
Abstract
Heat shock protein 32 (HSP32) is a stress response protein that can be induced by heat stress in the liver, and its induction can act as an important cellular defence mechanism against heat-induced liver injury. To investigate the functional role of HSP32 in protecting liver tissue against heat stress in mice and the mechanism by which it achieves this protective effect, HSP32 expression and carbon monoxide (CO) contents in a model of mice subjected to acute, transient heat exposure were examined. Furthermore, functional and histological parameters of liver damage and the possible involvement of oxidative stress to induce oxidative deterioration of liver functions and caspase-3 expression were also investigated in this study. We found that heat treatment of mice produced severe hepatic injury, whereas upregulation of HSP32 with hemin pretreatment prevented mice from liver damage. In contrast, addition of Sn-protoporphyrin (SnPP) to inhibit HSP32 expression completely reversed its hepatoprotective effect. It is concluded that upregulation of HSP32 by hemin could alleviate acute heat-induced hepatocellular damage in mice, and its by-product CO seems to play a more important role in hepatoprotective mechanism.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The heat shock response (HSR) is an ancient and highly conserved molecular response to disruptions of protein homeostasis (proteostasis) (Morimoto 2008, 2011; Åkerfelt et al. 2010). Heat shock proteins (HSPs), recognized as families of highly conservative stress proteins, were identified first as proteins overexpressed in response to heat stress (Ritossa 1962; Tissiéres et al. 1974). Many of the protective effects of the HSPs have been attributed to HSP32 (Willis et al. 1996; Otterbein et al. 1999; Petrache et al. 2000; Rensing et al. 1999; Shiraishi et al. 2000; Carchman et al. 2011), which is a member of the HSP family better known as heme oxygenase 1 (HO-1) and is one of the most widespread antioxidant defence enzymes.
HSP32 is a stress-responsive protein that catabolizes hemes into biliverdin, free iron and carbon monoxide (CO) and is upregulated under conditions of various stimuli such as hyperoxia, hypoxia, heat shock, endotoxemia, hydrogen peroxide, cytokines, ultraviolet-A radiation, heavy metals and nitric oxide (Camhi et al. 1995; Carraway et al. 1998, 2000; Lee et al. 1997; Keyse and Tyrrell 1987; Lautier et al. 1992; Maines and Ewing 1996; Eyssen-Hernandez et al. 1996). Moreover, its expression is much higher in the spleen and liver than in other tissues (Yao et al. 2009; Yumi et al. 2010). Induction of HSP32 occurs as an adaptive and beneficial response to various injurious stimuli, and this inducible nature of HSP32 signifies its importance in several pathophysiological states, such as liver diseases (Farombi and Surh 2006). Besides, there has been increasing evidence supporting the role of HSP32, and most notably, its reaction products in the hepatic stress response under acute and chronic pathophysiological conditions suggest that HSP32 could protect liver tissue against a wide array of noxious stimuli (Tsuchihashi et al. 2006; Sass et al. 2012). Our earlier research showed that the overexpression of HSP32 probably played a protective role in the liver of heat-exposed mice (Li et al. 2013a). However, the precise role of HSP32 in the protection of the liver subjected to heat stress and the underlying mechanisms still remains unclear.
In response to these findings, investigations into the role of HSP32 in protecting liver tissue against heat stress in mice and the mechanism by which it achieves this protective effect have great significance. The aim of the present study was to investigate the functional role of HSP32 induction in a mice model of liver injury caused by acute heat exposure and the possible protective mechanism of HSP32 in response to hyperthermia.
Materials and methods
Animals
Experiments were performed in 128 male Kunming mice (10-week-old) that were purchased from the Nanjing Qinglongshan Experimental Animal Factory. The mice were reared in separate cages under environmental conditions including free diet, 12–12 h light–dark cycles and a room temperature ranging from 23 to 25 °C. All experiments were carried out in accordance with the National Institutes of Health guidelines.
Animal heat treatment and tissue preparation
The treatment was based on previous reports (Redaelli et al. 2002) and our preliminary studies. Body temperature was monitored with a rectal thermometer; the acute, transient heat exposure consisted of 10-week-old mice that were placed in a perforated Perspex cage with cover and were exposed to a heat stress at 43 °C with 60 % relative humidity in FPQ multiband artificial climate chamber (Ningbo Lai Fu Technology Co., Ltd., Zhejiang, China). The body temperature was slowly elevated to 43 °C and maintained for 30 min.
Mice were randomly divided into four groups: (1) control group (C); (2) acute, transient heat-treated group (ATH); (3) hemin + ATH, hemin (30 μmol/kg body weight) was administered intraperitoneally to mice 24 h before the induction of liver injury and (4) Sn-protoporphyrin (SnPP) + ATH, SnPP (10 μmol/kg body weight) was administered intraperitoneally 24 h prior to acute, transient treatment. The last three groups were divided into five subgroups for sampling at different time points. The dose of hemin and SnPP adopted in this study was selected according to previous reports.
After heating, the mice were removed immediately and transferred to room temperature conditions. Untreated controls (C group) were reared normally and received no other treatment. At various time points 3, 6, 12, 24 and 36 h after the completion of the heat treatment, the mice were anaesthetized with pentobarbital sodium (4 mg/kg body weight, iv), then euthanized by cervical vertebra dislocation. Livers were collected and rinsed twice in phosphate-buffered saline, and then samples were immediately processed for measurements, fixed for histology evaluations or frozen in liquid nitrogen for subsequent experiments. Eight animals at each time point were tested in each group.
Immunohistochemistry and histology preparation and evaluation
To prepare slides for immunohistochemistry staining and histology evaluation, the fresh liver tissue samples were fixed in 10 % neutral-buffered formalin and tissue sections were subsequently dehydrated through steps of graded alcohol, cleared in xylene and imbedded in paraffin blocks. Sections of 6-μm thickness were cut from each block.
For immunohistochemical analysis, liver sections were deparaffinised, hydrated by successive series of ethanol, rinsed in phosphate-buffered saline (PBS) and then incubated in 3 % H2O2 in PBS for 10 min to quench endogenous peroxidase. For the mammalian study, primary antibodies included rabbit polyclonal HSP32 and caspase-3 (1:100) (Beyotime Biotechnology, China; diluted 1:100). Immunoreactivity was detected using biotinylated goat anti-rabbit IgG secondary antibody followed by avidin-biotinylated horseradish peroxidase complex visualized with diaminobenzidine tetrahydrochloride according to the manufacturer’s instructions. Slides were counterstained with haematoxylin. Negative and positive controls were run for every assay, and the primary antibody was substituted with the rabbit IgG in the negative controls.
For histological evaluation, the deparaffinised sections were stained with haematoxylin and eosin. Liver sections were then analysed under a microscope for evidence of injury.
ALT and AST measurements
Serum alanine aminotransferase (ALT) and transaminases as aspartate aminotransferase (AST) were measured in serum following the commercial kits (Jiancheng Bioengineering Inc., Nanjing, China) according to the manufacturer’s instructions. The results were expressed as U/L. AST and ALT are the sensitive indicators for hepatocellular damage which can reflect the condition of hepatocyte injury.
SOD and MDA measurements
Liver homogenates were prepared in PBS and centrifuged at 12,000g for 20 min at 4 °C. Total superoxide dismutase (SOD) activity in the hepatic tissue supernatant was determined. Samples were taken to detect absorbance at an absorbance of 550 nm. The results were expressed as units per milligram of protein. Malonaldehyde (MDA) content was measured using the thiobarbituric acid (TBA) method at an absorbance of 532 nm. The results were expressed as nanomoles per milligram of protein. Procedures were performed with assay kits according to the manufacturer’s instructions. SOD and MDA assay kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
CO content measurement
After centrifugation of the crude homogenates of liver tissue, endogenous CO content was determined using a carbon monoxide assay kit (Jiancheng Bioengineering Inc., Nanjing, China). The results were expressed as micromoles per gram.
RNA extraction and reverse transcription polymerase chain reaction analysis
Livers were perfused with PBS, frozen in liquid nitrogen and stored at −80 °C (frozen stock liver) until used as described previously. The level of HSP32 and caspase-3 mRNA in the liver were evaluated by reverse transcription polymerase chain reaction (RT-PCR), which was performed using an ABI Prism Sequence Detection System (Applied Biosystems). Total RNA was extracted from liver tissue using a Total RNA Isolation Kit (F. Hoffmann-La Roche Ltd., Shanghai, China) according to the manufacturer’s instructions. An aliquot of the total RNA was treated with DNase I, and then it was reverse transcribed into cDNA with random hexamer primers (TaKaRa, Dalian, China). The resulting cDNA was used for SYBR Green (Applied Biosystems) real-time PCR amplification of HSP32 and caspase-3. PCR reactions were performed with the following primers: 5′-TTT TCC ACG GCG ACT CAG-3′ and 5′-CAA CGG TAAACA ACA CGA TC-3′ for mouse HSP32; 5′-CTG GACTGT GGC ATT GAG AC-3′ and 5′-GCA AAG GGACTG GAT GAA CC-3′ for mouse caspase-3. All mRNA analyses were performed in triplicate, and expression levels of HSP32 and caspase-3 were corrected using an endogenous control (mouse GADPH, primers 5′-GGA TTTCCC TGG GTC TTC-3′ and 5′-TAA GAA AGG CAAACC AGA A-3′). The fold differences in mRNA expression of samples were relative to the internal control sample, which was included in all runs.
HSP32 quantitation by ELISA
Tissue HSP32 levels in tissue homogenates (50 μl) were determined using a commercially available ELISA (enzyme-linked immunosorbent assay) specific for the mice cytokine, purchased from R&D Systems (Shanghai, China); this kit shows a sensitivity of 0.78 ng/ml and ranges up to 25 ng/ml.
Statistical analysis
All results were expressed as mean ± SD. The statistical analysis was performed using a one-way analysis of variance. Results were considered statistically significant at P < 0.05.
Results
Morphology of heat-exposed liver
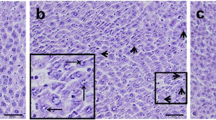
Histological evaluation of sections revealed that the liver morphology was normal in control animals (Fig. 1a), whereas, heat exposure groups demonstrated some hepatic injury, and the liver injury peaked after 24 h of transient heat exposure. The ATH mice showed variable hydropic degeneration and ballooning degeneration, accompanied by a small amount of inflammatory cell infiltration fatty (Fig. 1b). And the SnPP + ATH group showed more severe hepatocellular damage than that for the ATH group (Fig. 1d). However, only mild injury was present in the hemin + ATH group with little evidence of hydropic degeneration and ballooning degeneration (Fig. 1c).

Effect of pretreatment with hemin and Sn-protoporphyrin (SnPP) on the histological changes in the liver at 24 h after heat exposure. a The control mice, without any signs of liver damage. b The ATH mice, variable hydropic degeneration and ballooning degeneration, accompanied by a small amount of inflammatory cell infiltration fatty were shown in the sections. c The hemin + ATH mice, with little evidence of hydropic degeneration and ballooning degeneration. d The SnPP + ATH mice, more severe hepatocellular damage was observed than that for the ATH mice and accompanied by fatty degeneration. 1, 2, 3 magnifications ×100, ×200 and ×400
MDA content, SOD activity in the liver and Serum AST and ALT levels
The levels of MDA and serum ALT and AST in the ATH group started to increase at 3 h after heat exposure, and this elevation persisted until 24 h after heat treatment, which showed significant differences when compared with the control group (P < 0.05). The elevation in the levels of MDA, serum AST and ALT at all time points was markedly augmented by SnPP but attenuated by hemin treatment (Fig. 2a, c and d). In contrast, the hepatic SOD activity decreased significantly at 3 h after heat exposure and bottomed out at 24 h after heat treatment. These decreases in SOD activity were markedly augmented by SnPP but attenuated by hemin treatment (Fig. 2b).

Superoxide dismutase (SOD) activity, malonaldehyde (MDA) contents, serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels at all time points of each group. a Hepatic MDA contents in the mice. b SOD activity in the liver of mice. c Serum AST level in the mice. d Serum ALT level in the mice. 3, 6, 12, 24 and 36 h, time after heat exposure. The results are represented as the mean (±SD) of six mice. *P < 0.05 and **P < 0.01 compared to the control group, #P < 0.05 and ##P < 0.01 compared to the ATH group, n = 8
Caspase-3 expression in the liver
The expression of caspase-3 mRNA in the ATH group was increased at 6 h after heat exposure and peaked at 24 h after heat treatment, which was significantly higher than that in the control group (P < 0.05). However, in the hemin + ATH group, caspase-3 mRNA expression was significantly lower than that in the ATH group (P < 0.05), while the expression of caspase-3 mRNA in the SnPP + ATH group was significantly higher than that in the ATH group (P < 0.05) (Fig. 3a).

a The expression of caspase-3 mRNA in the liver of the acute heat-exposed mice. b Relative expression of heat shock protein 32 (HSP32) mRNA in mice exposed to different treatments c ELISA analysis of protein levels of HSP32 in the liver of mice. d Effect of HSP32 induction on CO contents in the liver of mice. 3, 6, 12, 24, 36 h, time after heat exposure. The results are represented as the mean (±SD) of six mice. *P < 0.05 and **P < 0.01 compared to the control group, #P < 0.05 and ##P < 0.01 compared to the ATH group, n = 8
To examine the protective effects of elevated HSP32 expression on the liver of heat-stressed mice, caspase-3 expression in the liver at 24 h after heat treatment was assessed by immunohistochemical staining. As indicated in Fig. 4d–f), positive caspase-3 expression was localized in the cytoplasm of hepatocytes.


Heat shock protein 32 (HSP32) and caspase-3 immunoreactivity in the liver of mice (24 h after heat treatment). a, b, c Localization of HSP32 protein expression in liver tissues, shown as brown cytoplasmic staining, mainly around the central veins where the liver injury was more serious. d, e, f The caspase-3 expression in the liver, positive caspase-3 expression was localized in the cytoplasm of hepatocytes. a, d The ATH group. b, e The hemin + ATH group. c, f The SnPP + ATH group
HSP32 expression in the liver
The mRNA and protein expression of HSP32 was increased significantly after transient heat exposure compared to the control group (P < 0.05). Compared with the ATH group, hemin administration significantly increased HSP32 expression at all time points, while addition of SnPP significantly inhibited the HSP32 mRNA and protein expression (Fig. 3b, c). Localization of HSP32 protein expression was determined in liver tissues at 24 h after heat exposure, shown as brown cytoplasmic staining, mainly around the central veins where the liver injury was more serious (Fig. 4a–c).
CO contents in the liver tissue supernatant
Parallel with increased HSP32 expression, CO contents in the ATH group was significantly increased in livers of mice subjected to acute, transient heat exposure (P < 0.05). This elevation in the level of CO at all time points was significantly augmented by hemin but attenuated by SnPP treatment (Fig. 3d).
Discussion
HSP32 is an inducible isoenzyme, which can be upregulated to provide an important protective response from many different noxious stimuli. Additionally, accumulating evidence shows that the induction of HSP32 can catalyse redundant heme to avoid heme-induced injury, and its by-products might also have anti-inflammatory, anti-apoptotic and anti-oxidation properties (Wen et al. 2007; Li et al. 2013b).
In the present study, we examined the effects of HSP32 induction, as well as the effects of HSP32 inhibition, on the outcome of liver injury caused by heat stress. The induction of HSP32 was made by pretreating mice with hemin, a well-known physiological substrate and potent inducer of HSP32. At the same time, SnPP as a competitive HSP32 inhibitor was administrated intraperitoneally in mice to inhibit hepatic HSP32 activity. The experiment results showed that expression levels of HSP32 started to increase within 3 h after transient heat exposure, and the levels remained high thereafter and reached a peak level at 24 h. What’s more, hemin pretreatment significantly increased HSP32 expression when compared with the ATH group. However, SnPP administration completely inhibited HSP32 expression in the livers of mice and remained at a low level throughout the entire heat exposure.
The results of this study clearly showed that HSP32 upregulation with enhanced HSP32 expression at the mRNA and protein levels following hemin administration attenuated the severity of heat-induced hepatic injury, as evidenced by analysis of functional and histological parameters of liver damage. Addition of SnPP completely abolished HSP32-induced hepatoprotective effect as confirmed by increased AST, ALT levels and more severe pathological lesions, suggesting that HSP32 induction in the liver of mice is essential for protection from acute liver damage.
MDA is a biomarker of lipid peroxidation (LPO); its concentration indicates the degree of oxidative degradation of polyunsaturated fatty acids (PUFA) and cellular deterioration in the liver (Drewa et al. 2002). SOD is considered the first line of defence against the deleterious effects of oxyradicals in the cell and plays a key role in the antioxidant system (Ha et al. 2010). The observations revealed that hemin pretreatment could decrease MDA concentrations and increase SOD activity in the liver of mice when compared with the heat-treated-alone mice, whereas SnPP administration concomitantly abolished this intervening effect, indicating that high expression of HSP32 induced by hemin could decrease the lipid peroxidation and play an anti-oxidative role in heat-induced liver injury, which might be partially responsible for the protective mechanisms.
Apoptosis has been identified as a mechanism of hepatic injury, and caspase-3 is probably the best understood of the mammalian caspases in terms of its specificity and roles in apoptosis. The activation of caspase-3 can be used as an improved index of apoptosis (Khan and Brown 2002). In the current study, to determine the effect of HSP32 on apoptosis, we carried out an immunohistochemical and RT-PCR analysis of caspase-3. Results showed that the expression of caspase-3 in the ATH group was increased significantly at 6 h after heat exposure and peaked at 24 h after heat treatment. After administrating hemin to induce HSP32 high expression, caspase-3 expression was significantly attenuated. Likewise, addition of SnPP abrogated the inhibitive effect of hemin. The results suggesting that upregulation of HSP32 may contribute to the suppression of apoptosis via inhibition of caspase-3 were consistent with research by Burger et al. (2009). Therefore, it might be another important mechanism responsible for HSP32-mediated protection from liver injury.
It has been proposed that HSP32-mediated hepatoprotection is mainly dependent on one or more of its reaction; the most recent work in the field implicates CO-related cell signalling as the key component of HSP32-mediated hepatoprotection (Ryter et al. 2002; Choi and Otterbein 2002; Morse et al. 2001). CO as a gaseous by-product of heme metabolism has long been thought to participate in many biological events and plays a pivotal role in mediating cytoprotection against oxidant-induced injury (Amersi et al. 2002; Ryter and Tyrrell 2000; Moore et al. 2005; Liu et al. 2010). Furthermore, recent evidence suggests that CO may additionally confer protective effects via anti-inflammatory and/or anti-apoptotic mechanisms (Farombi and Surh 2006). Physiological concentration of CO is mainly derived from intrinsic heme degradation by HSP32 under stress conditions (Ryter et al. 2006). In the present study, the experiment showed that CO contents were increased in livers of mice subjected to acute, transient heat exposure, and this elevation in the level of CO was augmented by hemin but attenuated by SnPP treatment; these results were in parallel with HSP32 expression. As noted above, it can be postulated that the protective effects of HSP32 may be mainly dependent on endogenous CO, which deserves more studies in the future.
In conclusion, the results obtained in this study indicate that acute heat treatment of mice produced severe hepatic injury, and the injury was the most serious at 24 h after heat exposure. Upregulation of HSP32 by hemin alleviates acute heat-induced hepatocellular damage in mice; the protection can probably be mediated through the antioxidant, anti-inflammatory and anti-apoptotic functions, and its by-product CO could play an important role in cytoprotective mechanism. In view of the above, our results will provide important clues for further exploration of the protective mechanism of HSP32 in response to noxious stimuli.
References
Åkerfelt M, Morimoto RI, Sistonen L (2010) Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol 11:545–555
Amersi F, Shen XD, Anselmo D, Melinek J, Iyer S, Southard DJ, Katori M, Volk HD, Busuttil RW, Buelow R, Kupiec-Weglinski JW (2002) Ex vivo exposure to carbon monoxide prevents hepatic ischemia/reperfusion injury through p38 MAP kinase pathway. Hepatology 35:815–823
Burger D, Xian F, Hammoud L (2009) Role of heme oxygenase-1 in the cardioprotective effects of erythropoietin during myocardial ischemia and reperfusion. J Physiol Heart Circ Physiol 296:84–93
Camhi SL, Alam J, Otterbein L, Sylvester SL, Choi AM (1995) Induction of heme oxygenase-1 gene expression by lipopolysaccharide is mediated by AP-1 activation. Am J Respir Cell Mol Biol 13:387–398
Carchman EH, Rao J, Loughran PA, Rosengart MR, Zuckerbraun BS (2011) Heme oxygenase-1-mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. J Hepatol 53:2053–2062
Carraway MS, Ghio AJ, Taylor JL, Piantadosi CA (1998) Induction of ferritin and heme oxygenase-1 by endotoxin in the lung. Am J Physiol 275:583–592
Carraway MS, Ghio AJ, Carter JD, Piantadosi CA (2000) Expression of heme oxygenase-1 in the lung in chronic hypoxia. Am J Respir Cell Mol Biol 278:806–812
Choi AM, Otterbein LE (2002) Emerging role of carbon monoxide in physiologic and pathophysiologic states. Antioxid Redox Signal 4:227–228
Drewa G, Krzyzyńska-Malinowska E, Woźniak A, Protas-Drozd F, Mila-Kierzenkowska C, Rozwodowska M, Czajkowski R (2002) Activity of superoxide dismutase and catalase and the level of lipid peroxidation products reactive with TBA in patients with psoriasis. Med Sci Monit 8:338–343
Eyssen-Hernandez R, Ladoux A, Frelin C (1996) Differential regulation of cardiac heme oxygenase-1 and vascular endothelial growth factor mRNA expressions by hemin, heavy metals, heat shock and anoxia. FEBS Lett 382:229–233
Farombi EO, Surh Y (2006) Heme oxygenase-1 as a potential therapeutic target for hepatoprotection. Int J Biochem Mol 39:479–491
Ha HL, Shin HJ, Feitelson MA, Yu DY (2010) Oxidative stress and antioxidants in hepatic pathogenesis. World J Gastroenterol 16:6035–6043
Keyse SM, Tyrrell RM (1987) Both near ultraviolet radiation and the oxidizing agent hydrogen peroxide induce a 32-kDa stress protein in normal human skin fibroblasts. J Biol Chem 262:14821–14825
Khan V, Brown I (2002) The effect of hyperthermia on the induction of cell death in brain, testis, and thymus of the adult and developing rat. Cell Stress Chaperones 7:73–90
Lautier D, Luscher P, Tyrrell RM (1992) Endogenous glutathione levels modulate both constitutive and UVA radiation/hydrogen peroxide inducible expression of the human heme oxygenase gene. Carcinogenesis 13:227–232
Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL, Choi AM (1997) Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J Biol Chem 272:5375–5381
Li CM, Li L, Bai JY, Wu J, Huang S, Wang GL (2013a) Correlation between heat shock protein 32 and chronic heat-induced liver injury in developing mice. J Therm Biol 38:513–519
Li L, Han ZY, Li CM, Jiang XQ, Wang GL (2013b) Upregulation of heat shock protein 32 in Sertoli cells alleviates the impairments caused by heat shock-induced apoptosis in mouse testis. Cell Stress Chaperones 1–19
Liu SH, Ma K, Xu XR, Xu B (2010) A single dose of carbon monoxide intraperitoneal administration protects rat intestine from injury induced by lipopolysaccharide. Cell Stress Chaperones 15:717–727
Maines MD, Ewing JF (1996) Stress response of the rat testis: in situ hybridization and immunohistochemical analysis of heme oxygenase-1 (HSP32) induction by hyperthermia. Biol Reprod 54:1070–1079
Moore BA, Overhaus M, Whitcomb J, Ifedigbo E, Choi AM, Otterbein LE, Bauer AJ (2005) Brief inhalation of low-dose carbon monoxide protects rodents and swine from postoperative ileus. Crit Care Med 33:1317–1326
Morimoto RI (2008) Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Gene Dev 22:1427–1438
Morimoto RI (2011) The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harb Sym 76:91–99
Morse D, Sethi J, Choi AM (2001) Carbon monoxide-dependent signaling. Crit Care Med 30:12–17
Otterbein LE, Kolls JK, Mantell LL, Cook JL, Alam J, Choi AM (1999) Exogenous administration of heme oxygenase-1 by gene transfer provides protection against hyperoxia-induced lung injury. J Clin Invest 103:1047–1054
Petrache I, Otterbein LE, Alam J, Wiegand GW, Choi AM (2000) Heme oxygenase-1 inhibits TNF-alpha-induced apoptosis in cultured fibroblasts. Am J Physiol-Lung C 278:312–319
Redaelli CA, Tian YH, Schaffner T, Ledermann M, Baer HU, Dufour JF (2002) Extended preservation of rat liver graft by induction of heme oxygenase-1. Hepatology 35:1082–1092
Rensing H, Bauer I, Datene V, Patau C, Pannen BH, Bauer M (1999) Differential expression pattern of heme oxygenase-1/heat shock protein 32 and nitric oxide synthase-II and their impact on liver injury in a rat model of hemorrhage and resuscitation. Crit Care Med 27:2766–2775
Ritossa FM (1962) A new puffing pattern induced by a temperature shock and DNP in Drosophila. Experientia 18:571–573
Ryter S, Tyrrell RM (2000) The heme synthesis and degradation pathways: role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic Biol Med 28:289–309
Ryter SW, Otterbein LE, Morse D, Choi AM (2002) Heme oxygenase/carbon monoxide signaling pathways: regulation and functional significance. Mol Cell Biochem 234–235:249–263
Ryter SW, Alam J, Choi AM (2006) Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 86:583–650
Sass G, Barikbin R, Tiegs G (2012) The multiple functions of heme oxygenase-1 in the liver. Z Geomorphol 50:34–40
Shiraishi F, Curtis LM, Truong L, Poss K, Visner GA, Madsen K, Nick HS (2000) Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am J Physiol Renal 278:726–736
Tissiéres A, Mitchell HK, Tracy UM (1974) Protein synthesis in salivary glands of Drosophila melanogaster: relation to chromosome puffs. J Mol Biol 84:389–398
Tsuchihashi SI, Livhits M, Zhai Y, Busuttil RW, Araujo JA, Kupiec-Weglinski JW (2006) Basal rather than induced heme oxygenase-1 levels are crucial in the antioxidant cytoprotection. J Immunol 177:4749–4757
Wen T, Wu ZM, Liu Y, Tan YF, Ren F, Wu H (2007) Upregulation of heme oxygenase-1 with hemin prevents d-galactosamine and lipopolysaccharide-induced acute hepatic injury in rats. Toxicology 237:184–193
Willis D, Moore AR, Frederick R, Willoughby DA (1996) Heme oxygenase: a novel target for the modulation of the inflammatory response. Nat Med 2:87–90
Yao P, Hao L, Nussler N, Lehmann A, Song F, Zhao J, Nussler A (2009) The protective role of HO-1 and its generated products (CO, bilirubin, and Fe) in ethanol-induced human hepatocyte damage. Am J Physiol-Gastr L 296:1318–1323
Yumi A, Yasuhiro S, Daigo S, Yoshito K (2010) Reduction of arsenic-induced cytotoxicity through Nrf2/HO-1 signaling in HepG2 cells. J Toxicol Sci 35:419–423
Acknowledgments
This work has received funding from the National Supporting Projects for Science and Techniques of China (2011BAD28B02, 2012BAD12B10).
Conflict of interest
There are no conflicts of interest, financial or otherwise declared by the author(s).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, Cm., Li, L., Wu, J. et al. Upregulation of heat shock protein 32 with hemin alleviates acute heat-induced hepatic injury in mice. Cell Stress and Chaperones 19, 675–683 (2014). https://doi.org/10.1007/s12192-014-0495-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12192-014-0495-6