard antiplatelet therapy without monitoring.
Conclusions
A network of interactions exists between platelets, inflammatory, and endothelial cells. Platelets actively participate in the early phases of endothelial dysfunction, which is the initial step of atherogenesis and plaque formation, and they are the mediators of the thrombotic complications that represent the most detrimental manifestations of coronary atherosclerosis. The role of platelets and of their interaction with vessel wall is even more important in patients treated with PCI, where mechanical stimuli directly induce vascular injury and cell activation. All these factors should be taken into account when choosing the appropriate antiplatelet treatment, in order to overcome the transient or persistent increase in platelet reactivity consequent to their interaction with vessel wall. Tailored strategies based on the extent of coronary atherosclerosis and on the type of intravascular procedures should be tested in future clinical trials.
References
Davi, G., & Patrono, C. (2007). Platelet activation and atherothrombosis. The New England Journal of Medicine, 357, 2482–94.
Ombrello, C., Block, R. C., & Morrell, C. N. (2010). Our expanding view of platelet functions and its clinical implications. Journal of Cardiovascular Translational Research, 3, 538–46.
Gawaz, M., Neumann, F. J., Dickfeld, T., Reininger, A., Adelsberger, H., Gebhardt, A., et al. (1997). Vitronectin receptor (alpha(v)beta3) mediates platelet adhesion to the luminal aspect of endothelial cells: implications for reperfusion in acute myocardial infarction. Circulation, 96, 1809–18.
Bombeli, T., Schwartz, B. R., & Harlan, J. M. (1998). Adhesion of activated platelets to endothelial cells: evidence for a GPIIbIIIa-dependent bridging mechanism and novel roles for endothelial intercellular adhesion molecule 1 (ICAM-1), alphavbeta3 integrin, and GPIbalpha. The Journal of Experimental Medicine, 187, 329–39.
Gawaz, M., Langer, H., & May, A. E. (2005). Platelets in inflammation and atherogenesis. The Journal of Clinical Investigation, 115, 3378–84.
May, A. E., Seizer, P., & Gawaz, M. (2008). Platelets: inflammatory firebugs of vascular walls. Arteriosclerosis, Thrombosis, and Vascular Biology, 28, s5–10.
Henn, V., Slupsky, J. R., Grafe, M., Anagnostopoulos, I., Forster, R., Muller-Berghaus, G., et al. (1998). CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature, 391, 591–94.
May, A. E., Kalsch, T., Massberg, S., Herouy, Y., Schmidt, R., & Gawaz, M. (2002). Engagement of glycoprotein IIb/IIIa (alpha(IIb)beta3) on platelets upregulates CD40L and triggers CD40L-dependent matrix degradation by endothelial cells. Circulation, 106, 2111–17.
Mayadas, T. N., Johnson, R. C., Rayburn, H., Hynes, R. O., & Wagner, D. D. (1993). Leukocyte rolling and extravasation are severely compromised in P selectin-deficient mice. Cell, 74, 541–54.
Subramaniam, M., Saffaripour, S., Watson, S. R., Mayadas, T. N., Hynes, R. O., & Wagner, D. D. (1995). Reduced recruitment of inflammatory cells in a contact hypersensitivity response in P-selectin-deficient mice. The Journal of Experimental Medicine, 181, 2277–82.
Husain, S., Andrews, N. P., Mulcahy, D., Panza, J. A., & Quyyumi, A. A. (1998). Aspirin improves endothelial dysfunction in atherosclerosis. Circulation, 97, 716–20.
Heitzer, T., Ollmann, I., Koke, K., Meinertz, T., & Munzel, T. (2003). Platelet glycoprotein IIb/IIIa receptor blockade improves vascular nitric oxide bioavailability in patients with coronary artery disease. Circulation, 108, 536–41.
Jakubowski, A., Chlopicki, S., Olszanecki, R., Jawien, J., Lomnicka, M., Dupin, J. P., et al. (2005). Endothelial action of thienopyridines and thienopyrimidinones in the isolated guinea pig heart. Prostaglandins, Leukotrienes, and Essential Fatty Acids, 72, 139–45.
Ziemianin, B., Olszanecki, R., Uracz, W., Marcinkiewicz, E., & Gryglewski, R. J. (1999). Thienopyridines: effects on cultured endothelial cells. Journal of Physiology and Pharmacology, 50, 597–604.
Heitzer, T., Rudolph, V., Schwedhelm, E., Karstens, M., Sydow, K., Ortak, M., et al. (2006). Clopidogrel improves systemic endothelial nitric oxide bioavailability in patients with coronary artery disease: evidence for antioxidant and antiinflammatory effects. Arteriosclerosis, Thrombosis, and Vascular Biology, 26, 1648–52.
Warnholtz, A., Ostad, M. A., Velich, N., Trautmann, C., Schinzel, R., Walter, U., et al. (2008). A single loading dose of clopidogrel causes dose-dependent improvement of endothelial dysfunction in patients with stable coronary artery disease: results of a double-blind, randomized study. Atherosclerosis, 196, 689–95.
Muller, O., Hamilos, M., Bartunek, J., Ulrichts, H., Mangiacapra, F., Holz, J. B., et al. (2010). Relation of endothelial function to residual platelet reactivity after clopidogrel in patients with stable angina pectoris undergoing percutaneous coronary intervention. The American Journal of Cardiology, 105, 333–38.
Hamilos, M., Muller, O., Ntalianis, A., Trana, C., Bartunek, J., Sarno, G., et al. (2011). Relationship between peripheral arterial reactive hyperemia and residual platelet reactivity after 600 mg clopidogrel. Journal of Thrombosis and Thrombolysis, 32, 64–71.
Patti, G., Grieco, D., Dicuonzo, G., Pasceri, V., Nusca, A., & Di Sciascio, G. (2011). High versus standard clopidogrel maintenance dose after percutaneous coronary intervention and effects on platelet inhibition, endothelial function, and inflammation results of the ARMYDA-150 mg (antiplatelet therapy for reduction of myocardial damage during angioplasty) randomized study. Journal of the American College of Cardiology, 57, 771–78.
Barbato, E., Bartunek, J., Wyffels, E., Wijns, W., Heyndrickx, G. R., & De Bruyne, B. (2003). Effects of intravenous dobutamine on coronary vasomotion in humans. Journal of the American College of Cardiology, 42, 1596–601.
Barbato, E., Piscione, F., Bartunek, J., Galasso, G., Cirillo, P., De Luca, G., et al. (2005). Role of beta2 adrenergic receptors in human atherosclerotic coronary arteries. Circulation, 111, 288–94.
Methia, N., Andre, P., Denis, C. V., Economopoulos, M., & Wagner, D. D. (2001). Localized reduction of atherosclerosis in von Willebrand factor-deficient mice. Blood, 98, 1424–28.
Egan, K. M., Wang, M., Fries, S., Lucitt, M. B., Zukas, A. M., Pure, E., et al. (2005). Cyclooxygenases, thromboxane, and atherosclerosis: plaque destabilization by cyclooxygenase-2 inhibition combined with thromboxane receptor antagonism. Circulation, 111, 334–42.
Pratico, D., Cyrus, T., Li, H., & FitzGerald, G. A. (2000). Endogenous biosynthesis of thromboxane and prostacyclin in 2 distinct murine models of atherosclerosis. Blood, 96, 3823–26.
Dixon, D. A., Tolley, N. D., Bemis-Standoli, K., Martinez, M. L., Weyrich, A. S., Morrow, J. D., et al. (2006). Expression of COX-2 in platelet-monocyte interactions occurs via combinatorial regulation involving adhesion and cytokine signaling. The Journal of Clinical Investigation, 116, 2727–38.
Barbato, E., Rubattu, S., Bartunek, J., Berni, A., Sarno, G., Vanderheyden, M., et al. (2009). Human coronary atherosclerosis modulates cardiac natriuretic peptide release. Atherosclerosis, 206, 258–64.
Jayachandran, M., Litwiller, R. D., Lahr, B. D., Bailey, K. R., Owen, W. G., Mulvagh, S. L., et al. (2011). Alterations in platelet function and cell-derived microvesicles in recently menopausal women: relationship to metabolic syndrome and atherogenic risk. Journal of Cardiovascular Translational Research, 4, 811–22.
Koyama, H., Maeno, T., Fukumoto, S., Shoji, T., Yamane, T., Yokoyama, H., et al. (2003). Platelet P-selectin expression is associated with atherosclerotic wall thickness in carotid artery in humans. Circulation, 108, 524–29.
Fateh-Moghadam, S., Li, Z., Ersel, S., Reuter, T., Htun, P., Plockinger, U., et al. (2005). Platelet degranulation is associated with progression of intima-media thickness of the common carotid artery in patients with diabetes mellitus type 2. Arteriosclerosis, Thrombosis, and Vascular Biology, 25, 1299–303.
Furman, M. I., Benoit, S. E., Barnard, M. R., Valeri, C. R., Borbone, M. L., Becker, R. C., et al. (1998). Increased platelet reactivity and circulating monocyte-platelet aggregates in patients with stable coronary artery disease. Journal of the American College of Cardiology, 31, 352–58.
Furman, M. I., Barnard, M. R., Krueger, L. A., Fox, M. L., Shilale, E. A., Lessard, D. M., et al. (2001). Circulating monocyte-platelet aggregates are an early marker of acute myocardial infarction. Journal of the American College of Cardiology, 38, 1002–06.
Keating, F. K., Whitaker, D. A., Kabbani, S. S., Ricci, M. A., Sobel, B. E., & Schneider, D. J. (2004). Relation of augmented platelet reactivity to the magnitude of distribution of atherosclerosis. The American Journal of Cardiology, 94, 725–28.
Mangiacapra, F., De Bruyne, B., Muller, O., Trana, C., Ntalianis, A., Bartunek, J., et al. (2010). High residual platelet reactivity after clopidogrel: extent of coronary atherosclerosis and periprocedural myocardial infarction in patients with stable angina undergoing percutaneous coronary intervention. JACC. Cardiovascular Interventions, 3, 35–40.
Tcheng, J. E., Lim, I. H., Srinivasan, S., Jozic, J., Gibson, C. M., O’Shea, J. C., et al. (2009). Stent parameters predict major adverse clinical events and the response to platelet glycoprotein IIb/IIIa blockade: findings of the ESPRIT trial. Circulation. Cardiovascular Interventions, 2, 43–51.
Wang, T. H., Bhatt, D. L., Fox, K. A., Steinhubl, S. R., Brennan, D. M., Hacke, W., et al. (2007). An analysis of mortality rates with dual-antiplatelet therapy in the primary prevention population of the CHARISMA trial. European Heart Journal, 28, 2200–07.
Mangiacapra, F., Barbato, E., Patti, G., Gatto, L., Vizzi, V., Ricottini, E., et al. (2010). Point-of-care assessment of platelet reactivity after clopidogrel to predict myonecrosis in patients undergoing percutaneous coronary intervention. JACC. Cardiovascular Interventions, 3, 318–23.
Mangiacapra, F., Patti, G., Peace, A., Gatto, L., Vizzi, V., Ricottini, E., et al. (2010). Comparison of platelet reactivity and periprocedural outcomes in patients with versus without diabetes mellitus and treated with clopidogrel and percutaneous coronary intervention. The American Journal of Cardiology, 106, 619–23.
Breet, N. J., van Werkum, J. W., Bouman, H. J., Kelder, J. C., Ruven, H. J., Bal, E. T., et al. (2010). Comparison of platelet function tests in predicting clinical outcome in patients undergoing coronary stent implantation. JAMA: The Journal of the American Medical Association, 303, 754–62.
Mangiacapra, F., Patti, G., Barbato, E., Peace, A. J., Ricottini, E., Vizzi, V., et al. (2012). A therapeutic window for platelet reactivity for patients undergoing elective percutaneous coronary intervention: results of the ARMYDA-PROVE (antiplatelet therapy for reduction of MYocardial damage during angioplasty-platelet reactivity for outcome validation effort) study. JACC. Cardiovascular Interventions, 5, 281–89.
Mehta, S. R., Yusuf, S., Peters, R. J., Bertrand, M. E., Lewis, B. S., Natarajan, M. K., et al. (2001). Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. The Lancet, 358, 527–33.
Steinhubl, S. R., Berger, P. B., Mann, J. T., Fry, E. T., DeLago, A., Wilmer, C., et al. (2002). Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA: The Journal of the American Medical Association, 288, 2411–20.
Saito, Y., Wada, H., Yamamuro, M., Inoue, A., Shimura, M., Hiyoyama, K., et al. (1999). Changes of plasma hemostatic markers during percutaneous transluminal coronary angioplasty in patients with chronic coronary artery disease. American Journal of Hematology, 61, 238–42.
Serrano, C. V. J., Ramires, J. A., Venturinelli, M., Arie, S., D'Amico, E., Zweier, J. L., et al. (1997). Coronary angioplasty results in leukocyte and platelet activation with adhesion molecule expression. Evidence of inflammatory responses in coronary angioplasty. Journal of the American College of Cardiology, 29, 1276–83.
Scharf, R. E., Tomer, A., Marzec, U. M., Teirstein, P. S., Ruggeri, Z. M., & Harker, L. A. (1992). Activation of platelets in blood perfusing angioplasty-damaged coronary arteries. Flow cytometric detection. Arterioscler Thromb, 12, 1475–87.
Gurbel, P. A., Samara, W. M., & Bliden, K. P. (2004). Failure of clopidogrel to reduce platelet reactivity and activation following standard dosing in elective stenting: implications for thrombotic events and restenosis. Platelets, 15, 95–99.
Matetzky, S., Shenkman, B., Guetta, V., Shechter, M., Beinart, R., Goldenberg, I., et al. (2004). Clopidogrel resistance is associated with increased risk of recurrent atherothrombotic events in patients with acute myocardial infarction. Circulation, 109, 3171–75.
Siller-Matula, J. M., Haberl, K., Prillinger, K., Panzer, S., Lang, I., & Jilma, B. (2009). The effect of antiplatelet drugs clopidogrel and aspirin is less immediately after stent implantation. Thrombosis Research, 123, 874–80.
Mangiacapra, F., Bartunek, J., Bijnens, N., Peace, A. J., Dierickx, K., Bailleul, E., et al. (2012). Peri-procedural variations of platelet reactivity during elective percutaneous coronary intervention. Journal of Thrombosis and Haemostasis, 10(12), 2452–61.
Price, M. J., Berger, P. B., Teirstein, P. S., Tanguay, J. F., Angiolillo, D. J., Spriggs, D., et al. (2011). Standard- vs high-dose clopidogrel based on platelet function testing after percutaneous coronary intervention: the GRAVITAS randomized trial. JAMA: The Journal of the American Medical Association, 305, 1097–105.
Collet, J. P., Cuisset, T., Range, G., et al. (2012). Bedside monitoring to adjust antiplatelet therapy for coronary stenting. The New England Journal of Medicine, 367(22), 2100–9.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mangiacapra, F., Barbato, E. Clinical Implications of Platelet—Vessel Interaction. J. of Cardiovasc. Trans. Res. 6, 310–315 (2013). https://doi.org/10.1007/s12265-012-9441-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-012-9441-0