
Abstract
A molecularly imprinted polymer (MIP) monolith has been prepared and characterized. Its application to the assay of thiamphenicol in milk with high-performance liquid chromatography–photodiodes array detector was validated. The newly developed MIP monolith was produced using an analogue to thiamphenicol as the template molecule to avoid major traditional drawback associated with MIPs of residual template bleeding. The MIP monolith synthesized in a micropipette tip could be connected with syringes in different sizes simply to perform solid-phase microextraction process without any other treatment. This molecularly imprinted polymer monolith microextraction (MIPMME) method showed high selectivity and enrichment ability for thiamphenicol (TAP). Several parameters affecting MIPMME were investigated, including the flow rate, volume, pH and salt concentration of sample, the type and volume of washing solution, and the type and flow rate of eluent. The recovery of this method for TAP was investigated and high recoveries of 93.5 ~ 96.8% from milk were obtained with relative standard deviations less than 6.3%.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
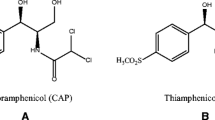
Thiamphenicol (TAP), Fig. 1, is an analogue of chloramphenicol in which the nitro group in the benzene ring is replaced with a methylsulfonic group. It has been reported that TAP showed particular therapeutic effect in respiratory infections, bacterial prostatitis, and venereal diseases. But TAP also showed hematological toxicity (Dumont et al. 2006). So, it is very important to develop a sensitive, rapid, and simple method for the determination of TAP in food commodities.

The molecular structures of TAP, CAP, EGDMA, and 4-VP
Analytical technique used in detection of TAP are mainly chromatography (Saeki 1992; Pfenning et al. 2000; Zhang et al. 2006b; Wrzesinski et al. 2003; Vue et al. 2002; Giorgi et al. 2000; Posyniak et al. 2003) and multiple methods of chromatography linked with mass spectrum (Zhang et al. 2008; Nagata and Oka 1996; Van de Riet et al. 2003; Bogusz et al. 2004; Forti et al. 2005; Peng et al. 2005). The complexity of food matrices and the presence of much potential interference require specific and selective methods for extracting and isolating analyte from food samples before detection. Solid-phase extraction (SPE) is routinely used for cleanup and preconcentration in the analysis of biological and environmental samples. Compared with liquid–liquid extraction (LLE), SPE has the advantages of simplicity, rapidity, and less consumption of organic solvents. However, generic sorbents usually lack selectivity and are easily subjected to interference by nontarget substances with similar characteristics. Although immunoaffinity chromatography is capable of differentially adsorbing target analytes, it still has some disadvantages such as lack of stability and high costs of antibody preparation. Recent research has been oriented towards the development of efficient, economical, and miniaturized sample preparation methods. As a result, solid-phase microextraction (SPME) (Bordagaray et al. 2011; Djozan et al. 2001; Mathure et al. 2011; Verzera et al. 2010, 2011) and liquid-phase microextraction (LPME) (He and Lee 1997) have been developed. Compared with LLE, SPME is a solvent-free process that includes simultaneous extraction and preconcentration of analytes from aqueous samples or the headspace of the samples. However, SPME is expensive, its fiber is fragile and has limited lifetime, and the sorbents usually lack selectivity. LPME is a solvent-minimized sample pretreatment procedure that is inexpensive, and since very little solvent is used, there is minimal exposure to toxic organic solvents. However, this method suffers from some disadvantages as follows: fast stirring would tend to format air bubble, extraction is time-consuming, and equilibrium cannot be attained after a long time in most cases (Ahmadi et al. 2006).
Due to their high selectivity, reusable, inexpensive to prepare, physiochemical stability, and applicability in harsh chemical media, molecularly imprinted polymers (MIPs) have been used as a sorbent in SPE and SPME to selectively extract analytes from complex matrices (Jiang et al. 2008; Xu et al. 2010; Hu et al. 2009; Barahona et al. 2011). Traditionally, MIPs were synthesized in bulk polymerization followed by a grinding and sieving process to acquire the desired particles in shape and size, which limited the extraction efficiency. To overcome these disadvantages, the MIP monolithic columns were prepared by in situ polymerization directly inside appropriate columns or capillaries (Liu et al. 2004, 2006). This strategy could avoid the tedious grinding and sieving procedures as well as the problems of costly particle loss, particle in homogeneity, and molecularly imprinted spots loss and could easily obtained a MIP monolith with good resolution and low back pressure at high flow rate. Polymer monolith microextraction (PMME) was a type of SPME in which the polymer monolith was used as the sorbent (Zhang et al. 2006a). The combination of MIP technology with PMME could exhibit excellent extraction selectivity in dealing with biological samples (Zheng et al. 2007, 2010). But, the MIP monolith synthesized in capillary was fragile, and tedious postpreparation was needed.
Template bleeding is considered to be the main drawback of the MIPs that can affect the analytical results, and it may generate false-positive results. Therefore, molecules that are structurally related to analytes were used as templates for polymer synthesis to avoid a major traditional drawback associated with MIPs of residual template bleeding (Schirmer and Meisel 2009). To the best of our knowledge, little attention has been paid to make use of MIP monolith and PMME for high selective extraction of TAP from complex matrices.
In this work, a MIP monolith was synthesized in a micropipette tip using chloramphenicol (CAP), an analogue of thiamphenicol, as the template, 4-vinylpyridine (4-VP) as the functional monomer, ethylene dimethacrylate (EGDMA) as the cross-linker, and the mixture of toluene–dodecanol as the porogenic solvent. This strategy could reduce the analysis cost in TAP extraction procedure for CAP was cheaper than TAP. The robust micromonolith could be connected with syringes in different sizes simply to perform the PMME process without any other treatment. The derivated MIP monolith showed high selectivity and enrichment ability for TAP. Further, an MIPMME–HPLC procedure has been employed for the determination of TAP by using the MIP monolith for the cleanup and preconcentration of TAP. The results indicated that this method could be applied for the selective and sensitive analysis of TAP in milk samples.
Experimental
Reagents, Materials, and Equipment
EGDMA purchased from Acros (New Jersey, USA) was extracted with 5% aqueous sodium hydroxide and water, then dried over using anhydrous magnesium sulfate. 2, 2′-azobisisobutyronitrile (AIBN) was obtained from Shanghai No.4 Chemical Reagent Corp. (Shanghai, China) and recrystallized in anhydrous ethanol before use. 4-VP was obtained from Acros (New Jersey, USA). Methacrylic acid (MAA), acrylic acid (AA), acrylamide (AM), toluene, and dodecanol purchased from Fuchen Chemical Reagent Company (Tianjin, China) were distilled under vacuum prior to use. TAP and CAP were purchased from Sigma (St Louis, MO, USA). Methanol and acetonitrile (HPLC grade) were obtained from Tedia Company Inc. (Ohio, USA). Sodium chloride, phosphoric acid, and other reagents used were all of analytical grade. The water used was purified on an Ultrapure Water System (Beijing, China).
The chromatographic analysis was carried out on a Dionex Summit U3000 HPLC system equipped with a manual injector and a photodiode array detector (PAD) (Dionex Technologies, USA). A personal computer equipped with a Chromeleon ChemStation program for LC was used to process chromatographic data. A amethyst-C18 column (4.6 × 250 mm, 5 μm) from Sepax Technologies Inc. (Newark, USA) was connected with a guard column (cartridge 2.1 × 12.5 mm, 5 μm, Agilent Technologies, PaloAlto, CA, USA) filled with the same packing material. The mobile phase was a mixture of methanol/water (45:55, v/v) and the flow rate was 1.0 mL/min. The column temperature was set at 25 °C by a temperature controller for column oven (Nuohai Technologies, China). The UV detector was set at a wavelength of 225 nm for analyte. All injections were performed manually with a 20.0-μL sample loop. A DZF-6021 vacuum drying oven (Yiheng Instrument Factory Co. Ltd., Shanghai, China) was used for polymerization. An LSP01-1A longer pump (Baoding Longer Precision Pump Co. Ltd., China) was used for pumping. A membrane (0.45 μm) was obtained from Xingya Scavenging Material Company (Shanghai, China). The microscopic morphology of the monolith was examined by a Model X-650 scanning electron microscope (Hitachi, Tokyo, Japan). The infrared spectrogram of CAP, 4-VP, MIP, and non-imprinted polymer (NIP) monolith was examined by a Fourier transform infrared spectrometer (Perkin Elmer, USA).
Standard Solutions and Milk Samples
The stock standard solution of TAP was prepared in methanol at a concentration of 1 mg/mL and stored at 4 °C in a refrigerator. Working standard solutions of analytes were prepared by appropriate dilution of the stock solution using purified water.
Preliminary analyses showed the milk samples purchased from the local retail market to be analyte free. Five grams of milk samples was spiked with known variable amounts of TAP. Then 5 mL of acetonitrile was added to the spiked milk samples. After being mixed with a vortex mixer (WH-3, Luxi Analysis Instrument Factory Co. Ltd., Shanghai, China), the samples were centrifuged at 7 °C for 10 min at 10,000 rpm (Xiangzhi Centrifuge Instrument Co. Ltd., Changsha, China). Then, the supernatant was completely transferred to another 50-mL volumetric flask. After evaporation of the solvent under a gentle nitrogen flow, the residue was redissolved in 20 mL purified water. Finally, the reconstituted solutions were stored at 4 °C and filtered through a 0.45-μm membrane filter prior to use. Blank samples were prepared in the same way as above but without the compound-spiking step.
Preparation of Molecularly Imprinted Monolith
For the preparation of the MIP monolith, the template molecule chloramphenicol (0.05 mmol) was dissolved in appropriate porogenic solvents (46 μL toluene, 0.2950 g dodecanol) in a clean PE tube and mixed with 4-VP (0.1 mmol) as the functional monomer. The mixture was surged ultrasonically for 4 h. Then, 1 mmol of cross-linker EGDMA and 8.5 mg of initiator AIBN were added and degassed by ultrasonication for about 10 min. Next, 50 μL of the homogeneous solution was filled into a micropipette tip which had been sealed at one end. Subsequently, the other end of the pipette tip was sealed with silicon rubber. After polymerization at 60 °C for 24 h, the silicon rubber was removed. The resultant MIP monolith was washed with methanol to remove the template molecules. A reference, NIP monolith, was prepared simultaneously as the same procedure including washing, but in the absence of the template molecule.
As shown in Fig. 2, the MIP monolith could be connected with syringes in different sizes simply without any other treatment. A syringe infusion pump (Baoding Longer Precision Pump Co. Ltd., China) was employed for the delivery of sample solution, washing solution, and desorption solvent.

Scheme of the novel MIPMME device
MIPMME Procedure
The MIP monolith was washing with 2.0 mL of methanol and 1.0 mL of water, respectively. Then, an aliquot of 4.0 mL pretreated sample solution (1.2 g NaCl was added) was loaded at a flow rate of 0.2 mL/min with the aid of an infusion pump. The MIP monolith was washed with 0.5 mL water at a flow rate of 0.2 mL/min to remove the matrix interferences. Then, the analytes were eluted with 0.1 mL of the mixture of methanol/water (55:45, v/v) at a flow rate of 0.1 mL/min. The eluent solution in the PE tube was removed using a 100 μL HPLC microsyringe and injected into the HPLC system for analysis directly. All experiments were performed repeatedly and means of results were used in plotting of curves or in tables.
Results and Discussion
In order to obtain the optimized extraction conditions, enrichment factor (EF) and extraction recovery (ER) were used to evaluate the extraction efficiency of MIP monolith under different conditions:
where C elu, n elu, and V elu are TAP concentration, number of moles in eluent, and the volume of eluent, respectively. C 0, n 0 and V aq are TAP concentration, number of moles in sample solution, and the volume of sample solution, respectively.
The imprinting factor (IF) was used to evaluate the recognition abilities of the MIP monolith:
where EFMIP and EFNIP are the enrichment factors of TAP extracted in MIP and NIP monoliths under the same conditions, respectively.
Optimization of Synthesis Conditions
To obtain higher specific recognition ability and extraction efficiency for the target analyte, the nature of functional monomer and porogenic solvent were investigated. For investigation of the synthesis conditions, 1 mL of 5 μg/mL TAP standard solution was passed through the MIP monolith at 0.2 mL/min. The analyte was eluted with 0.1 mL of methanol at a flow rate of 0.1 mL/min.
Different functional monomers will construct different binding site with template. To improve the recognition and selectivity property of MIP, four different functional monomers, including AA, MAA, AM, and 4-VP were investigated. The results showed that 4-VP has the higher specific recognition ability for TAP comparing with other functional monomers. A possible explanation for the result was that the molecular structure of TAP was similar to that of CAP. The hydrogen bonds were expected to be formed among the hydroxyl groups of TAP and the nitrogen atom of 4-VP. So, in our further work, 4-VP was chosen as the functional monomer.
The selection of the porogenic solvent is significant for the preparation of the molecularly imprinted monolith. Porogenic solvent can make all components into one phase in the polymerization process and played an important role in the morphology of the MIP in terms of specific surface area and pore size. In this study, methanol, acetonitrile, carbon tetrachloride, chloroform, toluene, dodecanol, and their mixed solutions as porogenic solvent were tested. The experimental results indicated the MIP with low polar mixture of toluene and dodecanol as the porogenic solvent displayed better extraction efficiency for TAP. Finally, the mixture of toluene and dodecanol was selected as the appropriate porogenic solvent. The experimental results also illustrated that 0.05 mmol of the template CAP in the presence of 0.1 mmol of functional monomer 4-VP and 1 mmol of cross-linker EGDMA resulted in a monolith with higher specific recognition ability and extraction efficiency for TAP.
The Characterization and Specificity Evaluation of the MIP Monolith
The MIP monolith morphological structure was investigated by a scanning electron microscope. As can be seen in Fig. 3, there were many macropores and flow through channels inlaid in the network skeleton of the MIP monolith which provided flow paths through the column. Due to the size and density of the macropore network, the monolith had a high external porosity and, consequently, a large permeability and low column hydraulic resistance. This pores allowed the mobile phase to flow through with low flow resistance.

SEM image of the MIP monolith (magnification = ×10000)
Figure 4 showed that the infrared spectrogram of MIP monolith was different from that of CAP and 4-VP. Comparing with the infrared spectrogram of 4-VP, the stretching vibration wide peaks of 3,000–3,300 cm−1 and the C=C stretch vibration peak of 1,633 cm−1 became weak in the infrared spectrogram of the associated complexes. Comparing with the infrared spectrogram of CAP, the O−H and N−H stretch vibration peaks of 3,000–3,500 cm−1 disappeared. There were the C=O stretch vibration peak of 1,729 cm−1 and the C−O stretch vibration peaks of 1,000–1,300 cm−1 (both originated from EGDMA) in the infrared spectrogram of MIP monolith. The NIP and MIP monoliths showed similar locations and appearances of the major bands. These results showed that the polymers have been successfully synthesized.

FT-IR spectra of CAP, 4-VP, MIP, and NIP
In order to evaluate the selectivity of the MIP monolith, 1 mL of 5 μg/mL TAP standard solution was passed through the MIP and NIP monoliths at 0.2 mL/min, respectively. The analyte was eluted with 0.1 mL of methanol at a flow rate of 0.1 mL/min. The eluent was analyzed by HPLC directly. The experimental results indicated that the MIP monolith had a higher affinity for TAP than NIP, where IF was 2.46.
To estimate the adsorption capacity of TAP on the MIP monolith, an adsorption experiment was carried out under optimized conditions. Five micrograms per milliliter TAP standard solutions were continuously passed through the MIP monolith at 0.05 mL/min until the peak area of TAP in the eluent was equal to that of standard solution. Then, the adsorption capacity of TAP was calculated on the base of the TAP concentrations and volumes of standard solution and eluent. The experimental results showed that the adsorption capacity of TAP on the MIP monolith (Q TAP) was 62.72 μg. These results demonstrated the good selectivity and high adsorption capacity of the synthesized MIP monolith for TAP. And, the MIP monolith could be used for cleanup and enrichment of TAP.
Based on the results above, hydrogen bonds were expected to be formed among TAP and monomers (4-VP) as a key interaction necessary for binding site construction. The hydroxyl groups of TAP acted as hydrogen bond donors. The higher hydrogen-bonding ability of the hydroxyl group in TAP and 4-VP enhanced the strength of the hydrogen bonding between TAP and monomers and thus yielded imprinted polymers with better recognition properties. The illustration of the MIP monolith and its molecular recognition was shown in Fig. 5.

The illustration of the MIP monolith and its molecular recognition
Optimization of MIPMME Conditions
Several parameters associated with the MIPMME efficiency, such as the flow rate, volume, pH and salt concentration of sample, the type and volume of washing solution, and the type and flow rate of eluent were optimized in this study. Sample solutions were spiked with TAP at 0.2 μg/mL to perform the experiments.
The flow rate of the sample solution was optimized in the range of 0.05 ~ 0.40 mL/min. The extraction efficiency decreased with the increasing of the flow rate from 0.2 to 0.5 mL/min. EF and ER increased slightly while changing the flow rate from 0.2 to 0.05 mL/min. This may be due to the plenitudinous mass transfer of the analyte from sample solution to MIP monolith at low flow rate. To achieve high extraction efficiency within a short time, 0.2 mL/min was chosen as the optimized flow rate of sample solution in the following experiments.
The washing solution was adjusted by optimizing the proportion of CH3OH in water. The experimental results indicated that EF and ER of TAP decreased obviously with increasing CH3OH content in the washing solution. And, there was no observed difference in EF and ER of TAP after washing with 0.5 and 1 mL of purified water. So, 0.5 mL of purified water was selected as the optimized washing solution.
The selection of an appropriate eluent is of high important for the PMME process. Considering the consistency to the mobile phase used in liquid chromatography, the eluent was limited to solvents such as methanol, acetonitrile, and purified water. The results indicated that methanol as eluent was better than acetonitrile and water. Then, different proportions of methanol with water as eluent were tested. The best extraction efficiency was achieved when a mixture of methanol/water (55:45, v/v) was used as the eluent. The experimental results also showed that both of the EF and ER decreased when acetic acid was added in the eluent. So, methanol/water (55:45, v/v) was selected as the desorption solvent in the following experiments.
The flow rate of the eluent was optimized in the range of 0.01 ~ 0.3 mL/min. The results showed that no significant change in the extraction efficiency was found when the flow rate of the eluent in the range of 0.01 ~ 0.1 mL/min. Then, the extraction efficiency decreased with the flow rate increasing from 0.1 ~ 0.3 mL/min. So, 0.1 mL/min was selected as the optimized flow rate of eluent in the following experiments.
The effect of sample volume was monitored by loading sample solution (containing 0.2 μg/mL of the analyte) from 2.0 to 10.0 mL at a constant flow rate. The eluent volume (methanol) was 0.1 mL. The results showed that EF of TAP increased with the increasing of sample volume from 2.0 to 10.0 mL. This indicated that the extraction capacity had not been reached even when 10.0 mL of sample solution was loaded. However, ER began to decrease when the sample volume increased. To achieve sufficient sensitivity within a short time, 4.0 mL of sample solution was selected in the PMME procedure.
The sample pH is a significant factor, which may affect the molecule form of the analyte and closely relate to the interaction between analytes and the MIP monolith. Considering that TAP is unstable when pH > 7, the effect of the sample pH on the extraction efficiency for TAP was investigated using several buffer solutions with pH 2–7. The experimental results showed that EF and ER decreased slightly when sample pH decreased from 7 to 2. This can be explained by the fact that the interaction between the analyte and the monolith was mainly based on the hydrogen binding. TAP likes to exist in positively charged form at low pH, resulting in the weakening of interaction between TAP and the polymer and thus poor extraction performance. Finally, no buffer solution was needed to adjust the sample pH in the subsequent experiments.
The effect of salt concentration of the sample on the extraction efficiency was also investigated. As can be seen from Fig. 6, EF and ER increased as the concentration of NaCl increased from 0% to 30% (w/v). Addition of salt into the sample solutions could lead to the salting-out effect, and more analyte molecules would be extracted onto the MIP monolith. To obtain high extraction efficiency, 30% NaCl (w/v) was added in the sample solution in the following experiments.

The effect of salt concentration on EF (a) and ER (b) of TAP
Evaluation of the Method
Under the optimized conditions, the method was applied for determination of TAP in milk samples. Blank milk samples were spiked at range of 0.02–20 μg/g with TAP. Then, the spiked samples were analyzed by the proposed MIPMME–HPLC method. The regression coefficient (r) was 0.9951. The limit of detection and limit of quantification, based on signal-to-noise ratios of 3 and 10, were 0.005 and 0.017 μg/g for TAP in milk, respectively.
The reproducibility of the method was determined by the within-day and between-day precisions at the concentration of 1 μg/g for TAP in spiked milk samples. The results showed that the within-day precision (RSD, n = 5) was 5.0%, while the between-day precision (RSD, n = 5) was 8.7%.
The chromatograms of spiking milk samples before and after treated by MIPMME were showed in Fig. 7. It can be seen that after treated by MIPMME, a majority of interfering substances in milk sample was eliminated, thus quantification of TAP can be successfully achieved. And, in comparison with the chromatogram of direct injection, a dramatic enrichment of the peak height was observed, which indicated the remarkable preconcentration ability of the MIP monolith for TAP.

The chromatograms of spiked milk samples (1), after treated by NIPMME (2), and after treated by MIPMME (3)
The MIP monolith showed high stability since no significant changes in the back pressure and extraction efficiency of the monolith were found in the experiment.
Real Samples Analysis
The developed MIPMME–HPLC technique was applied for the determination of TAP in milk to further elucidate the applicability and reliability of this method. Three batches of milk samples were collected from local supermarkets. The results showed that two milk samples were free of TAP residue; TAP residue in the third sample was 33.1 ng/g. To test the performance of this established method, the extraction recoveries were performed by spiking fresh milk samples with TAP standard solution. For each concentration level, three replicate experiments with the whole analysis process were made. The recoveries ranging between 93.5 ~ 96.8% were obtained (Table 1). Thus, the developed method is robust and reliable for routine analysis of TAP in complex milk sample.
Comparison of MIPMME–HPLC–UV with Other Methods
The efficiency of the presented MIPMME–HPLC–UV method for milk samples was compared with that of other reported methods. As listed in Table 2, the enrichment factors of SPE–DLLME–HPLC were obviously higher than other reported methods, the detection limits were lower, and the extraction time was relatively short. All these results revealed that the SPE–DLLME is a sensitive, simple, and reproducible technique that can be used for ultra preconcentration of carbamates from fruit and vegetable samples.
Conclusion
For the selective separation of the antibiotic thiamphenicol, a novel, durable MIP monolith was synthesized in a micropipette tip using an analogue to thiamphenicol as the template molecule to avoid major traditional drawback associated with MIPs of residual template bleeding. The monolith could be connected with syringes in different sizes simply without any other treatment to perform PMME process. The derivated MIP monolith showed high selectivity and enrichment ability for TAP. MIPMME followed by HPLC and PAD detection was developed as an analytical method for the sensitive and selective determination of TAP in milk sample. The optimum conditions of synthesis and extraction performance have been obtained. The experimental results revealed that this method provided high selectivity, lower solvent consumption, higher extraction efficiency, and good linearity over the investigated concentration range. The performance of this procedure in the analysis of TAP in milk sample was satisfactory.
References
Ahmadi F, Assadi Y, Hosseini MRM, Rezaee M (2006) J Chromatogr A 1101:307–312
Barahona F, Turiel E, Cormack PAG, Martín-Esteban A (2011) J Sep Sci 34:217–224
Bogusz MJ, Hassan H, Al-Enazi E, Ibrahim Z, Al-Tufail M (2004) J Chromatogr B 807:343–356
Bordagaray A, Garcia-Arrona R, Millán E (2011) Food Anal Methods 4:293–299
Chen XJ, Yang B, Liang N, Wang GJ (2006) J Pharm Biomed Anal 41:943–949
Djozan D, Assadi Y, Haddadi SH (2001) Anal Chem 73:4054–4058
Dumont V, Huet AC, Traynor I, Elliott C, Delahaut P (2006) Anal Chim Acta 567:179–183
Forti AF, Campana G, Simonella A, Multari M, Scortichini G (2005) Anal Chim Acta 529:257–263
Giorgi M, Romani M, Bagliacca M, Mengozzi G (2000) J Vet Pharmacol Ther 23:397–399
He Y, Lee HK (1997) Anal Chem 69:4634–4640
Hu XG, Pan JL, Hu YL, Li GK (2009) J Chromatogr A 1216:190–197
Jiang XM, Zhao CD, Jiang N, Zhang HX, Liu MC (2008) Food Chem 108:1061–1067
Kowalski P (2007) J Pharm Biomed Anal 43:222–227
Liu ZS, Xu YL, Yan C, Gao RY (2004) Anal Chim Acta 523:243–250
Liu XJ, Ouyang CB, Zhao R, Shangguan DH, Chen Y, Liu GQ (2006) Anal Chim Acta 571:235–241
Mathure SV, Wakte KV, Jawali N, Nadaf AB (2011) Food Anal Methods 4:326–333
Nagata T, Oka H (1996) J Agric Food Chem 44:1280–1284
Peng T, Li SJ, Chu XG, Cai YX, Li CG (2005) Chin J Anal Chem 33:463–466
Pfenning AP, Roybal JE, Rupp H, Turnipseed SBS, Gonzales A, Hurlbut JA (2000) J AOAC Int 83:26–30
Posyniak A, Zmudzki J, Niedzielska J (2003) Anal Chim Acta 483:307–311
Saeki N (1992) J Liq Chromatogr 5:2045–2056
Schirmer C, Meisel H (2009) Anal Bioanal Chem 394:2249–2255
Song JZ, Wu XJ, Sun ZP, Tian SJ, Wang ML, Wang RL (1997) J Chromatogr B 692:445–451
Van de Riet JM, Potter RA, Christie-Fougere M, Burns BG (2003) J AOAC Int 86:510–514
Verzera A, Condurso C, Romeo V, Tripodi G, Ziino M (2010) Food Anal Methods 3:80–84
Verzera A, Dima G, Tripodi G, Ziino M, Lanza CM, Mazzaglia A (2011) Food Anal Methods 4:141–149
Vue C, Schmidt LJ, Stehly GR, Gingerich WH (2002) J Chromatogr B 780:111–117
Wang L, Li YQ (2009) Chromatographia 70:253–258
Wrzesinski CL, Crouch LS, Endris R (2003) J AOAC Int 86:515–520
Xu ZX, Fang GZ, Wang S (2010) Food Chem 119:845–850
Zhang M, Wei F, Zhang YF, Nie J, Feng YQ (2006a) J Chromatogr A 1102:294–301
Zhang SX, Sun FY, Li JC, Cheng LL, Shen JZ (2006b) J AOAC Int 89:1437–1441
Zhang SX, Liu ZW, Guo X, Cheng LL, Wang ZH, Shen JZ (2008) J Chromatogr B 875:399–404
Zheng MM, Lin B, Feng YQ (2007) J Chromatogr A 1164:48–55
Zheng MM, Gong R, Zhao X, Feng YQ (2010) J Chromatogr A 1217:2075–2081
Acknowledgments
This work was supported by Education Department of Hubei Province (grant no. T201101), the National Nature Science Foundation of China (grant no. 20975030, 20835004), the Natural Science Foundation of Hubei province of China (grant no. 2009CDB364), and the Specialist Fund of Hubei University (020091130-ky2006004). The authors would like to thank their colleagues for their valuable technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ma, C., Chen, H., Sun, N. et al. Preparation of Molecularly Imprinted Polymer Monolith with an Analogue of Thiamphenicol and Application to Selective Solid-Phase Microextraction. Food Anal. Methods 5, 1267–1275 (2012). https://doi.org/10.1007/s12161-012-9368-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-012-9368-8