
Abstract
Phthalates represent a potential risk for humans, since they are ubiquitous environmental contaminants. Efficient extraction and purification procedures are demanded for the detection of low concentration levels of phthalates. In this work, a novel type of molecularly imprinted polymers coated onto the surface of vinyl functionalized multi-walled carbon nanotubes was synthesized and coupled with gas chromatography–mass spectrometry (GC/MS) for the selective separation and determination of dioctyl phthalate (DOP) in beverage samples. The morphology, structure property, and thermostability of the resultant polymers were characterized by transmission electron microscopy, Fourier transform infrared spectroscopy, and thermogravimetric analysis. The adsorption properties of the prepared polymers were investigated by equilibrium rebinding and competitive experiments. The resultant imprinted nanomaterials exhibited high capacity and favorable selectivity. In addition, the feasibility of the developed method using the obtained imprinted polymers as a solid-phase sorbent coupled with GC/MS for the selective isolation and determination of DOP in different beverage samples was demonstrated. Under optimal conditions, the limit of detection of the proposed method for DOP was 2.3 ng L−1. DOP spiked at three levels of concentration in beverage samples was extracted and determined through the application of the present method, with recoveries ranging from 88.6 to 93.0 % with relative standard deviations less than 5.6 %.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Phthalates (PAEs), such as dioctyl phthalate (DOP), dibutyl phthalate (DBP), and di-(2-ethylhexyl) phthalate (DEHP), are primarily utilized as plasticizers to improve the flexibility and extensibility of polymeric materials in plastic industry (Aragón et al. 2013; Del Carlo et al. 2008). Because PAEs are only physically bound to the polymer chains, they can be released from the polymer products to the environment, even to the food and beverage which are packed with plastic materials (Zhang and Chen 2014). Several studies have suggested that some PAEs are recognized as ubiquitous environmental contaminants and have adverse effects on human health due to their suspected carcinogenic, teratogenic, and endocrine disrupting action (Fu and Du 2011; Luo et al. 2012; Wang et al. 2013b). Therefore, it is of great significance to arouse public attentions on the use of PAEs and develop reliable analytical methods to monitor the residual PAEs in food samples.
Many approaches have been described for determination of PAEs in different samples, including high-performance liquid chromatography (HPLC) (Liang et al. 2008; Luo et al. 2014), gas chromatography (GC) (Nanni et al. 2011; Yan et al. 2010), and gas chromatography–mass spectrometry (GC/MS) (Cinelli et al. 2014; Liu et al. 2013; Wang et al. 2013c). Because of interference from the complex matrix of food samples, the PAE analysis requires extensive sample pre-treatment, for example, solid-phase extraction (SPE) (Del Carlo et al. 2008; Ji et al. 2013; Liu et al. 2013) and liquid-liquid micro-extraction (Guo and Lee 2014; Liang et al. 2008). Generally, sample pre-treatment is performed by SPE. This technique is faster, simpler, and more economical than traditional liquid-liquid extraction, but the materials used in SPE lack selectivity (Lv et al. 2012; Wen et al. 2014). Accordingly, there is considerable attention on developing an effective extraction method prior to the measurement. Novel imprinted polymers as sorbents represent an interesting tool to meet the requirements of high selectivity and easy separation (Du et al. 2013; Hao et al. 2015).
Molecularly imprinted polymers (MIPs) are synthetic materials possessing specific recognition ability to target molecules. The imprinted materials can be produced by polymerization of functional monomers and cross-linkers in the presence of template molecules. After removal of the templates, a number of pre-determinative binding cavities are left in the polymeric network and complementary to the shape, size, and functionality of the templates (Jung et al. 2010; Wang et al. 2013a). Most MIPs for PAEs have been prepared by traditional bulk polymerization, which suffer from drawbacks in some aspects, for example, a tedious process of grinding and sieving, poor site accessibility, low binding capacity, and slow binding kinetics (Qian et al. 2011; Tang et al. 2014). To solve these problems, the surface molecular imprinting technique has been developed to control the binding sites situated at the surface of support materials, which has advantages of high selectivity, enhanced adsorption, and fast mass transfer. During the past decade, surface imprinting had adopted various support nanomaterials, including silica nanoparticles, nanotubes, nanowires, quantum dots, and magnetic nanoparticles (Anirudhan et al. 2014; Gao et al. 2014; Wei et al. 2012; Wang et al. 2013d; Huang et al. 2008). Among these supports, multi-walled carbon nanotubes (MWNTs) have gained much attention due to their unique mechanical properties and extremely huge surface area (Tunckol et al. 2013). To date, some studies have proven successful the combination between MWNTs and MIPs, with a superior performance compared to the sole MIPs. Zhang and co-workers have prepared a novel composite of MWNTs-MIPs by using melamine as template molecules (Zhang et al. 2011). Xu and Xu have reported the MWNT/MIP nanocomposite for selective recognition of ribavirin (Xu and Xu 2012). Yang et al. have developed the core-shell MIPs for specific recognition of 2,4-dichlorophenoxyacetic acid (Yang et al. 2013). To our knowledge, the MIPs of DOP prepared by surface molecular imprinting technique, especially occurring at the surface of MWNTs, have not been explored.
Herein, we prepared core-shell nanocomposites consisting of a MWNT core surrounded by a thin imprinting layer for specific recognition of DOP for the first time. The morphology, chemical structure, and thermal stability of the obtained nanomaterials were characterized. The thin imprinting shell was approximately 15 nm and could enable the template recognition sites to be accessed easily, providing high adsorption capacity and satisfactory selectivity. Meanwhile, the resultant polymers coupled with GC/MS were successfully applied for the determination of trace DOP in beverage samples.
Experiment
Chemicals and Reagents
DOP), DBP, and DEHP were purchased from Crystal Pure Industrial Co., Ltd. (Shanghai, China). MWNTs (diameter 40–60 nm, length 15–5 μm) were purchased from Nanotech Port Co., Ltd. (Shenzhen, China). Ethylene glycol dimethacrylate (EGDMA), methacrylic acid (MAA), and allyl chloride were purchased from Aladdin Company (Shanghai, China). Azobisisobutyronitrile (AIBN), sodium ethylate, acetonitrile (ACN), methanol (MeOH), acetic acid (AA), nitric acid, and sulfuric acid were provided by Chemical Reagents Co., Ltd. (Xi’an, China). All the reagents used were of analytical grade except for acetonitrile of chromatographic grade. The ultrapure water (18.0 MΩ cm−1) was obtained from a WaterPro water system (Axlwater Corporation, TY10AXLC1805-2, China) and used throughout the experiments. Beverage samples (including juice—orange juice, dairy drinks—nutri-express, and carbonated drinks—sprite) were purchased from the local market (Xi’an, China).
Instrumentation and Conditions
PAE detection was performed using gas chromatography/mass spectrometer GC/MS-QP2010 (Shimadzu, Japan) equipped with a capillary column (RXi-5 ms, 30 m × 0.25 mm × 0.25 μm) and an auto-injector (Shimadzu, AOC-20i). Helium was used as the carrier gas at a flow rate of 1.2 mL min−1, and the injection volume was 1 μL. The injection port and detector temperature were 250 and 280 °C, respectively. The GC oven was initially held at 75 °C for 2 min; then, the temperature was increased to 280 °C at a rate of 20 °C min−1 and held for 8 min. The samples were injected at the split mode with the split ratio of 10:1. The solvent delay time was 2 min. The analytes were analyzed in selective ion monitoring mode for quantitative determination. The morphologies of the resultant polymers were evaluated by transmission electron microscope (TEM; Tecnai G2). The infrared spectra of the obtained nanomaterials in KBr were recorded using a Fourier transform infrared spectrometer (FT-IR; Nicolet AVATAR 330) with a resolution of 4 cm−1 and a spectral range of 4000–400 cm−1. The thermal stabilities of the products were analyzed by thermogravimetric analysis (TGA-209C; Netzsch, Germany) under nitrogen atmosphere, over the temperature of 20–600 °C at a heating rate of 20 °C min−1.
Pre-treatment of MWNTs
Preparation of multi-walled carbon nanotubes with carboxyl groups (denoted as MWNTs-COOH) was carried out according to a previous procedure (Yola et al. 2014). Briefly, crude MWNTs (0.5 g) were dispersed into 100 mL of concentrated sulfuric acid and nitric acid (3:1, v/v) by sonication for 10 min. The mixture was stirred continuously at 85 °C for 4 h under reflux. After cooling to the room temperature, the mixture was filtered through a 0.22-μm microporous membrane and washed thoroughly with ultrapure water several times until pH of the filtrate was neutral. Finally, the filtered solid was dried under vacuum at 80 °C for 24 h.
Vinyl group functionalized MWNTs (denoted as MWNTs-CH = CH2) were synthesized according to the published method (Hua et al. 2013). The obtained MWNTs-COOH (60 mg) and sodium ethoxylate (0.25 g) were suspended in 50 mL of anhydrous tetrahydrofuran and sonicated for 30 min at room temperature. Subsequently, 5 mL of 3-allyl chloride was added into the mixture and incubated on an oscillator at 60 °C for 2 h. The product was collected and dried in a vacuum desiccator at 80 °C for 24 h for further use.
Preparation of MWNTs-MIPs and MWNTs-nanomolecularly imprinted polymers
The MWNTs-MIPs were synthesized as follows: MWNTs-CH = CH2 (200 mg) was suspended into 25 mL of acetonitrile and mixed with 0.5 mmol of DOP and 2 mmol of MAA. The mixture was incubated for 2 h with magnetic stirring at room temperature. Then, 10 mmol of EGDMA and 20 mg of AIBN were added into this mixture and deoxygenated by nitrogen gas for 5 min. The sealed reaction was allowed to continue at 60 °C for 24 h under oscillation for polymerization. After that, the resulting products were eluted with the mixture solution of methanol and acetic acid (9:1, v/v) several times by a Soxhlet apparatus to remove the template. Finally, the obtained polymers were rinsed with highly purified water to remove the remaining acetic and dried in a vacuum desiccator at 80 °C for 24 h. As a reference, nanomolecularly imprinted polymers based on the surface of multi-walled carbon nanotubes (denoted as MWNTs-NIPs) were prepared using the above procedures in the absence of the template DOP.
Static Adsorption Test
To investigate the binding ability of the prepared nanocomposites, 20 mg of MWNTs-MIPs or MWNTs-NIPs were suspended into 5 mL of DOP standard solution at various concentrations ranging from 10 to 80 μg mL−1. The mixtures were shaken for 30 min at the room temperature and separated by centrifugation. The concentrations of DOP in the supernatants were determined by GC/MS. The amount of DOP bound to the nanomaterials could be calculated according to the following formula:
where Q (mg g−1) represents the adsorption capacity, C 0 and C (μg mL−1) are the initial and equilibrium concentration of DOP, respectively, V (mL) is the volume of the solution, and M (mg) is the mass of the polymers.
The Scatchard analyis (Ma and Chen 2013) was used to evaluate the number of binding sites. The equation is listed as follows:
where C (μg mL−1) is the free DOP concentration at equilibrium, Q (mg g−1) is the amount of DOP bound to MWNTs-MIPs at equilibrium, Q max (mg g−1) is the apparent maximum adsorption capacity, and K d is dissociation constant. The values of K d and Q max can be calculated from the slope and intercept of the linear line plotted in Q/C versus Q.
Competitive Adsorption Study
The selectivity test was also performed via comparing the rebinding amount of other phthalates (DBP and DEHP) with DOP on the MWNTs-MIPs and the MWNTs-NIPs. Twenty milligram of MWNTs-MIPs or MWNTs-NIPs was added into 5 mL of mixed acetonitrile solution of DBP, DEHP, and DOP at a concentration of 70 μg mL−1. The procedures were the same as static adsorption test.
Real Sample Analysis
Juice, dairy drinks, and carbonated drinks purchased from local supermarket (Xi’an, China) were spiked with DOP at three levels (0.05, 0.1, and 0.5 ng mL−1) for analysis. Twenty milliliter of three beverage samples were put into centrifuge tubes containing 10 mL of acetonitrile and thoroughly mixed by ultrasonic vibration for 30 min, respectively. The mixtures were centrifuged at 4000 rpm for 10 min. The insoluble fractions of the mixtures were discarded, and the supernatants were filtered through a 0.22-μm filter. The filtrate was dried with a stream of nitrogen and dissolved in 20 mL of acetonitrile. Sixty milligram of MWNTs-MIPs was dispersed into 20 mL of the above treated beverage samples containing DOP, and the mixtures were incubated for 30 min on an oscillator at room temperature. After centrifugation, the MWNTs-MIPs which absorbed DOP were eluted with a mixture of methanol-acetic acid (9:1, v/v), and then, the elution was collected and evaporated to dry under a nitrogen stream. Finally, the residue of the elution was dissolved in 0.2 mL of acetonitrile and measured by GC/MS.
Result and Discussion
Preparation of MWNTs-MIPs
The synthesis of MWNTs-MIPs via a surface imprinting process is illustrated in Fig. 1. The procedure involves modification of vinyl groups on the surface of MWNTs, anchoring imprinting films on MWNTs by selective polymerization with vinyl groups and final extraction of template molecules to generate the specific recognition sites. First, the crude MWNTs were oxidized by sulfuric acid and nitric acid (3:1, v/v) to obtain carboxyl-modified MWNTs. Next, vinyl groups were functionalized on the surface of carboxyl-modified MWNTs with allyl chloride. Then, the imprinting films were coated on the vinyl groups functionalized MWNTs by molecular imprinting technique. The films were prepared in the presence of template molecule (DOP), functional monomer (MAA), cross-linking agent (EGDMA), and initiator (AIBN). Finally, after removal of the template molecule with the mixture of methanol and acetic acid (9:1, v/v), the MWNTs-MIPs were obtained.

The scheme of the synthetic route for MWNTs-MIPs
Characterization
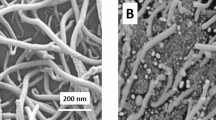
The morphologies of the MWNTs and MWNTs-MIPs were examined by TEM. As displayed in Fig. 2, the MWNTs are in the form of individual tube (Xiao et al. 2013) with the diameter of 40–60 nm (Fig. 2a). After the process of molecular imprinting, the diameter of MWNTs-MIPs increases to 70–90 nm, corresponding to a 15-nm-thick imprinting film covering the MWNTs (Fig. 2b). The thickness of this imprinted polymer layer will be effective for mass transport between the solution and the surface of MWNTs-MIPs.

TEM images of MWNTs (a) and MWNTs-MIPs (b)
In order to characterize the chemical structures of the synthetic materials, FT-IR spectra of MWNTs, MWNTs-COOH, MWNTs-CH = CH2, and MWNTs-MIPs are shown in Fig. 3. The crude MWNTs (curve a) almost have no obvious adsorption bands (Gao et al. 2011). The characteristic peaks at 3489 and 1645 cm−1 (curve b) represent the stretching vibrations of −OH and C = O, indicating that the −COOH groups have been modified onto MWNTs through oxidation. In the spectrum of MWNT-CH = CH2 (curve c), the absorption bands at 1625 and 1718 cm−1 correspond to stretching vibrations of C = C and C = O, suggesting that the vinyl groups have been grafted on the surface of MWNTs. For curve d, the typical peaks at 1728, 1226, and 1155 cm−1 come from C = O stretching vibration of carboxylic (MAA) and C-O stretching vibration of esters (EGDMA) (Jin et al. 2013), demonstrating the formation of MWNTs-MIPs.

FT-IR spectra of MWNTs (a), MWNTs-COOH (b), MWNTs-CH = CH2 (c), and MWNTs-MIPs (d)
To study the thermal stabilities of MWNTs, MWNTs-COOH, MWNTs-CH = CH2, and MWNTs-MIPs, the TGA measurements were carried out. As shown in Fig. 4, the pristine MWNTs (curve a) and MWNTs-COOH (curve b) exhibit no considerable weight loss below 600 °C. The MWNTs-CH = CH2 (curve c) have a slight weight loss of 10.9 % between 125 and 600 °C, probably due to the thermal degradation of the grafted vinyl groups and the residual unreacted oxygen-containing substances. The MWNTs-MIPs (curve d) display an obvious weight loss about 57.5 % between 340 and 420 °C; this could be because of the thermal decomposition of the imprinted polymers. The results indicate that the MIPs account for about 46.6 % of the total weight, which could further state that the imprinted layers have been successfully coated on the surface of MWNTs.

TGA curves of MWNTs (a), MWNTs-COOH (b), MWNTs-CH = CH2 (c), and MWNTs-MIPs (d)
Equilibrium Rebinding Study and Scatchard Analysis
The binding capacities of MWNTs-MIPs and MWNTs-NIPs for DOP were investigated in different initial concentrations ranging from 10 to 80 μg mL−1 (Fig. 5a). The amounts of DOP bound to MWNTs-MIPs and MWNTs-NIPs increased along with increasing the initial concentration, and the adsorption reached saturation when the initial concentration was above 70 μg mL−1. The amount of DOP bound to MWNTs-MIPs was dramatically higher than that of MWNTs-NIPs at the same initial concentration. The results demonstrated that the recognition sites on the surface of MWNTs-MIPs had better chemical and steric matching with the template DOP. On the contrary, the MWNTs-NIPs had no imprinted sites and non-specific adsorption had a dominant effect. Thus, the adsorption capacity of MWNTs-NIPs was lower.

Equilibrium adsorption of MWNTs-MIPs and MWNTs-NIPs for DOP (a) and Scatchard analysis to estimate the binding properties of MWNTs-MIPs (b)
The saturation binding data were further processed with the Scatchard equation to estimate the binding properties of MWNTs-MIPs. The Scatchard equation is
where Q is the amount of DOP bound to MWNTs-MIPs at equilibrium, Q max is the apparent maximum adsorption capacity, [DOP] is the free analytical concentration at equilibrium, and K D is the dissociation constant. The values of K D and Q max can be calculated from the slope and intercept of the linear plot of Q/[DOP] versus Q.
As presented in Fig. 5b, the Scatchard plots consisted of two distinct linear sections with different slopes and intercepts. The linear regression equations for the left and right parts of the biphasic curve were Q/[DOP] = −52.93 + 500.4Q (r = 0.9956) and Q/[DOP] = −13.20 + 363.9Q (r = 0.9924). The values of K D are 18.9 and 75.8 μg mL−1, and Q max are 9.5 and 27.5 mg g−1, respectively. The results illustrated that there were two kinds of binding sites in the imprinting materials.
Competitive Adsorption
Two other PAEs (DBP and DEHP) were selected as structural analogues to evaluate the selectivity of MWNTs-MIPs and MWNTs-NIPs. The results are shown in Fig. 6. The adsorption capacity of DOP on MWNTs-MIPs was about 1.5 and 2 times higher than that of DBP and DEHP, respectively. In contrast, the adsorption capacities were of little differences between DOP and its structural analogues on MWNTs-NIPs. This fact could be explained considering that the cavities left in MWNTs-MIPs were complementary in size and shape to the template DOP, while there were no imprinted sites in MWNTs-NIPs. These results further verified that the imprinted polymers could specifically recognize DOP from complicated matrices.

Selective adsorption of DOP, DBP, and DEHP on MWNTs-MIPs and MWNTs-NIPs
Optimization of the Eluting Conditions for Molecularly Imprinted Solid-Phase Extraction
In order to obtain high recovery of DOP, the elution step is crucial to ensure that the template molecules are completely removed. The factors affecting the elution efficiency, including solvents, volume, and time, were investigated (Fig. 7).

The effects of elution solvents (a), volume (b), and time (c) on the extraction performance of DOP
Elution Solvent
Effect of elution solvent on the extraction performance of MWNTs-MIPs was studied by comparing the elution efficiencies of ACN, ACN-AA (95:5, v/v), MeOH, MeOH-AA (95:5, v/v), and MeOH-AA (90:10, v/v). The results (Fig. 7a) indicated that MeOH-AA (90:10, v/v) had relatively higher eluting ability for DOP absorbed by MWNTs-MIPs than other elution solvents. The major reason might be that MeOH-AA (90:10, v/v) had the strongest ability to damage the interactions between DOP and MWNTs-MIPs. According to the results, MeOH-AA (90:10, v/v) was chosen as optimal elution solvent.
Elution Volume
To investigate the effect of elution volume, different volumes of elution solvent were examined. As shown in Fig. 7b, we could see that the recovery of DOP absorbed by MWNTs-MIPs exhibited an upward trend with increasing the volume of eluent and attained a pinnacle at the volume of 5 mL. Further increase in volume of eluent did not result in a significant increment in recovery but only in the waste volume. Thus, on the basis of the elution efficiency and solvent consumption, 5 mL was selected as optimal elution volume for this experiment.
Elution Time
Similarly, elution time is an important factor affecting recovery; therefore, it was investigated as well. The collected MWNTs-MIPs were eluted with MeOH-AA (90:10, v/v, 5 mL) by constant oscillation. Different time intervals (3–40 min) were tested. Figure 7c shows that 20 min was sufficient to achieve the better elution efficiency. Therefore, the optimal elution time of 20 min was used in the experiments.
Method Validation
Under optimal extraction conditions, the presented molecularly imprinted solid-phase extraction (MISPE)-GC/MS method was validated in terms of linearity, limits of detection (LODs), and limit of quantification (LOQ). The calibration curve was constructed by plotting the chromatographic peak areas versus corresponding concentrations. Good linearity (R 2 = 0.9990) was obtained in the range of 0.01–20 ng mL−1. The LOD and LOQ based on the signal-to-noise ratio of 3 and 10 were 2.3 and 7.7 ng L−1, respectively, indicating that the developed method was of satisfactory sensitivity.
Different methods for determination of DOP were summarized briefly in Table 1. As can be seen, compared with other analytical methods (He et al. 2010; Jin et al. 2013; Luo et al. 2012; Özer and Güçer 2011; Yan et al. 2012), the present method offered a relatively wide linear range and a lower LOD, suggesting that the approach developed in this work is sensitive.
Analysis in Beverage Samples
To evaluate the accuracy and practical applicability of the developed method, the beverage samples spiked with three levels (0.05, 0.1, and 0.5 ng mL−1) of DOP were analyzed and each concentration was measured three times. As shown in Table 2, the recoveries of DOP in juice, dairy drink, and carbonated drink samples ranged from 89.4 to 91.6, 88.6 to 91.0, and 90.6 to 93.0 %, respectively. The RSDs were less than 5.6 %. The results demonstrated that the MWNTs-MIPs coupled with GC/MS could satisfy the need of selective isolation and determination of trace DOP in beverage samples.
The typical chromatograms of juice (Fig. 8a), dairy drink (Fig. 8b), and carbonated drink (Fig. 8c) samples spiked with DOP at the concentration of 0.1 ng mL−1 and elution of adsorbed MWNTs-MIPs are displayed. As seen from the chromatograms in Fig. 8, the peaks of DOP could not be observed in the spiked samples (Fig. 8a-a, b-a, c-a). However, after the pre-treatment of spiked beverage samples with MWNTs-MIPs, and washing with methanol and acetic acid (9:1, v/v), the peaks of DOP appeared distinctly at the appropriate retention time (15.36 min) and other interference peaks were almost eliminated (Fig. 8a-b, b-b, c-b). The results confirmed that DOP in the spiked beverage samples could be selectively isolated by the MWNTs-MIPs and enriched by the washing step.

GC/MS chromatograms of the juice (a), dairy drink (b), and carbonated drink (c) samples spiked with DOP at the concentration of 0.1 ng mL−1 (a) and elution of absorbed MWNTs-MIPs (b)
Conclusion
In summary, we prepared a type of nanomolecularly imprinted polymers used as sorbents for the selective separation and enrichment of trace DOP in beverage samples. TEM images, FT-IR spectra, and TG analysis were used to prove that the MIPs were coated on the surface of functionalized MWNTs. The as-prepared nanomaterials possessed high adsorption capacity and selectivity for template DOP. Furthermore, the obtained imprinted polymers were successfully used as SPE adsorbents coupled with GC/MS for specific isolation and detection of DOP from beverage samples. The proposed approach showed the features of selectivity, simplicity, and sensitivity. At the same time, good recoveries and low LODs were obtained. The results demonstrated the potential application of the MWNTs-MIPs as solid-phase extractant for highly efficient and cost-effective sample analysis.
References
Anirudhan TS, Alexander S, Lilly A (2014) Surface modified multiwalled carbon nanotube based molecularly imprinted polymer for the sensing of dopamine in real samples using potentiometric method. Polymer 55:4820–4831
Aragón M, Borrull F, Marcé RM (2013) Thermal desorption-gas chromatography–mass spectrometry method to determine phthalate and organophosphate esters from air samples. J Chromatogr A 1303:76–82
Cinelli G, Avino P, Notardonato I, Centola A, Russo MV (2014) Study of XAD-2 adsorbent for the enrichment of trace levels of phthalate esters in hydroalcoholic food beverages and analysis by gas chromatography coupled with flame ionization and ion-trap mass spectrometry detectors. Food Chem 146:181–187
Del Carlo M, Pepe A, Sacchetti G, Compagnone D, Mastrocola D, Cichelli A (2008) Determination of phthalate esters in wine using solid-phase extraction and gas chromatography–mass spectrometry. Food Chem 111:771–777
Du W et al (2013) Dummy-template molecularly imprinted solid phase extraction for selective analysis of ractopamine in pork. Food Chem 139:24–30
Fu XW, Du QZ (2011) Uptake of di-(2-ethylhexyl) phthalate of vegetables from plastic film greenhouses. J Agric Food Chem 59:11585–11588
Gao RX, Su XQ, He XW, Chen LX, Zhang YK (2011) Preparation and characterisation of core-shell CNTs@MIPs nanocomposites and selective removal of estrone from water samples. Talanta 83:757–764
Gao RX, Mu XR, Hao Y, Zhang LL, Zhang JJ, Tang YH (2014) Combination of surface imprinting and immobilized template techniques for preparation of core-shell molecularly imprinted polymers based on directly amino-modified Fe3O4 nanoparticles for specific recognition of bovine hemoglobin. J Mater Chem B 2:1733–1741
Guo L, Lee HK (2014) Automated dispersive liquid-liquid microextraction-gas chromatography–mass spectrometry. Anal Chem 86:3743–3749
Hao Y, Gao R, Shi L, Liu D, Tang Y, Guo Z (2015) Water-compatible magnetic imprinted nanoparticles served as solid-phase extraction sorbents for selective determination of trace 17beta-estradiol in environmental water samples by liquid chromatography. J Chromatogr A 1396:7–16
He J, Lv RH, Zhu J, Lu K (2010) Selective solid-phase extraction of dibutyl phthalate from soybean milk using molecular imprinted polymers. Anal Chim Acta 661:215–221
Hua J, Wang ZG, Xu L, Wang X, Zhao J, Li FF (2013) Preparation polystyrene/multiwalled carbon nanotubes nanocomposites by copolymerization of styrene and styryl-functionalized multiwalled carbon nanotubes. Mater Chem Phys 137:694–698
Huang J, Wei Z, Chen J (2008) Molecular imprinted polypyrrole nanowires for chiral amino acid recognition. Sensor Actuators B-Chem 134:573–578
Ji L, Liao Q, Wu L, Lv W, Yang M, Wan L (2013) Migration of 16 phthalic acid esters from plastic drug packaging to drugs by GC/MS. Anal Methods 5:2827–2834
Jin YF, Zhang YJ, Zhang YP, Chen J, Zhou XM, Bai LY (2013) Synthesis and evaluation of molecularly imprinted polymer for the determination of the phthalate esters in the bottled beverages by HPLC. J Chem 2013:1–9
Jung BM, Kim MS, Kim WJ, Chang JY (2010) Molecularly imprinted mesoporous silica particles showing a rapid kinetic binding. Chem Commun 46:3699–3701
Liang P, Xu J, Li Q (2008) Application of dispersive liquid-liquid microextraction and high-performance liquid chromatography for the determination of three phthalate esters in water samples. Anal Chim Acta 609:53–58
Liu YP, Wang SH, Wang L (2013) Development of rapid determination of 18 phthalate esters in edible vegetable oils by gas chromatography tandem mass spectrometry. J Agric Food Chem 61:1160–1164
Luo YB, Yu QW, Yuan BF, Feng YQ (2012) Fast microextraction of phthalate acid esters from beverage, environmental water and perfume samples by magnetic multi-walled carbon nanotubes. Talanta 90:123–131
Luo X, Zhang FF, Ji SL, Yang BC, Liang XM (2014) Graphene nanoplatelets as a highly efficient solid-phase extraction sorbent for determination of phthalate esters in aqueous solution. Talanta 120:71–75
Lv YK, Wang LM, Yang L, Zhao CX, Sun HW (2012) Synthesis and application of molecularly imprinted poly(methacrylic acid)-silica hybrid composite material for selective solid-phase extraction and high-performance liquid chromatography determination of oxytetracycline residues in milk. J Chromatogr A 1227:48–53
Ma GF, Chen LG (2013) Determination of chlorpyrifos in rice based on magnetic molecularly imprinted polymers coupled with high-performance liquid chromatography. Food Anal Methods 7:377–388
Nanni N, Fiselier K, Grob K, Di Pasquale M, Fabrizi L, Aureli P, Coni E (2011) Contamination of vegetable oils marketed in Italy by phthalic acid esters. Food Control 22:209–214
Özer ET, Güçer S (2011) Determination of some phthalate acid esters in artificial saliva by gas chromatography–mass spectrometry after activated carbon enrichment. Talanta 84:362–367
Qian K, Fang GZ, Wang S (2011) A novel core-shell molecularly imprinted polymer based on metal-organic frameworks as a matrix. Chem Commun 47:10118–10120
Tang KJ, Gu XH, Luo QS, Chen SW, Wu LY, Xiong JH (2014) Preparation of molecularly imprinted polymer for use as SPE adsorbent for the simultaneous determination of five sulfonylurea herbicides by HPLC. Food Chem 150:106–112
Tunckol M, Fantini S, Malbosc F, Durand J, Serp P (2013) Effect of the synthetic strategy on the non-covalent functionalization of multi-walled carbon nanotubes with polymerized ionic liquids. Carbon 57:209–216
Wang DS, Xie DY, Shi WB, Sun SD, Zhao CS (2013a) Designing a photoresponsive molecularly imprinted system on a silicon wafer substrate surface. Langmuir 29:8311–8319
Wang J, Luo YM, Teng Y, Ma WT, Christie P, Li ZG (2013b) Soil contamination by phthalate esters in Chinese intensive vegetable production systems with different modes of use of plastic film. Environ Pollut 180:265–273
Wang SF, Yang WF, Shi MX, Sun XL, Pang WY, Wang GQ (2013c) GC/MS assisted with chemometric methods applied for investigation of migration behavior of phthalate plasticizers in fatty foods simulant. Chromatographia 76:529–534
Wang Y, Zang D, Ge S, Ge L, Yu J, Yan M (2013d) A novel microfluidic origami photoelectrochemical sensor based on CdTe quantum dots modified molecularly imprinted polymer and its highly selective detection of S-fenvalerate. Electrochim Acta 107:147–154
Wei FD, Liu XP, Zhai MJ, Cai Z, Xu GH, Yang J, Du SH, Hu Q (2012) Molecularly imprinted nanosilica solid-phase extraction for bisphenol A in fish samples. Food Anal Methods 6:415–420
Wen YY, Chen L, Li JH, Liu DY, Chen LX (2014) Recent advances in solid-phase sorbents for sample preparation prior to chromatographic analysis. TrAC Trends Anal Chem 59:26–41
Xiao DL, Dramou P, Xiong NQ, He H, Yuan DH, Dai H, Li H, He XM, Peng J, Li N (2013) Preparation of molecularly imprinted polymers on the surface of magnetic carbon nanotubes with a pseudo template for rapid simultaneous extraction of four fluoroquinolones in egg samples. Analyst 138:3287–3296
Xu L, Xu ZF (2012) Molecularly imprinted polymer based on multiwalled carbon nanotubes for ribavirin recognition. J Polym Res 19:9942–9947
Yan HY, Liu BM, Du JJ, Row KH (2010) Simultaneous determination of four phthalate esters in bottled water using ultrasound-assisted dispersive liquid-liquid microextraction followed by GC-FID detection. Analyst 135:2585–2590
Yan HY, Cheng XL, Yang GL (2012) Dummy molecularly imprinted solid-phase extraction for selective determination of five phthalate esters in plastic bottled functional beverages. J Agric Food Chem 60:5524–5531
Yang WJ, Jiao FP, Zhou L, Chen XQ, Jiang XY (2013) Molecularly imprinted polymers coated on multi-walled carbon nanotubes through a simple indirect method for the determination of 2,4-dichlorophenoxyacetic acid in environmental water. Appl Surf Sci 284:692–699
Yola ML, Eren T, Atar N (2014) Molecularly imprinted electrochemical biosensor based on Fe@Au nanoparticles involved in 2-aminoethanethiol functionalized multi-walled carbon nanotubes for sensitive determination of cefexime in human plasma. Biosens Bioelectron 60:277–285
Zhang XX, Chen Z (2014) Observing phthalate leaching from plasticized polymer films at the molecular level. Langmuir 30:4933–4944
Zhang HB, Zhang ZH, Hu YF, Yang X, Yao SZ (2011) Synthesis of a novel composite imprinted material based on multiwalled carbon nanotubes as a selective melamine absorbent. J Agric Food Chem 59:1063–1071
Acknowledgments
The authors are grateful for financial support from the National Natural Science Foundation of China (No. 21305107), the Fundamental Research Funds for the Central Universities (Nos. xjj2013041 and 08142034), and China Postdoctoral Science Foundation (No. 2014 M562388).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by the National Natural Science Foundation of China (No. 21305107), the Fundamental Research Funds for the Central Universities (Nos. xjj2013041 and 08142034), and China Postdoctoral Science Foundation (No. 2014 M562388).
Conflict of Interest
Jiaojiao Du declares that she has no conflicts of interest. Ruixia Gao declares that she has no conflicts of interest. Hui Mu declares that she has no conflicts of interest. The institutes where the authors work agree to the submission of this paper to the journal. All authors have read and approved this version of the article.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable.
Rights and permissions
About this article

Cite this article
Du, J., Gao, R. & Mu, H. A Novel Molecularly Imprinted Polymer Based on Carbon Nanotubes for Selective Determination of Dioctyl Phthalate from Beverage Samples Coupled with GC/MS. Food Anal. Methods 9, 2026–2035 (2016). https://doi.org/10.1007/s12161-015-0383-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-015-0383-4