
Abstract
S-adenosyl-L-homocysteine hydrolase of Plasmodium falciparum (PfSAHH) is a potential drug target against malaria, and selective inhibition of PfSAHH is the excellent strategy to prevent the growth of parasite inside the host. Therefore, a comparative analysis of human S-adenosyl-L-homocysteine hydrolase (HsSAHH) and PfSAHH has been performed to explore the structural differences. Structural superimposition of PfSAHH and HsSAHH has generated the RMSD of 0.749 Å over 394 alpha carbon pairs. Residues of PfSAHH from position Tyr152 to Lys193 aligned with insertion/deletion region in HsSAHH, and these extra residues results in an extent of variation in cavity region of PfSAHH. Nicotinamide adenine dinucleotide (NAD) was observed to form hydrogen bonding with Thr201, Thr202, Thr203, Asn235, Val268, Glu287, Asn322, Ile343, Asn391, Lys473, and Tyr477 and also forms hydrophobic interactions with Val268, Ile288, and Thr320 of PfSAHH. In comparison to HsSAHH, Asn322, Lys473, and Tyr477 residues of PfSAHH are unique in interaction with NAD. 2-Fluoroaristeromycin and other analogues of aristeromycin have shown the good binding affinity for both enzymes. Structural differences between PfSAHH and HsSAHH might be employed to design the potential inhibitor of PfSAHH. To find the target enzyme responsible for an anti-malarial effect, molecular docking and interaction analysis of curcumin were performed with 34 drug targets of P. falciparum. Curcumin shows high affinity for binding with HGPRT of PfHGPRT, and an anti-malarial effect of curcumin might be due to binding with PfHGPRT.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
S-adenosyl-L-homocysteine hydrolase (SAHH) is an enzyme that catalyzes the hydrolysis of S-adenosyl-L-homocysteine (SAH) to adenosine (ADN) and homocysteine in vivo. SAHH play a very important role in the regulation of biological methylation [1]. SAH is a potent feedback inhibitor of S-adenosyl-L-methionine (SAM)-dependent biological methylation. SAM is a co-factor that helps in methylation of proteins, lipids, nucleic acids, and small molecules. The eukaryotic mRNA must possess a methylated 5′-cap structure for stability against enzymes phosphatases and ribonucleases for proper binding to the ribosome and for the promotion of splicing. Therefore, inhibition of SAHH prevents normal protein expression by synthesizing an uncapped mRNA [2]. SAHH has been reported as a therapeutic target to cure malaria; cancer; and inflammatory, viral, and cardiovascular diseases. Inhibitor of SAHH prevents the methylation process of nucleic acids, proteins, and other small molecules. Conventional drugs are not very much effective against malaria due to the emergence of new strains of Plasmodium falciparum. PfSAHH is homo-tetramer of identical subunits, each of which comprises 479 amino acid residues, and each subunit is consists of two large domains separated by a cleft-containing deep pocket [3]. Structural comparison of PfSAHH with that of HsSAHH reveals that the selectivity of inhibitors depends on the difference of single residue in the binding region, Cys59 in PfSAHH vs. Thr60 in HsSAHH [4]. HsSAHH is tetrameric protein, and each subunit is comprised of 432 amino acid residues. Active site residues Glu156, Asp190, Asn181, Lys186, and Asn191of HsSAHH have been mutated by alanine (Ala). It was observed that Asn181 and Asn191 play a role in localizing the flexible side chain of Lys186 in a catalytically effective position, and residue Glu156 also maintain the Lys186 in the desired protonation state [5].
Noraristeromycin analogues were synthesized, and their inhibitory activity against HsSAHH and PfSAHH was investigated. Among them, 3′,4′-anhydronoraristeromycin has shown inhibitory activity against HsSAHH [6]. 2-Modified aristeromycin derivatives and its analogues were also synthesized to study their inhibitory role against HsSAHH and PfSAHH. 2-Fluoroaristeromycin has shown a strong inhibitory activity against PfSAHH and has shown complete resistance to adenosine deaminase. 2-Fluoronoraristeromycin also possesses significant inhibitory activity against PfSAHH [7]. In vitro screens of nucleoside analogues results in moderate but selective inhibition of recombinant PfSAHH, by 2-position substituted adenosine analogues [8]. Similar selectivity, in the growth inhibition assay of cultured cells, has been reported. Among the analogues of 3-deaza-adenosine (DZA) including neplanocin A, 3-deaza-(±)-aristeromycin (DZAri) is the most potent inhibitor of the PfSAHH and inhibits the in vitro growth of P. falciparum parasites [9]. A comprehensive phylogenetic study performed by Bujnicki et al., 2003 infers that SAHH evolved in the common ancestor of Archaea and Eukaryota and subsequently horizontally transferred to Bacteria.
Curcumin is a yellow-colored pigment obtained from the rhizome of turmeric. It has a wide range of therapeutic activities and a plethora of drug targets. Curcumin is a potential candidate for the development of adjunctive therapy for cerebral malaria [10]. Curcumin and its derivatives have been found to have a higher binding affinity with PfSAHH than noraristeromycin. Curcumin binds with Leu53, His54, Thr56, Lys230, Gly397, His398, and Phe407 residues of PfSAHH, and these residues also observed in interaction with noraristeromycin. Curcumin and noraristeromycin utilize the same site of PfSAHH for binding [11, 12]. P. falciparum is responsible for millions of deaths each year. This work is aimed to develop potential inhibitors of PfSAHH against malaria. Structural differences between HsSAHH and PfSAHH offer opportunities for chemotherapy of malaria.
Material and methods
Databases
The available X-ray structures (PDB file) of SAHH for Homo sapiens (1LI4), P. falciparum (1V8B), Mycobacterium tuberculosis (2ZJO), Burkholderia pseudomallei (3GLQ), Trypanosoma brucei (3H9U), and Bradyrhizobiu elkanii (4LVC) were retrieved from RCSB protein database (http://www.rcsb.org). The PubChem (https://pubchem.ncbi.nlm.nih.gov) and ChEMBL (https://www.ebi.ac.uk/chembl) databases were used for retrieving 2D/3D structural properties and bioassay of inhibitors. IC50 of six inhibitors for PfSAHH and HsSAHH was retrieved from ChEMBL database (Table 1). Amino acid sequence of uracil-DNA glycosylase (gi||23497213|gb|AAN36760.1) was retrieved from NCBI database (http://www.ncbi.nlm.nih.gov).
Structural superimposition
MatchMaker module of UCSF Chimera 1.9 was used to create a superposition of PDB structures [14]. It superimposes structures pair-wise by first aligning their sequences and then fitting the alpha carbons of residues in the same columns of the sequence alignment. This program has the facility of interactive visualization and analysis of molecular structures, sequence alignments, docking results, trajectories, and conformational ensembles (http://www.cgl.ucsf.edu/chimera). It is often used to superimpose related structures for structural comparison and analysis [14]. In general, more closely related proteins are easier to superimpose. The six SAHH structure from human (1LI4), P. falciparum (1V8B), M. tuberculosis (2ZJO), B. pseudomallei (3GLQ), T. brucei (3H9U), and B. elkanii (4LVC) were superimposed to find the structural differences in terms of RMSD.
Cavity, binding site analysis
Molegro Molecular Visualizer (MMV) was used for analyzing the cavities presents in PfSAHH and HsSAHH enzyme. This tool was also used to visualize the hydrogen bonding, an electrostatic and steric interaction of coenzyme NAD with PfSAHH and HsSAHH. Top three cavities of PfSAHH and HsSAHH were predicted to compare the size and volume of cavities in both the targets. Cavity associated with the binding of NAD and its spatial position also mapped. Interaction of NAD with HsSAHH and PfSAHH was explored using PoseView tool [15].
Modeling
The 3D structure of uracil-DNA glycosylase of P. falciparum 3D7 was modeled using SWISS-MODEL tool (ProMod Version 3.70) [16]. The primary amino acid sequence, for which templates searched and models built, is given in FASTA format.
>gi|23497213|gb|AAN36760.1| uracil-DNA glycosylase, putative [Plasmodium falciparum 3D7]
MNNPTIQKTIDQFFKVKRKSSILSGEIEKKRKKVILEEVEEKSLEGSLKEENVNILKTKKLMNNDEDIEKMGTISNISMSTSTIDNEINNNVKQNVCEQGYMEEIKKLMHIEWYELLKDELKKNYFKNMYLKIKEERKTKVIYPPEQLVFNAFLKTPLSNIKVVIVGQDPYHQKDQAMGLCFSVPIGVKIPPSLKNILKEMKQKSNHGNLISWSEQGVFLLNTSLTVEENKPASHKNYGWETFTDTVINIINRQKEKIIFMLWGNFAIKKCKNIDINKHFILKAGHPSPLSIKHFENCNHFAKCNKILAQHNLTPIKWELPQ
Template search with BLAST and HHBlits has been performed against the SWISS-MODEL template library for evolutionary-related structures matching the target. The quality of template predicted on the basis of features obtained from target-template alignment and a high-quality template selected for model generation. Models were built based on the target-template alignment using Promod. Coordinates for the conserved regions of the template assigned from the template to the target. Insertions and deletions were remodeled using a fragment library. Side chains are built using rotamer libraries extracted from high resolution X-ray structures. Finally, the geometry of the resulting model was regularized by using a force field.
Docking
Molegro Virtual Docker (MVD) 2007.2.0.0 was used for docking study. MVD requires a 3D structure of protein and ligand for docking. MVD performs flexible docking, and the optimal geometry of ligand determined during the docking of the ligand with protein [17]. The candidates with the best conformational and energetic results were selected. MVD calculates the interaction energies between ligand and protein from the 3D structure of protein and ligand. The MolDock Score, an adaptation of the differential algorithm, was used for energy calculation.
admetSAR
The admetSAR is a most comprehensive, curated resource for diverse chemicals associated with known absorption, distribution, metabolism, excretion, and toxicity profiles [18]. admetSAR provides a interface to search for ADMET by name, CASRN and similarity search. admetSAR will be helpful for in silico screening ADMET profiles of drug candidates and environmental chemicals. admetSAR is useful in optimizing ADMET profiles of hits or leads in drug discovery and also make a smart decision during drug discovery process.
Results and discussion
Structural variations of SAHH in Plasmodium, human, and other pathogenic organism
The motivation behind structural superimposition was to find out the structural difference of HsSAHH with SAHH of other pathogens. Structural superimposition has explored the structural similarity and differences between PfSAHH and HsSAHH (Fig. 1). Sequence composition, similarity statistics, and superimposition results indicate that HsSAHH is structurally dissimilar to PfSAHH. SAHH of M. tuberculosis (2ZJO), B. pseudomallei (3GLQ), T. brucei (3H9U), and B. melkanii (4LVC) has shown high RMSD with PfSAHH than HsSAHH (Table 2). T.brucei is a salivary trypanosome which causes African trypanosomiasis (sleeping sickness). In comparison to SAHH of other pathogens, HsSAHH has shown good structural similarity with the SAHH of T. brucei. A good alignment score of 1772.9 between HsSAHH and SAHH from T. brucei was obtained with a low RMSD of 0.523 Å over 412 alpha carbon pairs. It indicates that SAHH of T.brucei could not be used, as a potential drug target for the cure of African trypanosomiasis. Root-mean-square distance (RMSD) is calculated using one point per residue CA in amino acids. RMSD represents the spatial variation between at least two associated structure residues. M. tuberculosis is pathogenic bacteria and causes tuberculosis. Structural comparison of HsSAHH with SAHH of M. tuberculosis has also resulted in a good alignment score (1575.3) and low RMSD value (0.599 Å) over 401 alpha carbon pairs. Structural differences between HsSAHH and PfSAHH are sufficient to cause the selective inhibition of Plasmodium.

a Structural superimposition of HsSAHH with SAHH of other organism. b Structural superimposition of PfSAHH with SAHH of other organism, SAHH of human is shown by 1LI4 (golden), 1V8B (light black), 2ZJO (blue), 3GLQ (pink), 3H9U (green), and 4LVC (violet-red)
To investigate the structural differences, PfSAHH compared with SAHH from M. tuberculosis, B. pseudomallei, T. brucei, and B. elkanii. B. pseudomallei is a Gram-negative, rod-shaped, aerobic bacterium. It causes melioidosis disease by infecting humans and animals. B. pseudomallei is also capable of infecting plants. B. elkanii is a species of legume-root nodulating, microsymbiotic nitrogen-fixing bacterium. If a good structural similarity exists between drug targets, then a single inhibitor can be designed to cure diseases caused by the different pathogen. 5′-Noraristeromycin and its analogues have shown a wide range of anti-viral effects [19, 20]. The anti-trypanosomal activity of 7-deaza-5′-noraristeromycin reported with IC50 values ranging from 0.16 to 5.30 μM against four strains of African trypanosomes [21]. The 3-deaza-(±)-aristeromycin was reported to induce differentiation and inhibit proliferation of HL-60 cells [22]. These studies validate that noraristeromycin and its analogues have been tested and recommended for treatment of diseases other than malaria. A sequence alignment score of 1559.2, between PfSAHH and SAHH of M. tuberculosis, was found which is higher than the alignment score obtained for B. pseudomallei, T. brucei, and B. elkanii. Superimposition of PfSAHH with SAHH of M. tuberculosis generates the RMSD of 0.601 Å over 437 alpha carbon pairs. Sequence similarity and structural alignment data indicate few structural differences between PfSAHH and SAHH of M. tuberculosis. Therefore, keeping in mind these structural differences, a single potential inhibitor can be designed to cure the disease caused by both pathogens.
An alignment score of 1405.4 was obtained between HsSAHH and PfSAHH. These two structures have generated the RMSD of 0.749 Å over 394 alpha carbon pairs. Structurally aligned residues of HsSAHH and PfSAHH have been represented by red boxes and labels that correspond to these locations are highlighted in the superimposition diagram by green color (Fig. 2). Sequence variations observed between PfSAHH and HsSAHH due to deletion of a long region in HsSAHH. The region that corresponds to deletion/insertion has constituted a helix in PfSAHH and results in a structural difference in terms of RMSD. Amino acid residues of PfSAHH from Tyr152 to Lys193 are aligned with gap region in HsSAHH. The extent of sequence variations observed between two proteins is related to the extent of structural differences. Residues that correspond to region of variation are present in PfSAHH and absent in HsSAHH. These positions are related to the point of insertion/deletion which occurred during evolution in PfSAHH and HsSAHH. These regions might be useful in designing inhibitors that selectively target the positions of variation in PfSAHH.

a Sequence alignment of PfSAHH with HsSAHH showing highly variable region by dotted lines and matched region by red boxes (UCSF Chimera 1.9). Residues from Tyr152 to Lys193 of PfSAHH that are present in PfSAHH and absent in HsSAHH might be selectively used for designing inhibitors specific to PfSAHH. b Structural alignment of HsSAHH (blue) with PfSAHH (dark red) generates a RMSD of 0.749 (Å3) over 394 atoms, which is very high as compared to RMSD for alignment of HsSAHH with SAHH of other organisms. Structurally aligned regions are highlighted by green color
Cavity, energy map, and NAD interaction analysis
A comparative account of cavities present in HsSAHH and PfSAHH has been performed to illustrate the 3D structural differences. The difference in the area and volume of cavities also provides a clue about structural differences between PfSAHH and HsSAHH. In addition, the specificity of inhibitors depends on the amino acid composition of the binding site. The volume of top three cavities present in HsSAHH was 402.944, 101.376, and 28.672 (Å3). Cavity with the largest volume of 402.944 Å3 found associated with binding of NAD. Similarly, the largest cavity (263.168 Å3) in PfSAHH was also occupied by NAD. A remarkable difference observed in the volume of largest cavities for pfSAHH and HsSAHH. In PfSAHH, two other cavities of volume 226.304 and 130.560 Å3 were also measured. Because of sequence variation around the largest cavity in both the enzyme, structural differences in shape and size of the cavity has been observed. Energy map represents the distribution of positive and negative electrostatic field, hydrogen bond donor and acceptor, and steric field around the binding pocket of enzymes HsSAHH and PfSAHH (Fig. 3). Structures of PfSAHH and HsSAHH compared with the aim of illustrating the differences in the distribution of energy map. The binding site of HsSAHH is mainly surrounded by positively charged, electrostatic residues, and a small region occupied by hydrogen donor favorable, hydrogen acceptor favorable, and steric favorable residues. In PfSAHH, the binding site is occupied by a more prominent steric favorable region and negative electrostatic region. In comparison to the binding site of HsSAHH, the binding site of PfSAHH is surrounded by more steric favorable residues. The binding cavity of PfSAHH differs from HsSAHH in volume, cavity composition, and energy map distribution. These findings support the designing of potential inhibitor for PfSAHH.

a Energy map for NAD in the largest cavity (light green mesh) of HsSAHH. b Energy map for NAD in the largest cavity (light green mesh) of PfSAHH. Energy map was generated by using MVD tool. Coloring scheme used for this map: positive electrostatic interaction (red), negative electrostatic interaction (blue), hydrogen donor favorable (yellow), hydrogen acceptor favorable (blue-green), and steric favorable (dark green)
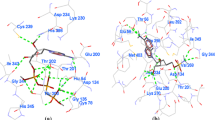
A comparative interaction map of NAD with HsSAHH and PfSAHH has been generated and represents the residues involved in hydrogen-bonding and hydrophobic contacts (Fig. 4). Co-factor NAD forms hydrogen bonding with amino acid residues Thr157, Thr158, Thr159, Asn191, Asp223, Val224, Glu243, Ile299, His301, and Asn346 of HsSAHH. Hydrophobic interactions of NAD detected with Asn191, Val224, Ile244, and Thr276 residues of HsSAHH. NAD shows hydrogen bonding with Thr201, Thr202, Thr203, Asn235, Val268, Glu287, Asn322, Ile343, Asn391, Lys473, and Tyr477 of PfSAHH. Val268, Ile288, and Thr320 of PfSAHH involved in hydrophobic interaction with NAD. It is necessary to emphasize that the same set of residues involved in hydrogen bonding with both the target enzymes. A similar set of residues Asn191, Val224, Ile244, and Thr276 of HsSAHH and Val268, Ile288, and Thr320 of PfSAHH were observed in hydrophobic interaction with NAD. It is interesting to note that three additional and distinct residues Asn322, Lys473, and Tyr477 of PfSAHH involved in hydrogen-bonding interaction with NAD. In comparison to HsSAHH, Asn322, Lys473, and Tyr477 residues of PfSAHH are unique in interaction with NAD, and these residues can be targeted for designing of selective inhibitor. Potential analogue of NAD can also be used as an inhibitor of PfSAHH.

a Interaction of NAD with HsSAHH. b Interaction of NAD with PfSAHH. Residues involved in hydrogen-bonding and hydrophobic interactions with NAD are labeled with their position and chain. Black dashed lines—hydrogen bonds; green solid lines—hydrophobic interactions. Covalent bonds to protein are not displayed. Interaction of NAD with HsSAHH and PfSAHH was visualized using PoseView [15]. This tool display hydrogen bonding without distances and hydrophobic contacts
Binding and interaction analysis of known inhibitors with HsSAHH and PfSAHH
P. falciparum completes its asexual life cycle in human. The presence of SAHH enzyme in human and Plasmodium is a challenge for designing the selective inhibitor of PfSAHH. Therefore, the comparative binding study of known inhibitors (Supplementary Fig. 1) was carried out for both drug targets. PDB file of HsSAHH (1LI4) and PfSAHH (1V8B) was retrieved from PDB database. Chain A of each enzyme was used for docking purpose by removing the already bound ligand and heteroatoms from PDB. In docking study, cavity with the largest volume was specified for docking. Six known inhibitors of PfSAHH were docked into binding site of PfSAHH and HsSAHH, and their binding affinity toward these targets was calculated. Docking results indicate that all inhibitors were docked into the same binding site of HsSAHH; a similar binding mode and a nearly similar set of interacting residues were determined. Thr57, Glu59, Asp131, Glu156, and Asp190 of HsSAHH are the most common residues that were seen in interaction with all inhibitors (Table 3). Inhibitor CHEMBL261619 forms a stable, low energy complex (−153.55 Kcal/mol) with HsSAHH and has shown interaction with Leu54, His55, Thr57, Glu59, Thr60, Asp131, Glu156, Thr157, Lys186, Asp190, His301, Met351, Gly352, His353, and Met358. Thr157 and His353 of HsSAHH which has been observed in binding with NAD were also seen in interaction with inhibitors. It is clear that inhibitors prefer their binding near NAD binding region in HsSAHH, and they may compete with the NAD for its binding with HsSAHH.
CHEMBL261619 and other inhibitors have shown a higher affinity for its binding with HsSAHH than PfSAHH. In silico binding affinity is not in good agreement with earlier reported IC50. These inhibitors have shown energetically favorable and good hydrogen-bonding interaction with PfSAHH. CHEMBL261619 forms a stable, low energy complex (−145.31 Kcal/mol) with PfSAHH and have shown binding interaction with Leu53, His54, Thr56, Glu58, Asp134, Glu200, Thr201, Lys230, Asp234, Thr396, Gly397, His398, Met403, and Phe407 (Table 4). Residues of PfSAHH that were common in interaction with all inhibitors are Thr56, Glu58, Asp134, Glu200, Thr201, Asp234, Thr396, and Gly397. Inhibitors bind near NAD binding region of PfSAHH and two common residues, Thr201 and His398, of PfSAHH observed in interaction with both NAD and inhibitors. Therefore, it can be assumed that aristeromycin and its analogues generate an anti-malarial effect by replacing the NAD for its binding with PfSAHH.
2-Fluoroaristeromycin forms a favorable binding energy complex (−144.22 Kcal/mol) with HsSAHH, and it interacts with Leu54, His55, Thr57, Glu59, Thr60, Asp131, Glu156, Thr157, Lys186, Asp190, Met351, Gly352, His353, Met358, and Phe362 residues of HsSAHH. 2-Fluoroaristeromycin forms 12 hydrogen bonds (−26.48 Kcal/mol) with Thr57, Glu59, Asp131, Glu156, Thr157, Lys186, Asp190, Met351, and His353 of HsSAHH (Fig. 5a). 2-Fluoroaristeromycin has shown highly favorable docking into the binding site of PfSAHH. 2-Fluoroaristeromycin show a favorable binding energy (−145.29 Kcal/mol) with PfSAHH and it interacts with Leu53, His54, Thr56, Glu58, Asp134, Glu200, Thr201, Lys230, Asp234, Thr396, Gly397, Met403, and Phe407. 2-Fluoroaristeromycin forms 12 hydrogen bonds (−25.81 Kcal/mol) with Thr56, Glu58, Asp134, Glu200, Thr201, Lys230, Asp234, Thr396, and Gly397 of PfSAHH (Fig. 5b). This study indicates that 2-fluoroaristeromycin has the good binding affinity for both HsSAHH and PfSAHH and also prefers the similar set of amino acids in the binding interaction with both the targets. However, because of enough similarity in the structure of PfSAHH and HsSAHH, a similar binding mode, affinity, and interacting residues were observed in the docking. The additional residues from Tyr152 to Lys193 are present in PfSAHH and absent in HsSAHH might be used for designing inhibitors of PfSAHH. It is important to note that these residues were neither involved in the interaction of NAD with PfSAHH nor in docking interaction of inhibitors with PfSAHH. Though, the additional residues were not involved in interaction with PfSAHH, but might play an important role in shaping the pocket/binding site of PfSAHH.

a Interaction of 2-fluoroaristeromycin with HsSAHH. b Interaction of 2-fluoroaristeromycin with PfSAHH residues are labeled with its position and hydrogen bonding are shown by green dashed lines
IC50 is a quantitative measurement which indicates how much a particular drug or inhibitor is required to inhibit an enzyme or biological process. Docking predicts the optimized conformation of an inhibitor or molecule in the binding site of target enzyme. Docking score is an energy parameter which predicts the binding energy of a ligand-protein complex. Therefore, IC50 and docking score of an inhibitor are two different parameters. The conflicting results have been obtained on comparison of IC50 with docking score. The experimental IC50 of CHEMBL261619 for the target enzyme HsSAHH has been reported 97,500 nmol, whereas the docking score for CHEMBL261619 with HsSAHH was predicted −153.55 Kcal/mol. IC50 value indicates CHEMBL261619 as a poor inhibitor but docking indicates a good affinity for the HsSAHH. In the case of PfSAHH, four compounds (2-fluoroaristeromycin, 5-methylenearisteromycin, noraristeromycin, and CHEMBL261619) have shown good correlation between IC50 values and MolDock score. IC50 values for two compounds (aristeromycin and 2-fluoronoraristeromycin) were exceptionally very high, and they do not fit in co-linearity with the rest four compounds. Actually, in silico binding energies are the theoretical calculation, whereas IC50 values are determined by the experimental study. Inhibitors have shown very high IC50 values for enzyme HsSAHH than PfSAHH except in the case of aristeromycin and noraristeromycin. The IC50 of 2-fluoroaristeromycin is reported 1980 nM for target PfSAHH, and it is lower than the other inhibitors. Docking score and IC50 value of 2-fluoroaristeromycin for target PfSAHH is in good agreement. Therefore, 2-fluoroaristeromycin can be considered as better inhibitor among all known analogues of aristeromycin.
ADMET study of inhibitors
ADMET properties such as blood brain barrier (BBB), human intestinal absorption (HIA), Caco-2 permeability (Caco), P-glycoprotein inhibitor (PGI), CYP450 (CYP) substrate, CYP inhibitor, Ames toxicity (AT), and carcinogens (CG) were predicted for all six inhibitors (Table 5). BBB and HIA for all the compounds were positive (very good), and positive probability of BBB and HIA property was more than 0.83 %. Excellent value of BBB and HIA was found in the case of CHEMBL261619. All these compounds have good capability to cross BBB and can be easily absorbed in intestine. Caco value for all the compounds were predicted as negative with a moderate probability. Caco-2 cells are a human colon epithelial cancer cell line used as a model of human intestinal absorption of drugs. All these inhibitors are non-substrate (NS) and non-inhibitor (NI) of P-glycoprotein. These inhibitors were also predicted as NS for CYP2C9, CYP2D6, and CYP3A4. These inhibitors also passed the AT with good probability of NT. AT prediction are used for measuring the mutagenic capability of the inhibitor or chemical compound. An ADMET study suggests that these compounds do not inhibit drug-metabolizing enzymes such as CYP1A2, CYP2C9, CYP2D6, CYP2C19, and CYP3A4. These compounds have shown high probability to act as non-carcinogen (NCG).
Structure modeling of uracil-DNA glycosylase
The 3D structures of 33 drug targets, available in P. falciparum, were retrieved from RCSB database. The 3D structure of uracil-DNA glycosylase was modeled, due to the unavailability of X-ray-/NMR-verified structure. The structure of uracil-DNA glycosylase of P. falciparum was modeled using uracil-DNA glycosylase of Leishmania naiffi (PDB ID: 3CXM, X-ray, 1.50 Å) as a template (268 residues). Template sequence has shown 42.41 identities with the target. A total of 43 templates were found against query sequence. The quality of the model evaluated in terms of QMEAN4 (−4.29). QMEAN4 is a reliability score for a modeled protein structure which can be used to compare alternative models of the same protein. The QMEAN4 score for best quality models ranges between 0 and 1 with higher values for better models. Modeled structure of uracil-DNA glycosylase is composed of the helix, sheet, and coil (Fig. 6). Uracil-DNA glycosylase plays an important role in base excision repair and damaged DNA cannot be transcribed without prior repair. Therefore, uracil-DNA glycosylase may be used as a drug target for designing new anti-malarial compounds. Uracil-derived compounds 1-methoxyethyl-6-(p-n-octylanilino) uracil and 6-(phenylhydrazino) uracil have inhibited the growth of P. falciparum with IC50 of 15.6 and 12.8 μmol, respectively [23]. Computational interaction study between curcumin and modeled uracil-DNA glycosylase was conducted to predict a potential curcumin-related anti-malarial activity.

3D structure of uracil-DNA glycosylase from P. falciparum modeled using uracil-DNA glycosylase of Leishmania naiffi (3CXM) as a template. Secondary structure view of modeled enzyme was visualized using UCSF Chimera
Docking of experimentally validated drug targets of Plasmodium with curcumin
Being herbal in nature curcumin has the low risk of toxicity and side effects. Anti-malarial effect of curcumin reported in some literature and a few drug targets hypothesized in P. falciparum. But, its exact drug target has not been validated yet. Curcumin has shown potential role in the treatment of cerebral malaria [10]. Curcumin in combination with artemisinin and piperine has resulted in anti-plasmodial activity [24, 25]. Therefore, the curcumin was docked with experimentally validated anti-malarial drug targets available in P. falciparum. MolDock score, hydrogen-bonding (H-bond) score, residues in interaction, and hydrogen-bonding residues were determined for all 34 drug targets (Table 6). MolDock score is the sum of ligand-protein interaction energy and internal energy of the ligand. Best ligand forms protein-ligand complex with the lowest MolDock score. Curcumin has shown very favorable MolDock score (−175.97 Kcal/mol) for hypoxanthine-guanine-xanthine phosphoribosyltransferase (PfHGPRT), which indicates its high affinity for binding with of HGPRT. Curcumin observed in close interaction with Lys114, Tyr116, Asn118, Glu144, Ile146, Thr149, Gly150, Lys151, Thr152, Leu153, Leu203, Asp204, and Asn206 residues of PfHGPRT. H-bond score for binding of curcumin with PfHGPRT was −14.96 Kcal/mol, and it involves Lys114, Thr149, Gly150, Lys151, Leu153, and Asn206 residues in interaction (Fig. 7a). HGPRT catalyze the conversion of 6-oxopurine bases to their respective mononucleotides. Sequence identity and similarity between PfHGPRT and human HGPRT was 44 and 76 %, respectively. Despite the high degree of sequence and structural similarity, human HGPRT and PfHGPRT differ significantly in the substrate specificity [26]. PfHGPRT catalyzes the phosphoribosylation of xanthine, in addition to hypoxanthine and guanine. Purine analogues probably serve as substrates for HGPRT and have been shown lethal to P. falciparum in culture [27]. Curcumin has shown highest affinity for PfHGPRT among 34 drug targets of P. falciparum. Therefore, PfHGPRT is considered to be the most promising drug target of curcumin, and the anti-malarial effect of curcumin might be because of its binding with PfHGPRT.

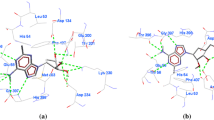
a Docked view of curcumin with PfHGPRT. b Docked view of curcumin with PfSpdS. Interacting residues of both targets with curcumin are labeled with amino acid and its position, whereas hydrogen-bonding interaction between residues and atom of curcumin has been shown by green dash lines
Curcumin has also shown a favorable binding with enzyme spermidine synthase and adenylosuccinate synthetase (AdSS) of P. falciparum. MolDock score for curcumin-spermidine synthase complex found −174.85 Kcal/mol and involves interaction of Gln72, Leu88, Gln93, Leu94, Tyr102, His103, Gly124, Gly125, Asp127, Cys146, Glu147, Ile148, Glu177, Asp178, Ala179, Asp196, Ser197, Pro203, Ala204, Thr206, Leu207, and Tyr264 residues of P. falciparum spermidine synthase (PfSpdS) with curcumin (Fig. 7b). Gln72, Gln93, Tyr102, His103, Gly125, Asp178, Ala179, Thr206, and Tyr264 residues of PfSpdS observed in hydrogen bonding with curcumin and contributed a total H-bond score of −17.04 Kcal/mol. PfSpdS catalyzes the synthesis of spermidine and spermine. It is a major polyamine flux-determining enzyme and prerequisite for the post-translational activation of P. falciparum eukaryotic translation [28]. PfSpdS can be a unique drug target against malaria because polyamine metabolism of the P. falciparum is distinct from the human.
AdSS catalyzes the formation of adenylosuccinate from inositol monophosphate (IMP) and aspartate. P. falciparum adenylosuccinate synthetase (PfAdSS) differs from mammalian homologs. Fructose-1, 6-bisphosphate, a potent inhibitor of the mammalian enzyme, serves as an activator of PfAdSS [29]. These structural differences are promising in terms of species-specific drug design, targeting this essential enzyme in the P. falciparum. PfAdSS forms a binding energy complex (−173.20 Kcal/mol) with curcumin and involves Asp26, Asn51, Ala52, Gly53, Asp124, Gly139, Thr140, Thr141, Ile145, Gly146, Val281, Thr307, Thr308, and Arg311 residues of PfAdSS in binding. Ala52, Gly53, Thr140, Gly146, Thr307, Thr308, and Arg311 residues of pfAdSS contributes in hydrogen bonding with a good H-bond score (−13.70 Kcal/mol) (Fig. 8a). Interaction of curcumin with PfSAHH performed with the assumption that an anti-malarial effect of curcumin may be due to its binding with PfSAHH. Here, curcumin forms an energy complex (−138.30 Kcal/mol) with PfSAHH. His54, Gly135, Thr201, Thr202, Thr203, Cys239, Val268, Gly321, Ile343, Gly344, His345, Phe346, Leu389, Leu392, and Asn391 residues of PfSAHH observed in interaction with curcumin. His54, Thr202, Thr203, and Asn391 residues of PfSAHH involved in hydrogen bonding with curcumin and relatively a low H-bond score (−9.28 Kcal/mol) was measured (Fig. 8b). These in silico results indicate that curcumin has a higher affinity for pfHGPRT, pfSpdS, and PfAdSS targets than PfSAHH. But, suitability of these drug targets depends on the sub-cellular location of these enzymes and bioavailability of curcumin to these enzymes. PfHGPRT hypothesized as a drug target for curcumin against malaria by virtually screening the 34 anti-malarial drug targets available in P. falciparum. Therefore, pfHGPRT can be used as a drug target for malaria, and potential inhibitors of pfHGPRT can be designed using curcumin as a lead compound.

a Docked view of curcumin with pfAdSS. b Docked view of curcumin with PfSAHH. Interacting residues of enzyme targets with curcumin are labeled with amino acid and its position, whereas hydrogen-bonding interaction between residues and atom of curcumin has been shown by green dash lines
Conclusion
Though there is enough sequence and structural similarity between PfSAHH and HsSAHH, yet slight structural differences in the composition of residues involved in the binding site may result in selective inhibition of PfSAHH. Docking study reveals that the 2-fluoroaristeromycin and other analogues of aristeromycin are not only selective to PfSAHH but also have an affinity for HsSAHH. 2-Fluoroaristeromycin can generate an anti-malarial effect on binding with PfSAHH, but its affinity toward HsSAHH might be toxic to human. There is a need to consider the information of target binding site as well pharmacophore of known inhibitor while designing potential inhibitor for PfSAHH. Virtual screening of curcumin with reported anti-malarial drug targets of P. falciparum indicate that anti-malarial effect of curcumin might be due to its binding with PfHGPRT. Virtual screening of anti-malarial drug targets indicates that curcumin has more energetically favorable interaction with pfHGPRT, pfSpdS, and PfAdSS drug targets than PfSAHH. But, suitability of these drug targets depends on the sub-cellular location of these enzymes and bioavailability of curcumin to these drug targets.
References
Yuan CS, Saso Y, Lazarides E, Borchardt RT, Robins MJ (1999) Recent advances in S-adenosyl-L-homocysteine hydrolase inhibitors and their potential clinical applications. Expert Opin Ther Patents 9:1197–1206
Kitade Y, Kozaki A, Miwa T, Nakanishi M, Yatome C (2000) Synthesis of carbocyclic nucleosides and their SAH hydrolase inhibitory activities. Nucleic Acids Symp Ser 44:111–112
Tanaka N, Nakanishi M, Kusakabe Y, Shiraiwa K, Yabe S, Ito Y, Kitade Y, Nakamura KT (2004) Three-dimensional structure of S-adenosyl-L-homocysteine hydrolase from Plasmodium falciparum. Nucleic Acids Symp Ser (Oxf) 48:281–282
Tanaka N, Umeda T, Kusakabe Y, Nakanishi M, Kitade Y, Nakamura KT (2013) Structural biology for developing antimalarial compounds. Yakugaku Zasshi 133:527–537
Elrod P, Zhang J, Yang X, Yin D, Hu Y, Borchardt RT, Schowen RL (2002) Contributions of active site residues to the partial and overall catalytic activities of human S-adenosylhomocysteine hydrolase. Biochemistry 41:8134–8142
Kojima H, Yamaguchi T, Kozaki A, Nakanishi M, Ueno Y, Kitade Y (2002) Synthesis of noraristeromycin analogues possessing SAH hydrolase inhibitory activity for the development of antimalaria agents. Nucleic Acids Res Suppl 2:141–142
Kitade Y, Kojima H, Zulfiqur F, Kim HS, Wataya Y (2003) Synthesis of 2-fluoronoraristeromycin and its inhibitory activity against Plasmodium falciparum S-adenosyl-L-homocysteine hydrolase. Bioorg Med Chem Lett 13:3963–3965
Nakanishi M (2007) S-adenosyl-L-homocysteine hydrolase as an attractive target for antimicrobial drugs. Yakugaku Zasshi 127:977–982
Bujnicki JM, Prigge ST, Caridha D, Chiang PK (2003) Structure, evolution, and inhibitor interaction of S-adenosyl-L-homocysteine hydrolase from Plasmodium falciparum. Proteins 52:624–632
Mimche PN, Taramelli D, Vivas L (2011) The plant-based immunomodulator curcumin as a potential candidate for the development of an adjunctive therapy for cerebral malaria. Malar J 15(10 Suppl 1):S10
Singh DB, (2014) Success, limitation and future of computer aided drug designing translational medicine doi:10.4172/2161-1025.1000e127
Singh DB, Gupta MK, Singh DV, Singh SK, Misra K (2013) Docking and in silico ADMET studies of noraristeromycin, curcumin and its derivatives with Plasmodium falciparum SAH hydrolase: a molecular drug target against malaria. Interdiscip Sci 5:1–12
Ando T, Iwata M, Zulfiqar F, Miyamoto T, Nakanishi M, Kitade Y (2008) Synthesis of 2-modified aristeromycins and their analogs as potent inhibitors against Plasmodium falciparum S-adenosyl-L-homocysteine hydrolase. Bioorg Med Chem 16:3809–3815
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Stierand K, Rarey M (2010) Drawing the PDB: protein-ligand complexes in two dimensions. ACS Med Chem Lett 1:540–545
Benkert P, Biasini M, Schwede T (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27:343–350
Thomsen R, Christensen MH (2006) MolDock: a new technique for high-accuracy molecular docking. J Med Chem 49:3315–3321
Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, Lee PW, Tang Y (2012) admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model 52:3099–3105
Matiugina ES, Seley-Radtke KL, Andronova VL, Galegov GA, Kochetkov SN, Khandazhinskaia AL (2010) Synthesis and antiviral evaluation against Vaccinia virus of new N1-oxide analogues of 5′-noraristeromycin. Bioorg Khim 36:797–801
Das SR, Schneller SW, Balzarini J, De Clercq E (2002) A mercapto analogue of 5′-noraristeromycin. Bioorg Med Chem 10:457–460
Seley KL, Schneller SW, Rattendi D, Bacchi CJ (1997) (+)-7-Deaza-5′-noraristeromycin as an anti-trypanosomal agent. J Med Chem 40(4):622–624
Aarbakke J, Miura GA, Prytz PS, Bessesen A, Slørdal L, Gordon RK, Chiang PK (1986) Induction of HL-60 cell differentiation by 3-deaza-(+/−)-aristeromycin, an inhibitor of S-adenosylhomocysteine hydrolase. Cancer Res 46:5469–5472
Suksangpleng T, Leartsakulpanich U, Moonsom S, Siribal S, Boonyuen U, Wright GE, Chavalitshewinkoon-Petmitr P (2014) Molecular characterization of Plasmodium falciparum uracil-DNA glycosylase and its potential as a new anti-malarial drug target. Malar 13:149
Tagboto S, Townson S (2001) Antiparasitic properties of medicinal plants and other naturally occurring products. Adv Parasitol 50:199–295
Rasoanaivo P, Wright CW, Willcox ML, Gilbert B (2011) Whole plant extracts versus single compounds for the treatment of malaria: synergy and positive interactions. Malar J 15(10 Suppl 1):S4
Raman J, Ashok CS, Subbayya SI, Anand RP, Selvi ST, Balaram H (2005) Plasmodium falciparum hypoxanthine guanine phosphoribosyltransferase. Stability studies on the product-activated enzyme. FEBS J 272:1900–1911
Queen SA, Vander JDL, Reyes P (1989) Characterization of adenine phosphoribosyltransferase from the human malaria parasite, Plasmodium falciparum. Biochim Biophys Acta 996:160–165
Burger PB, Williams M, Sprenger J, Reeksting SB, Botha M, Müller IB, Joubert F, Birkholtz LM, Louw AI (2015) A novel inhibitor of Plasmodium falciparum spermidine synthase: a twist in the tail. Malar J 14:54
Raman J, Mehrotra S, Anand RP, Balaram H (2004) Unique kinetic mechanism of Plasmodium falciparum adenylosuccinate synthetase. Mol Biochem Parasitol 138:1–8
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(GIF 136 kb)
Rights and permissions
About this article

Cite this article
Singh, D.B., Dwivedi, S. Structural insight into binding mode of inhibitor with SAHH of Plasmodium and human: interaction of curcumin with anti-malarial drug targets. J Chem Biol 9, 107–120 (2016). https://doi.org/10.1007/s12154-016-0155-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12154-016-0155-7