
Abstract
To obtain a highly selective material for the antibiotic chloramphenicol, which has several harmful side effects in humans, different molecularly imprinted polymers (MIPs) were prepared. In order to avoid a major traditional drawback associated with MIPs of residual template bleeding, molecules that are structurally related to chloramphenicol were used as templates for polymer synthesis. Chromatographic evaluation indicated that the employed template imparted a significant influence on the recognition properties of the corresponding polymer. A strong retention of chloramphenicol under nonpolar elution conditions (k = 68.03, IF = 17.72) and under aqueous elution conditions (k = 92.44, IF = 1.35) was achieved. After chromatographic evaluation, the MIP was utilized as the recognition sorbent in a solid-phase extraction to determine chloramphenicol using either an organic or aqueous washing solvent. Recoveries of nearly 100% from the chloramphenicol standard solution and nearly 90% from honey samples spiked with chloramphenicol were attained. Furthermore, the applicability of the MIP for sample cleanup was demonstrated.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Chloramphenicol (CAP) is a broad-spectrum antibiotic for the treatment of several infectious diseases including bacterial meningitis and typhoid fever. However, it has been shown to possess several harmful side effects in humans, such as Grey syndrome, bone marrow suppression, and fatal aplastic anemia [1, 2]. Due to these health concerns, the application of CAP in food production has been prohibited in the EU since 1994 [3]. Due to its broad spectrum of activity, ready availability, and relatively low cost, the use of CAP in veterinary medicine is still attractive in certain countries. Therefore, sensitive and reproducible detection techniques are needed to control and monitor CAP residues in food.
Several reported methods are based on gas chromatography [4, 5] or liquid chromatography [4, 6–8], with mass spectrometric and tandem mass spectrometric detection, respectively. Certain mass spectrometric detection methods involve extraction procedures, such as solid-phase extraction (SPE), for sample cleanup and preconcentration of the target analyte.
Molecularly imprinted polymers (MIPs), which imitate natural molecular recognition, are capable of meeting the demands of the SPE (for reviews, see [9–11]). The imprinting process is performed by copolymerizing functional and cross-linking monomers in the presence of a template molecule that corresponds to a certain target analyte or has a similar molecular structure. The subsequent removal of the imprint molecule reveals binding sites in the polymer network that are complementary to the template in size, shape, and position of the functional groups. This allows for the highly selective rebinding of the target analyte. Additionally, MIPs are reusable, are inexpensive to produce, exhibit high mechanical and chemical stability, and are applicable under a number of different operating conditions.
Chloramphenicol is retained on the MIPs under nonpolar elution conditions via selective hydrogen bonding [12–15]. The 1,3-diol moiety of CAP is mainly responsible for the selectivity. Nevertheless, the particular structure of CAP also contributes to the interactions since the substitution of the nitro group (in thiamphenicol) and the dichloroacetyl group (in azidamphenicol) results in a significant decrease in the affinity for the MIP. The retention of chloramphenicol under aqueous elution conditions is mainly a result of nonselective hydrophobic interactions [15, 16]. The use of MIP-SPE in combination with nonpolar washing solvents [15] or aqueous washing solvents [15–19] for the determination of CAP has been previously described in different sample matrices.
Previously [15], we described the preparation of CAP-imprinted polymers synthesized from different functional monomers. The polymer synthesized from 2-vinylpyridine was applied as a sorbent in the SPE of honey samples prior to high-performance liquid chromatography (HPLC). This polymer was able to remove matrix components from a honey sample, and it allowed for the extraction of CAP under nonpolar and aqueous elution conditions. Template bleeding is considered to be the main drawback of the MIPs that can affect the analytical results, and it may generate false-positive results. In order to further increase the utility of the MIP-SPE, we investigated several polymers that were imprinted with analog molecules of CAP.
Experimental
Chemicals
The functional monomer 2-vinylpryridine, the cross-linker ethylene glycol dimethacrylate, and the templates CAP, florfenicol (FF), and thiamphenicol (TAP) were purchased from Sigma-Aldrich Chemie GmbH (Steinheim, Germany). Azidamphenicol (AAP) was obtained from Laborchemie Apolda GmbH (Apolda, Germany). The free radical initiator α,α′-azoisobutyronitrile was provided by Fluka Chemie GmbH (Buchs SG, Switzerland). Acetonitrile, methanol, and ethyl acetate were gradient grade for liquid chromatography, and chloroform, tetrahydrofuran, trifluoroacetic acid (TFA), and sodium chloride were gradient grade for analysis. All were supplied by Merck KGaA (Darmstadt, Germany). Water was demineralized and purified by Seralpur PRO 90C, Seral (Ransbach-Baumbach, Germany).
Polymer preparation
A prepolymerization solution consisting of 0.42 mmol (0.12 g AAP, 0.15 g FF, or 0.15 g TAP) or 0.84 mmol (0.25 g AAP, 0.30 g FF, or 0.30 g TAP) template, 5 mmol (0.54 ml) 2-vinylpryridine, 25 mmol (4.72 ml) ethylene glycol dimethacrylate, 0.33 mmol (0.05 g) α,α′-azoisobutyronitrile, and 7 ml chloroform or tetrahydrofuran was prepared in a screw-capped glass vial (Table 1). The template to monomer molar ratio for the prepared MIPs was 1:12 or 1:6. The molar ratio of the functional monomer and the cross-linking monomer was 1:5. The solution was sonicated for 5 min and then purged with a stream of nitrogen for 5 min. The polymerization was thermally initiated at 60°C and maintained in a warm-water bath for 24 h. The resulting polymer was a solid block, which was ground and sieved with water. Particles with an average size below 25 µm were collected. In order to remove the template and nonreactive species residues, the polymer was transferred to a Soxhlet apparatus and refluxed over methanol for 6 h. Nonimprinted polymers (NIP) were prepared simultaneously under the same conditions, without the addition of the template.
Liquid chromatography
The HPLC system consisted of an HPLC pump P4000 and a UV6000 LP detector from Thermo Separation Products (San Jose, CA, USA). Data were recorded and processed on a Chrom Quest™ Chromatography workstation from Thermo Quest Inc. (San Jose, CA, USA).
The prepared polymers were slurry-packed into stainless steel columns 60 × 4.6 mm ID (CS-Chromatorgraphie Service GmbH, Langerwehe, Germany) using a Hitachi-Pump 655A-12 (Merck KGaA, Darmstadt, Germany). Acetonitrile/water (70:30, v/v) was used as the pushing solvent. A number of mixed mobile phases were investigated as suitable chromatography eluents, including acetonitrile/water and acetonitrile/water containing 0.1% TFA. The mixture ratios were between 100:0 and 10:90 (v/v). The analyses were performed at a flow rate of 1 ml min−1 in an isocratic mode, with a detection wavelength of 275 nm. A sample volume of 0.01 ml was injected. The retention factors k = (t r − t 0)/t 0 (t r = retention time of a given analyte; t 0 = retention time of the void volume marker acetone) and the imprinting factors IF = k MIP/k NIP (k MIP = retention factor of the MIP; k NIP = retention factor of the NIP) were calculated.
For the CAP quantification, the dried product obtained after SPE was redissolved in 0.2 ml of acetonitrile/water 30:70 (v/v), and the analysis of a 0.01-ml sample was performed on a 125 × 4.6 mm ID Superspher®100 RP-18 end-capped column (Merck KGaA, Darmstadt, Germany) at a flow rate of 1 ml min−1 and a detection wavelength of 275 nm. The recovery rates of CAP were determined by comparing the analyte peak area ratios with those of an external standard (0.01 µmol ml−1; calibration range 0.0001–0.5 µmol ml−1, correlation coefficient r = 0.9999, n = 15).
SPE
Into empty 10-ml SPE cartridges (Isolute-XL (G), Separtis GmbH, Grenzach-Wyhlen, Germany), 0.1 g of the dry polymer FF_6_C was packed between two polyethylene frits (Separtis GmbH, Grenzach-Wyhlen, Germany). Standard solutions containing 0.01 µmol CAP (3.23 µg), or each with 0.01 µmol CAP (3.23 µg), AAP (2.95 µg), FF (3.58 µg), and TAP (3.56 µg), were used. Prior to the extraction, the polymer was conditioned by thorough wetting with 2-ml washing solvent. Standard solutions (0.1 ml) or required sample volumes (1 ml) solubilized in washing solvent were applied to the cartridges, which were then washed with 5 ml of the corresponding washing solvent. Finally, elution was carried out with 3 ml of methanol at an approximate flow rate of 0.5 ml min−1. The elution fractions were collected and evaporated to dryness for further analysis.
Preparation of honey samples
The details of the sample preparation have been described previously [7]. One gram (±0.05 g) of a homogenized commercial polyflora honey was weighed into a 10-ml centrifuge tube spiked with 0.1 ml of a standard solution containing 0.01 µmol (3.23 µg) CAP, dissolved in 2 ml 4% sodium chloride solution, and combined with 5 ml ethyl acetate. This solution was vortexed for 15 min and then centrifuged at 10,000×g for 5 min. Next, the ethyl acetate was removed from the tube and evaporated to dryness. The residue was redissolved in 1 ml of a mixture of SPE washing solvent (ethyl acetate, acetonitrile, or methanol/water 5:95, v/v). This mixture was then loaded onto the SPE cartridge.
Results and discussion
Chromatographic evaluation
Several bulk polymers were synthesized in order to separate the antibiotic CAP. To avoid template bleeding, the polymers were imprinted with the molecules azidamphenicol (AAP), florfenicol (FF), or thiamphenicol (TAP), which are structurally related to CAP (Fig. 1). Polymer syntheses with analogs of chloramphenicol are mentioned in the literature [18, 19]. Unfortunately, the details for the prepolymer mixture and the template used are not given.

Molecular structure of chloramphenicol and structurally related molecules
HPLC analyses were performed to evaluate the imprinting effect of the MIPs. Previous studies identified the influence of the mobile phase composition on the retention time of CAP [15].
The template used for the synthesis of the corresponding polymers had a significant influence on the resulting recognition properties when acetonitrile was used as the mobile phase. As shown in Fig. 2, the polymers imprinted with florfenicol showed higher retention capabilities and imprinting effects towards CAP than the polymers imprinted with thiamphenicol. The retention of CAP in acetonitrile on the polymers imprinted with azidamphenicol was relatively low. The substitution of the nitro group (florfenicol, thiamphenicol) at the template affected the recognition of CAP to a lesser extent than the replacement of the dichloroacetyl group (azidamphenicol). The fluorine atom of the florfenicol obviously generated a more appropriate imprinting cavity for the recognition of CAP.

a, b Retention factors k (a) and imprinting factors IF (b) of CAP (0.1 µmol ml−1) on the polymers imprinted with chloramphenicol analogs, using acetonitrile as the mobile phase
The identity of the porogen employed in corresponding polymer syntheses showed significant effects on the recognition properties. Polymers synthesized with chloroform showed higher retention capabilities and imprinting effects towards CAP than the polymers synthesized with tetrahydrofuran. Thus, chloroform promoted the interactions between the template and the functional monomer 2-vinylpyridine.
The polymers prepared with a 1:6 template to functional monomer molar ratio showed higher retention and imprinting factors than those prepared with a 1:12 template to functional monomer molar ratio. Thus, an increased number of imprinted cavities resulted in improved recognition properties for chloramphenicol. Because of the limited solubility of the template thiamphenicol in the porogen chloroform, a polymer with a template to functional monomer molar ratio of 1:6 could not be prepared.
Under aqueous elution conditions, the MIP imprinted with florfenicol showed slightly higher retention factors than the MIPs imprinted with azidamphenicol or thiamphenicol (Fig. 3a). Slight imprinting effects with imprinting factors up to 1.35 on the florfenicol-imprinted polymers and up to 1.15 on the thiamphenicol imprinted polymers were achieved (Fig. 3b). No imprinting effect was observed on the polymers imprinted with azidamphenicol (IF below 1.05). The identity of the porogen and the molar ratio of template to functional monomer employed in the corresponding polymer syntheses showed minor effects on the recognition properties under aqueous elution conditions.

a, b Retention factors k (a) and imprinting factors IF (b) of CAP (0.1 µmol ml−1) on the polymers imprinted with chloramphenicol analogs, using acetonitrile/water (10:90, v/v) as the mobile phase
Addition of 1% water (k between 3.5 and 7.9, IF between 0.2 and 4.2) or 0.1% TFA (k between 0.4 and 0.5, IF between 1.0 and 1.2) to acetonitrile resulted in a drastic decrease in the retention and imprinting factors on all MIPs. Thus, CAP was retained on the AAP-, FF-, and TAP-imprinted polymers under nonpolar elution conditions, via CAP-selective hydrogen bonding and other nonspecific interactions such as on a CAP-imprinted polymer [15].
Using aqueous-rich mobile phases on the AAP- and TAP-imprinted polymers, the addition of 0.1% TFA had no influence on the retention behavior of CAP. Therefore, the retention of CAP under aqueous elution conditions was mainly based on additional nonselective hydrophobic interactions with the polymer matrix. However, the addition of 0.1% TFA to aqueous-rich mobile phases resulted in a 20% decrease in the retention (k between 66.7 and 73.6) and imprinting factors (IF between 1.0 and 1.1) on the FF-imprinted polymer. Thus, the higher retention of CAP on FF-imprinted polymers under aqueous elution conditions most probably resulted from a disturbance of selective hydrophobic interactions due to the protonation of the NH group.
MIP-SPE of CAP from standard solutions
On the basis of the chromatographic evaluations described above, the MIP synthesized from florfenicol and chloroform, with a template to functional monomer molar ratio of 1 to 6, was applied as a sorbent in manual SPE for the selective separation and concentration of CAP. The effects of different washing solvents on the extractions were investigated, and CAP was quantified in each eluted fraction by RP18-HPLC. Ethyl acetate and acetonitrile were investigated as SPE washing solvents because they do not interfere with the specific hydrogen bonds formed between CAP and the functional monomer residues within the imprinted cavities. Furthermore, to most effectively harness the nonselective hydrophobic interactions, different mixtures of methanol/water and acetonitrile/water were tested as SPE washing solvents.
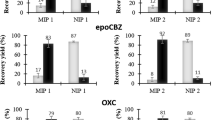
CAP recoveries of 59.2% (CAP standard solution) or 45.5% (standard solution including CAP, AAP, FF, and TAP) were achieved after washing with 5 ml acetonitrile. After washing with 5 ml ethyl acetate, CAP recoveries of approximately 100% were obtained (Table 2). The recoveries were reproducible with standard deviations below 0.7. We propose that ethyl acetate facilitates the formation of hydrogen bonds within the imprinting cavities more effectively than acetonitrile because it is less polar. The addition of 0.1% methanol or 0.1% water to ethyl acetate resulted in a decrease of nearly 5% or 25%, respectively. When the addition of methanol or water amounted to 1% or 5%, CAP recoveries below 10% were obtained, and 0.1% TFA in ethyl acetate resulted in a complete loss of CAP retention during the washing step. Thus, the addition of minimal amounts of a protic solvent or an acid resulted in a drastic loss of CAP recovery during SPE.
The elution strength of the aqueous washing solvent increased with the increasing content of methanol or acetonitrile due to a disruptive effect on the nonselective hydrophobic interactions (Fig. 4). With up to 20% methanol or 5% acetonitrile, CAP was strongly retained on the MIP, and recoveries were generally above 90%. Methanol disrupted the hydrophobic interactions to a lesser extent than acetonitrile. The addition of 0.1% TFA to the washing solvent resulted in a slight decrease in the recovery. The existence of CAP analogs in the sample insignificantly affected the retention of CAP during the washing step.

MIP-SPE of CAP from 0.1-ml standard solution (solid lines = 0.01 µmol CAP ml−1, dashed lines = each with 0.01 µmol CAP, AAP, FF, TAP per milliliter) using various organic solvents (A) in mixtures with water (B) as washing solutions, n = 3
MIP-SPE of CAP from honey matrix
The effects of the matrix on the retention of CAP were investigated by MIP-SPE of honey samples spiked with CAP. The extractions of the honey samples were performed under the same conditions as the extractions of the standard solutions. After washing of the honey sample with 5 ml ethyl acetate, a CAP recovery of 87.6 ± 2.0 (n = 3) was obtained. Similarly, with an aqueous solvent containing up to 20%, methanol recoveries of approximately 90% were achieved (Table 3). With the continuing increase of the methanol content above 20%, the recovery of CAP decreased in the elution fractions and increased in the washing fractions. As expected, the addition of 0.1% TFA to the aqueous washing solvent resulted in a slight decrease in the recovery of CAP. Obviously, the honey matrix marginally interfered with the recognition of CAP and consequently caused only a slight decrease in the recovery of CAP compared to the SPE of CAP from standard solutions. Thus, separation of the target analyte CAP could be performed under nonpolar as well as polar elution conditions.
The chromatograms obtained by HPLC after MIP-SPE of a honey sample spiked with CAP are shown in Fig. 5 (a–d). A baseline separation of the CAP peak from the honey matrix could be achieved when ethyl acetate was used as the washing solvent for the sample cleanup with MIP-SPE (Fig. 5, a). The water-based washing step revealed an insufficient cleanup of the sample because of the unspecific retention of the matrix components (Fig. 5, b–d). Consequently, less honey matrix was found in the aqueous washing fractions compared to the washing fraction of ethyl acetate (chromatograms of the washing fractions are not shown). With increasing methanol content in the aqueous washing solvent, a decreased retention of the honey matrix during the washing step was observed (Fig. 5, b and c). The addition of 0.1% TFA to the aqueous washing solvent had no influence on the retention of the matrix from the target analyte (Fig. 5, c and d). The improved CAP separation from the honey matrix under nonpolar conditions further highlights the selective interactions of CAP with the MIP imprinted with CAP analogs.

a–d Chromatograms (RP18-HPLC) obtained by MIP-SPE of 1-g honey spiked with 0.01 µmol CAP using different washing solvents: (a) washing with 5 ml ethyl acetate; (b) washing with 5 ml methanol/water 5:95 (v/v); (c) washing with 5 ml methanol/water 20:80 (v/v); (d) washing with 5 ml methanol/water + 0.1% TFA 20:80 (v/v); conditions for MIP-SPE and HPLC: see legend of Table 3
Conclusion
For the selective separation of the antibiotic chloramphenicol, we have described the synthesis and subsequent application of bulk polymers imprinted with molecules that are structurally related to chloramphenicol. The results demonstrate that chloramphenicol was strongly retained from the analog-imprinted polymers. Moreover, the polymer imprinted with florfenicol (FF_6_C) showed higher retention capabilities and imprinting effects towards chloramphenicol (nonpolar elution: k = 68.0 and IF = 17.7, aqueous elution: k = 92.4 and IF = 1.4) than the polymer imprinted with chloramphenicol (nonpolar elution: k = 58.7 and IF = 15.3, aqueous elution: k = 46.9 and IF = 1.1) [15] because a higher template amount of florfenicol than of chloramphenicol could be used for the polymer synthesis due to the limited solubility of chloramphenicol in the porogen. Further analyses have described the applicability of the florfenicol-imprinted polymer as a sorbent in an SPE of chloramphenicol from standard solutions and honey samples. A careful choice of the solvent composites was necessary for an efficient cleanup of CAP prior to the quantitative analysis. The cleanup of honey samples could be performed prior to the quantitative analysis under aqueous and in particular under nonpolar elution conditions. Template bleeding cannot cause false-positive results during the quantification of chloramphenicol because the templates used for polymer synthesis were molecules that are structurally related to the target analyte chloramphenicol. The efficiency and selectivity particularly under nonpolar elution conditions of the MIP-SPE enable the simple and efficient monitoring of the harmful antibiotic chloramphenicol.
References
Yunis AA (1988) Ann Rev 28:83–100
Holt D, Harvey D, Hurley R (1993) Adverse Drug React Toxicol Rev 12:83–95
EEC (1994) Council regulation (EEC) no. 2377/90. Amending regulation no. 1430/94 of 22 June 1994. Off J Eur Commun L156 23 (6)
Impens S, Reybroeck W, Vercammen J, Courtheyn D, Ooghe S, DeWash K, Smedts W, DeBrabander H (2003) Anal Chim Acta 483:153–163
Xie MX, Liu Y, Qiu YM, Han J, Liu YZ (2005) Chin J Anal Chem 33:1–4
Ortelli D, Edder P, Corvi C (2004) Chromatographia 59:61–64
Grotewahl D (2006) Entwicklung von Methoden zur Bestimmung von Chloramphenicol in Bienenprodukten mittels LC–MS. Cuvillier, Göttingen
Nicolich RS, Werneck-Barroso E, Marques MAS (2006) Anal Chim Acta 565:97–102
Sellergren B (2001) J Chromatogr A 906:227–252
Ye L, Mosbach K (2001) J Incl Phenom Macrocycl Chem 41:107–113
Cormack PAG, Elorza AZ (2004) J Chromatogr B 804:173–182
Levi R, McNiven S, Piletsky SA, Cheong SH, Yano K, Karube I (1997) Anal Chem 69:2017–2021
McNiven S, Kato M, Levi R, Yano K, Karube I (1998) Anal Chim Acta 365:69–74
Suárez-Rodríguez JL, Díaz-García ME (2001) Biosens Bioelectron 16:955–961
Schirmer C, Meisel H (2008) Anal Bioanal Chem 392:223–229
Schirmer C, Meisel H (2006) J Chromatogr A 1132:325–328
Mena ML, Agüí L, Martines-Ruiz P, Yáñez-Sedeño P, Reviejo AJ, Pingarrón JM (2003) Anal Bioanal Chem 376:18–25
Boyd B, Björk H, Billing J, Shimelis O, Axelsson S, Leonora M, Yilmaz E (2007) J Chromatogr A 1174:63–71
Mohamed R, Richoz-Payot J, Gremaud E, Mottier P, Yilmaz E, Tabet JC, Guy PA (2007) Anal Chem 79:9557–9565
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Schirmer, C., Meisel, H. Chromatographic evaluation of polymers imprinted with analogs of chloramphenicol and application to selective solid-phase extraction. Anal Bioanal Chem 394, 2249–2255 (2009). https://doi.org/10.1007/s00216-009-2898-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-009-2898-2