
Abstract
S-adenosyl-l-homocysteine hydrolase (SAHH) is a ubiquitous enzyme that plays a significant role in methylation-based processes by maintaining the intracellular balance between S-adenosylhomocysteine and S-adenosylmethionine. In the past years, some analogs and derivatives of aristeromycin have been reported as a potential inhibitor of Plasmodium falciparum’s SAHH (PfSAHH), but no effective therapy has been developed yet. In our previous studies, molecular dynamics simulation study of 2-fluoroaristeromycin in complex with PfSAHH was carried out, and a stable complex with favorable binding energy and interaction was observed. In the presented work, 2-fluoroaristeromycin was used as a central compound for finding the vast set of similar compounds using PubChem database search (65 compounds), pharmacophore-based search (1219 compounds) and ZINC database search for biogenic compounds (approximately 1, 82000 compounds). All these compounds were docked with PfSAHH drug target to screen compounds with energetically favorable binding and stable conformation. Binding energy and different ADMET based parameters were used for screening some potential compound from each set. Binding affinity and interaction of top scoring 15 compounds from the biogenic subset were again evaluated using other docking tools such as AutoDock and AutoDock Vina. These top scoring compounds satisfy the binding and most of the ADMET parameters, and their activity can be further optimized to find a more potent inhibitor of PfSAHH.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Malaria is a life-threatening disease in many tropical and subtropical countries. Malaria is caused by the protozoan parasite Plasmodium. The malaria parasite is transmitted by female anopheles mosquitoes. There are many drugs available in the market for the treatment of malaria. The motivation behind selecting this problem is because of a large number of deaths occur every year due to malaria globally including one child in every 30 s (Talsania et al. 2012). The burden of malaria in India has been underestimated. So there is a need for an effective therapeutic approach for the treatment of malaria. Conventional drugs are not very much effective against malaria due to an emergence of new strains of malaria parasite, i.e., Plasmodium falciparum. Therapeutic efficacy of drugs should be monitored because of the emergence of the drug resistance. There are many proteins/enzymes recommended and used as a drug target against malaria. Drug designing against malaria often focuses on blood stage of Plasmodium because they are responsible for the symptoms of the disease. Despite research, a significant hurdle is the market failure of the drug companies to invest in the discovery and designing of new anti-malarial drugs. Plasmodium falciparum hypoxanthine guanine phosphoribosyl transferase (PfHGPRT), an essential enzyme for purine nucleotide synthesis has also been recommended as drug target for the treatment of malaria (Verma et al. 2016).
Methylation is an important process in cellular pathways, including embryonic development, X-chromosome inactivation, genomic imprinting, and preservation of chromosome stability (Phillips 2008). S-adenosylhomocysteine hydrolase (SAHH) is a ubiquitous enzyme that plays a significant role in methylation-based processes by maintaining the intracellular balance between S-adenosylhomocysteine (SAH) and S-adenosylmethionine (SAM) (Reddy et al. 2008). SAHH is an enzyme that catalyzes the reversible hydrolysis of SAH into free adenosine (ADO) and l-homocysteine (HCY). SAH is a strong feedback inhibitor of many SAM-dependent methyltransferases. SAHH of Plasmodium falciparum (PfSAHH) has been validated as a potential drug target against malaria. In the past years, some analogs and derivatives of aristeromycin have been reported as a potential inhibitor of PfSAHH (Kojima et al. 2002; Kitade et al. 2003), but no effective therapy has been developed yet. Meanwhile, consistent progress has been made in discovering more inhibitors and drug targets against malaria. However, there is still a lack of potential drug in the market that can be effectively used for the treatment of malaria.
Structural comparison of human SAHH (HsSAHH) and PfSAHH has been performed by computational analysis. SAHH is present in both human (host) and Plasmodium falciparum (parasite). PfSAHH is not a unique target against malaria. Sequence composition, similarity statistics, and root mean square deviation (RMSD) indicate that enough structural difference exists in between HsSAHH and PfSAHH. The volume of the largest cavity in PfSAHH was measured 263.168 Å3 and associated with NAD binding. There is a remarkable difference in volume of the largest cavities in pfSAHH and HsSAHH (402.944 Å3) (Singh et al. 2013; Singh and Dwivedi 2016a). Because of variation in residues comprising the largest cavity in both the enzymes, structural differences in shape and size of the cavity were observed. NAD shows hydrogen bonding with Thr201, Thr202, Thr203, Asn235, Val268, Glu287, Asn322, Ile343, Asn391, Lys473, and Tyr477 of PfSAHH. Val268, Ile288, and Thr320 of PfSAHH were seen in hydrophobic interaction with NAD (Singh and Dwivedi 2016a). In comparison to HsSAHH, Asn322, Lys473, and Tyr477 of PfSAHH are unique in interaction with NAD, and these residues can be employed for selective inhibition of PfSAHH.
The stability of PfSAHH-2-fluoroaristeromycin complex was evaluated through molecular dynamics (MD) simulation. In MD simulations, solvating water molecules were considered for evaluating the stability of the protein–inhibitor complex. MD simulations provide a dynamic evolution of a given system using Newtonian mechanics through a period of time while molecular docking predicts binding interactions and affinities of two molecules without a time dimension. Implicit waters, on the other hand, can be taken into account in both approaches. Docking and MD simulation studies reveal that the binding of 2-fluoroaristeromycin with PfSAHH is stable under physiological conditions (Singh and Dwivedi 2016b).
Experts believe that more emphasis should be given on breaking the cycle of malaria transmission rather than treatment. Existing drugs such as primaquine and ivermectin can be used to break the transmission cycle. The result of RTS, S vaccine against malaria is also encouraging and radiation-attenuated sporozoite vaccine is under investigation (Burrows et al. 2017). A combination of two or more active molecules can play an important role to combat the drug resistance. Drug 8-aminoquinoline tafenoquine has demonstrated high activity against malaria (Beck et al. 2016). There is a need to develop compounds with similar efficacy to tafenoquine but without risk of hemolysis in malaria patients. tafenoquine has shown anti-hypnozoite activity. New molecules such as KAF156, DSM265, MMV048, and DDD498 were identified by their activity against hepatic schizonts (Wells et al. 2015). KAE609 clears parasites more rapidly than artesunate and could cure severe malaria (White et al. 2014). ATPase PfATP4 is a new anti-malarial drug target. PfATP4 serves as the primary Na+ -efflux pump in Plasmodium falciparum. Cipargamin inhibits the Na+-dependent ATPase activity present in Plasmodium falciparum membranes and results in an anti-malarial effect (Rosling et al. 2018). In trophozoite-stage, an addition of cipargamin or spiroindolones results in an increase in Na+ content of the parasite. Researchers are also working in the different directions to find the potential and safe therapeutic approach for against malaria.
The aim behind this study is to see the affinity of known inhibitors for the target PfSAHH. This can tell us about the binding mode of the known inhibitor on PfSAHH enzymes. The existing inhibitors can be modified or optimized provided they show better binding to PfSAHH. Absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of known inhibitors were also predicted to estimate the response of these compounds in the human body. There are many drug targets available for the treatment of malaria. Some drug targets malaria has been well studied and still work is going in the direction of anti-malarial drug designing. There are many drug targets and inhibitors available against malaria. However, still, effective cure of malaria is challenging in our country.
2 Materials and methods
2.1 Biological databases
The protein data bank (PDB) was established at Brookhaven National Laboratory (BNL) in 1971. PDB contains information about the 3D structure of proteins, nucleic acids, and complex structures that help in the study of all aspects of genomics, proteomics, biomedicine, and agriculture, from protein synthesis to health and disease. PDB database contains approximately 1, 21, 654 macromolecular structures (https://www.rcsb.org/). The available X-ray structures (PDB file) of SAHH for Homo sapience (1LI4), Plasmodium falciparum (1V8B) were retrieved from Research Collaboratory for Structural Bioinformatics (RCSB) protein database. RCSB curates and annotates the PDB data.
The PubChem compound database contains validated information about chemical compounds. Structures stored within PubChem compounds have been pre-clustered and cross-referenced on the basis of identity and similarity group (http://www.ncbi.nlm.nih.gov/pccompound). The PubChem is also used for searching the compounds similar to query molecules. The known inhibitors of PfSAHH were retrieved from PubChem database.
ZINC is an open-access database of commercially available compounds for the purpose of docking or virtual screening. ZINC contains over 35 million chemical compounds in different structural 3D file formats (http://zinc.docking.org/). ZINC has a different subset of compounds such as lead-like, fragment-like, drug-like, clean drug-like, leads now, all now, drugs now (Irwin et al. 2012). Compounds can be searched using SMILES, ZINC Id, ring name, catalog and vendor name or target.
ChEMBL is an open-access database containing ligand binding, bioassay and ADMET properties for a large set of drug-like compounds (Gaulton et al. 2012). This is a curated and standardized database in the field of chemical biology and drug discovery. ChEMBL also contains structures of the food and drug administration (FDA) approved drugs (https://www.ebi.ac.uk/chembl/). ChEMBL provides broad coverage of a diverse set of targets, organisms, and bioactivity of compounds reported in the literature. The 2D/3D structural properties, bioassay of inhibitors, IC50 of six inhibitors for PfSAHH and HsSAHH were retrieved from the ChEMBL database.
The Binding database (BindingDB) is a public, web-accessible database of measured binding affinities, focusing chiefly on the interactions of protein considered to be drug targets with small, drug-like molecules (https://www.bindingdb.org/bind/index.jsp). The information about inhibitors that target the PfSAHH was retrieved from the binding database.
2.2 ZincPharmer
Pharmacophore-based search approaches are used as a major tool in drug discovery. Various ligand-based and structure-based tools have been developed for pharmacophore modeling and applied in lead optimization, de novo design, compound searching, and virtual screening. ZINCPharmer is a pharmacophore search tool for screening the subset of the ZINC database. ZINCPharmer can import MOE pharmacophore and LigandScout definitions, and identify pharmacophoric features directly from the structure (Koes and Camacho 2012). Filtering parameters such as maximum hit per conformations, maximum hit per molecules, maximum total hit, RMSD, molecular weight and a number of rotatable bonds can be set to achieve the desired output. It uses the Pharmer open source pharmacophore search approach to search a large database of fixed conformers for pharmacophore matches (http://zincpharmer.csb.pitt.edu). Ligand-based pharmacophore modeling is recommended for drug discovery in the absence of protein structure. It is performed by extracting common pharmacophoric features from 3D structures of a set of known ligands. In general, pharmacophore modeling involves two main steps: creating the conformational space for each ligand, and aligning the multiple ligands in the training set and determining the essential common chemical features (Yang 2010).
2.3 Molegro virtual docker (MVD)
There are different docking tools available in the market and they have different performance. Some docking tools provide the solution to flexible docking problem. MVD 2007.2.0.0 was used for docking study. MVD requires a 3-D structure of protein and ligand for docking. MVD performs flexible docking, and the optimal geometry of ligand determined during the docking of the ligand with protein (Thomsen and Christensen 2006). The default parameters of MVD tool were used during the study. The candidates with the best conformational and energetic results were selected. MVD calculates the interaction energies between ligand and protein from the 3-D structure of protein and ligand. The MolDock Score is based on a differential algorithm and was used for energy calculation. Molegro molecular visualizer (MMV) was used for analyzing the cavities presents in PfSAHH and HsSAHH enzyme. This tool was also used to visualize the hydrogen bonding, an electrostatic and steric interaction of coenzyme nicotinamide adenine dinucleotide (NAD) with PfSAHH and HsSAHH.
2.4 AutoDock
AutoDock and AutoDock Vina are also docking tools. This was used for comparison, verification, and evaluation of docking results and poses obtained from MVD. Docking study of 2-fluoroaristeromycin with PfSAHH was also performed using AutoDock Tool 4 (Morris et al. 2009). All water molecules were removed from the protein structure. The hydrogen is added to the structure of PfSAHH and partial atomic charges were assigned. A three-dimensional grid box (dimensions: 126 × 64 × 54 unit in the number of grid points, grid spacing: 0.408 Å) centered at the ligand defining the search space was used. Thr54 and Asp134 residues of PfSAHH were defined as reference residue for docking as the reference residue. Twenty runs of Lamarckian Genetic Algorithm were set to optimize the ligand–protein interactions. The results were clustered according to the root mean standard deviation values and ranked by the binding free energy. Five top poses were observed. Interactions of one best pose with PfSAHH were visualized by Maestro of Schrodinger and MMV.
2.5 AutoDock Vina
AutoDock Vina is open-source software for molecular docking. It was designed and implemented by Dr. Oleg Trott in the Molecular Graphics Lab at The Scripps Research Institute. AutoDock Vina has been reported to improve the prediction accuracy of the ligand binding mode compared to AutoDock 4 (Trott and Olson 2010). A comparative docking study of some top scoring compounds with PfSAHH was performed by both AutoDock and AutoDock Vina.
2.6 admetSAR
The admetSAR is the most comprehensive, curated resource for diverse chemicals associated with known Absorption, Distribution, Metabolism, Excretion and Toxicity profiles (Cheng et al. 2012). It provides an interface to search for ADMET by name, CASRN and similarity search. admetSAR will be helpful for in silico screening ADMET profiles of candidate drug and other chemical compounds (http://lmmd.ecust.edu.cn/admetsar1). admetSAR is useful in optimizing ADMET profiles of Hits or Leads in drug discovery and also make a smart decision during the drug discovery process. The quantitative estimation of ADMET prediction can be a decisive step in computer-aided drug discovery (CADD). There is a demand for early information on ADMET to ensure further synthesis and testing. There are different medium and high-throughput in vitro ADMET screens are in use (Van de Waterbeemd and Gifford 2003). There is also a need of tools with good predictive accuracy to estimate these properties. The main objective is to optimize the screening and testing for the discovery of most promising compounds. ADMET parameters such as blood brain barrier (BBB), human intestinal absorption (HIA) Caco-2 permeability (Caco), P-glycoprotein inhibitor (PGI), CYP450 (CYP) substrate, CYP inhibitor (CIP), Ames toxicity (AT) and carcinogens (CG) were predicted for the purpose of compound screening.
3 Results and discussion
3.1 PubChem search, virtual screening and ADMET
2-Fluoroaristeromycin was used as a central compound for searching the similar compounds in PubChem database. Because 2-fluoroaristeromycin has shown better binding energy and H bonding score than other known inhibitors. Total 65 compounds selected and their structural data file (SDF) file downloaded from PubChem. The SDF file of these compounds converted into a Mol2 file using Open Babel tool. These compounds were docked with PfSAHH target to find the top scoring inhibitors. Docking parameters were kept default except runtime ten and returned conformation five. The MolDock score and hydrogen bonding contribution for these 65 compounds were tabulated. Finally, the top five scoring compounds were selected for interaction study. These compounds are CID5311019, CID44352200, CID58761799, CID70340123 and CID101638021. All these compounds have shown good binding energy and H bond contribution. The good number of hydrogen bonds was seen in the interaction of these compounds with PfSAHH. These top scoring inhibitors also bind nearly at the same site of the binding region as most of the residues were observed commonly in interaction with known inhibitors.
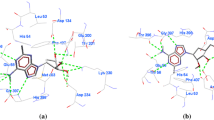
CID5311019 has shown highly favorable binding with MolDock score of − 176.85 Kcal/mol and a very good H bond score of − 30 Kcal/mol. CID5311019 interacts with 15 different residues His54, Cys78, Asp134, Gly135, Glu200, Thr201, Thr202, Thr203, Lys230, Asp234, Cys239, His398, Ile343, Gly344 and His345 of PfSAHH (Supplementary Table 1). From this set of 15 interacting residues, 11 residues His54, Cys78, Asp134, Thr201, Thr202, Thr203, Cys239, His398, Ile343, Gly344 and His345 contributes in the formation of 18 hydrogen bonds (Fig. 1a). Second, the energetically favorable compounds selected from the set of 65 was CID44352200, which was obtained with a MolDock score of − 146.80 Kcal/mol and H bond score of − 24.09. CID44352200 interacts with Thr56, Glu58, Asp134, Glu200, Thr201, Val268, Lys230, Asp234, Ile343, Gly344, Leu392, His398, and Met403 of PfSAHH. Thr56, Glu58, Glu200, Thr201, Lys230, Asp234, Ile343, Gly344 and His398 residues of PfSAHH were observed in 11 hydrogen bond interactions with CID44352200 (Fig. 1b). Rest of the compounds have shown MolDock score (energy) approximately − 146 Kcal/mol and interacts with the similar set of residues of PfSAHH. One another compound CID58761799 has shown very good H bond score of (− 41.59 Kcal/Mol) with a total MolDock score of − 146.78.

a Interaction of CID5311019 with PfSAHH. (b) Interaction of CID44352200 with PfSAHH. Residues are labeled with its position and hydrogen bonding is shown by green dash line
ADMET properties of these five compounds (Fig. 2) were predicted using admetSAR. It requires SMILES notation of compounds for ADMET prediction. SMILES notation of these compounds retrieved from PubChem database and submitted to prediction tool as input. Predicted ADMET properties listed in Table 1 along with their outcome and probability of an event. The CID5311019 has shown poor intestinal absorption and Caco permeability but it has shown good binding energy and interaction with target enzyme PfSAHH. The BBB for the CID5311019 was satisfactory but its probability of human intestinal absorption was predicted high with a negative outcome. Similarly, CID101638021 was predicted with a poor BBB and Caco permeability. So, it can be concluded it may not cross the BBB and intestinal membrane. For most of the compounds, Caco permeability was predicted negative with a moderate probability. ADMET properties for CID44352200 were predicted favorable except Caco permeability, but its docking score with PfSAHH was found lower than CID5311019. All these compounds predicted were predicted as non-inhibitor (NI) of P-glycoprotein with a good probability. The selected compounds were observed with a high probability of low cytochrome inhibitory promiscuity. These screened compounds were also predicted to have non-toxic (NT) and non-carcinogenic (NCG) property.

2-D view of top scoring compounds screened from PubChem search
3.2 Pharmacophoric features, pharmacophore-based searching and virtual screening of compounds
ZincPharmer provides facility to map all the pharmacophoric features present in compounds. It also enables to map the ligand features over the receptor by uploading the receptor structural file. Pharmacophoric features present in all known inhibitors were selected as input parameters for pharmacophore-based searching. It is interesting to note that all inhibitor share a much similar core region. Pharmacophoric features present in 2-fluoroaristeromycin were mapped, visualized and their coordinated tabulated (Supplementary Table 2). Here, Pharmacophore models were developed using a structure-based approach and examining the interactions of known inhibitors with PfSAHH. Finally, features that were thought most significant features for the interaction with PfSAHH were selected. ZincPharmer derives the pharmacophore models from a given protein–ligand interaction structure. A pharmacophore defines the spatial arrangement of the essential features of a biological interaction, such as the hydrogen bond, hydrophobic, charged, or aromatic features. Then, a minimum features present in all the compounds were selected as input for searching Zinc purchasable compounds. Quantitative structure–activity relationship (QSAR) models are used to establish a relationship between chemical structures and biological activity in a data set of compounds. The developed QSAR models are used to predict the biological activities of new compounds. QSAR study for the known inhibitors of PfSAHH can also be performed to find a predictive equation for estimating the biological activity. However, a very small dataset of inhibitor with known IC50 is available. QSAR tools require a good number of compounds to generate a good predictive equation.
The pharmacophoric feature selected for compound searching has been visualized (Fig. 3) and represented in Table 2. During the search, all the parameters were kept default except root mean square deviation (RMSD) value between ligand pharmacophore, which was kept 0.50 Å. Compounds matching with the assigned pharmacophore were then energy minimized and ranked with respect to scoring function. Searching results provide the information about ZINC Id, RMSD, Mass and rotatable bonds. It was observed that most of the screened compounds have RMSD below 0.40 Å. Finally, 1219 compounds were obtained as a hit from the total set of 22, 273, 923 compounds. These 1219 compounds were obtained as a result of pharmacophore-based searching was downloaded in SDF file. These compounds were again docked with the target enzyme PfSAHH to ensure the fitness of these compounds on the binding site of PfSAHH. Pharmacophore hypothesis validated if the compounds achieve good docking score (lower energy of docked complex) after docking.

Pharmacophore features selected for searching compounds against zinc database; representation of pharmacophore: blue circle—aromatic ring; yellow boll—H acceptor; white boll—H donor and dark green circle—hydrophobic. (Color figure online)
Top scoring five compounds were screened on the basis of docking score from a set of 1219 docked compounds (Fig. 4). These five compounds are ZINC02394600, ZINC02374138, ZINC02398002, ZINC13513114 and ZINC05046981. MolDock score for these compounds ranges between − 170 and − 190.86 Kcal/mol (Table 3). These compounds have also shown good hydrogen bond contribution in the total energy of docked complex. All these compounds have shown docking interaction with a similar set of residues. It indicates that all these compounds have preferred their binding near NAD and substrate-binding region. Compound ZINC02394600 has shown best docking energy (− 190.86 Kcal/mol) in the pharmacophore-based selection set. It interacts with Cys78, Asn79, Thr201, Thr202, Thr203, Asp235, Gly266, Asp267, Val268, Thr320, Gly321, Ile343, Gly344, His345 and Phe346 residues of PfSAHH and also forms 11 hydrogen bonds (− 27.24 Kcal/mol) in interaction with Thr201, Thr202, Asp267, Thr320, Gly321 and Phe346. ADMET property for this compound was predicted well with positive probability for BBB (0.86) and positive HIA (0.99). It was also predicted non-inhibitor of PGI and non-carcinogenic with a probability of 0.62 and 0.62, respectively. A moderately high probability for CIP (0.65) and AT (0.63) was observed (Table 4). Excellent ADMET values were predicted for ZINC13513114 with BBB (+ 0.86), HIA (+ 1.0), Caco (+ 0.50), non-inhibitor of PGI (0.85), non-toxic in Ames test (0.64) and non-carcinogenic (0.76). The docking profile of this compound was also good with MolDock score of − 181.25 Kcal/mol and H bond score of − 35.16 Kcal/mol.

2-D view of top scoring compounds screened from pharmacophore-based search
3.3 Screening of compounds from biogenic subset of ZINC database, virtual screening and ADMET prediction
ZINC biogenic compounds include non-human metabolites, endogenous (primary metabolites observed in human), primary metabolites of any species, natural products (secondary metabolites) and primary and secondary metabolites made by nature. Natural products are biological in origin and derived from cells, tissues, and secretions of animals, microbes, and plant. Nature offers a broad range of novel and structurally diverse compounds (Singh 2018). Total approximately 1, 82,000 compounds (biogenic) were downloaded from the zinc database in 16 different files. All these compounds were docked with PfSAHH drug target. The docking parameters were default except docking run 10 and returned conformation one. Here, the largest cavity that was associated with the interaction of NAD was defined as a binding site. After docking top 50 compounds from were screened from the set of approximately 1, 82,000 compounds on the basis of MolDock score and H bond score (Table 5). A very excellent MolDock score (− 254.639 Kcal/mol) was obtained for the compound ZINC95100383 for its binding with PfSAHH. Compound ZINC95100382, ZINC95100378, ZINC95100381, ZINC70707332 and ZINC08214433 have shown energetically favorable binding for PfSAHH with a MolDock score of − 232.907, − 232.033, − 230.258, − 220.35, − 231.332 Kcal/mol, respectively.
These top 50 were subjected to ADMET related study using their SMILES notations. The ADMET properties such as BBB, HIA, Caco, PGI, CIP, AT and CG for these 50 compounds are listed in Table 6. Compound ZINC95100382 that has achieved a good docking score, has shown a negative outcome in BBB with a moderate probability of 0.56. HIA for the ZINC95100382 was predicted positive with a probability of 0.98. A very high probability of CIP (0.93) was observed for ZINC95100382, with a moderate non-toxic probability of 0.50 and a non-carcinogenic probability of 0.87. Further, top 15 screened on the basis of docking energy of complex and ADMET related properties. Compounds ZINC95100382, ZINC95100378 and ZINC95100381 have shown high probability to pass BBB and HIA along with good docking scores. These compounds were also predicted as non-inhibitor of P-glycoprotein and non-carcinogenic. However, Caco permeability, Cytochrome450 inhibitory promiscuity, and Ames toxicity results are not in favor of ZINC95100382, ZINC95100378 and ZINC95100381. Caco permeability was predicted negative with high or moderate probability in all the cases, except for compound ZINC68583591. But this compound ZINC68583591 was predicted inhibitor of P-glycoprotein with a high probability of 0.84. ZINC68583591 has favorable BBB and HIA with a probability of 0.90 and 0.99, respectively.
ZINC70707332 and ZINC08214433 have shown energetically favorable binding for PfSAHH with a MolDock score of − 232.907, − 232.033, − 230.258, − 220.35, − 231.332 Kcal/mol, respectively. The favorable BBB, HIA, Caco, and CIP were found for the compound ZINC85867831 with a probability of 0.75, 0.70, 0.95 and 0.60, respectively. But this compound was predicted inhibitor of PGI with a high probability of 0.91. Compound ZINC85867831 was also non-toxic in AT and non-carcinogenic with a high probability of 0.90 and 0.90, respectively. Many other compounds which have shown lower docking score, but has been predicted as a non-inhibitor of PGI with a lower probability were screened for the further study. Compounds ZINC02095684, ZINC02095149, ZINC08791059, and ZINC70707332 were predicted non-inhibitor of PGI and have a lower probability of CIP along with good BBB and HIA.
Top 15 compounds screened from the set of 50 on the basis of binding energy and ADMET, were again subjected to docking at run 10 and 5 conformations to explore the conformational flexibility. Docked conformations of these 15 compounds were analyzed, and interacting residues, H bonding residues and number of H bonding interactions were represented in Table 7. Seven compounds ZINC95100383, ZINC95100382, ZINC95100378, ZINC95100381, ZINC8214433, ZINC85867831 and ZINC2119660 have shown very favorable docking with a MolDock score more than − 200 Kcal/Mol. ZINC95100382 forms very low energy complex (−265.82 Kcal/mol) with PfSAHH which indicates very good docking affinity for this compound. It interacts with 17 residues Thr202, Thr203, Lys210, Gly264, Gly266, Asp267, Val268, Gly269, Lys270, Thr286, Glu287, Cys292, Cys319, Thr320, Gly321, Asn322 and Val325 residues of PfSAHH (Fig. 5a). It also forms 11 hydrogen bonds with the H bond score of − 15.74 Kcal/mol. Total six residues Lys210, Gly266, Asp267, Gly269, Lys270, Thr286, and Thr320 were seen in hydrogen bond interaction with ZINC95100382.

a Interaction of ZINC95100382 with PfSAHH. b Interaction of ZINC95100383 with PfSAHH, residues are labeled with its position and hydrogen bonding is shown by green dash line. (Color figure online)
The second most favorable compound observed in docking was ZINC95100383 which forms a low energy complex of − 259.31 Kcal/mol by interacting with 17 residues Asp134, Thr201, Thr202, Thr203, Leu206, Asn225, Asp226, Gln231, Gly266, Asp267, Cys319, Gly321, Gly344, His345, Leu389, Leu392, and His398 of PfSAHH (Fig. 5b). Total four hydrogen bonds with the H bond score of − 7.75 Kcal/mol were observed in the interaction of ZINC95100383. The four residues observed in hydrogen bonding interaction in ZINC95100383-PfSAHH complex are Thr202, Thr203, Gln231 and Asp267. These 15 compounds docked nearly at the same site of binding that was observed in docking interaction 2-fluroaristeromycin and other known inhibitors. This study also suggests that these screened inhibitors have a higher affinity to dock at the same binding site than known inhibitors. Residues Thr201, Thr202, Thr203, Asn235, Val268, Glu287, Asn322, Ile343, Asn391, Lys473, and Tyr477 were seen in hydrogen bond interaction with PfSAHH. In screened inhibitors, some residues were involved either in overall interaction or hydrogen bonding. It may be also an important concluding remark that screened inhibitor may interrupt the interaction of NAD with PfSAHH as they utilize nearly the same site for bonding with a higher affinity.
Binding affinity and interaction of these 15 compounds (Fig. 6) were again evaluated using other docking tools such as AutoDock and AutoDock Vina. In AutoDock study, good binding energy was obtained for the seven compounds Zinc68583591, Zinc95100383, Zinc95100378, Zinc95100381, Zinc8917968, Zinc2095149 and Zinc2095149, which was more than 8.5 Kcal/mol (Table 8). Docking energies calculated by AutoDock and AutoDock Vina are nearly in the same range. For most of the compounds, docking energies calculated by different tools are in good correlation. The difference in the calculated energies of compounds by different tools might be due to different algorithms and parameters used by these docking tools. However, docking energies calculated by AutoDock and AutoDock Vina are in good correlation with few exceptions.

2-D view of some top scoring compounds from Biogenic dataset
The AUTODOCK has predicted lowest binding energy (− 13.42 Kcal/mol) for the compound Zinc68583591 in interaction with His54, Cys78, Asn79, Asp134, Thr201, Thr202, Asn225, Lys230, Asp234, Cys239, Cys319, Thr320, Gly344, His345, Phe346, Leu389, Leu392, and His398. AutoDock study indicates four hydrogen bonds with three residues Thr201, Thr202 and Asn225 of PfSAHH. Another compound Zinc8917968 has shown a binding energy of -11.33 Kcal/mol and interacts with Thr56, Glu58, Asp134, Gly135, Glu200, Thr201, Thr203, Lys230, Asp235, Val268, Ile343, Gly344, His345, Phe346, Leu389, Asn391, Leu392, His398, and Met403 residues of PfSAHH (Supplementary Table 3). Compounds Zinc68583591, Zinc8917968 and some others were predicted in similar interaction by AutoDock as retrieved by MVD visualizer tool. Compound ZINC95100382 and ZINC95100383 were found to dock with PfSAHH with a binding energy of − 7.78 and − 8.5 Kcal/Mol, respectively.
In AutoDock study, these two compounds ZINC95100382 and ZINC95100383 have also preferred docking at a different site other than site observed by MVD tool. Here, compound ZINC95100382 was predicted to docked with Gly136, Thr139, Leu140, His143, Lys144, Glu147, Tyr148, Glu169, Phe172, Leu173, Thr203, Leu206, Arg207, Met211 and Glu216 and only two residues Tyr148 and Arg207 were seen in hydrogen bond interaction. Similarly, a different set of residues Leu140, Lys165, Asn166, Glu169, Phe172, Leu173, Thr203, Leu206, Arg207, Lys210, Met221, Gly321, Asp348 were seen in AutoDock predicted interaction of ZINC95100383 with PfSAHH. This compound did not form any hydrogen bonding in AutoDock study, whereas the same compound ZINC95100383 forms four hydrogen bonds with Thr202, Thr203, Gln231 and Asp267 of PfSAHH. In mammalian SAHH, the substrate-binding domain of SAHH comprises residues 1–181 and 355–385 (Kusakabe et al. 2015). The cofactor-binding domain comprises residues 197–351. The basic element of the secondary structure of this domain is a six-stranded parallel β-sheet in the center of the domain that is sandwiched by two arrays of three α-helices each. Our docking results indicate that most of the screened inhibitor prefers NAD binding site along with the interaction with residues of substrates binding site. It may be concluded that these inhibitors might cause inhibition of PfSAHH by interrupting or deforming the shape of the NAD binding site along with the shape of the substrate-binding site.
An interaction fingerprint has been generated from the docking interaction data (Table 9). Interacting residues are shown by “1” while residues that are in interaction with at least 4 compounds have been represented by bold “1”. A diverse set of residues are involved in interaction for different compounds, while some of the residues are common. Screened compounds are structurally diverse and this is one of the main reasons behind the major difference in the interacting residues for different compounds. Residues that are involved in interaction with at least four compounds are Leu140, Lys144, Tyr148, Lys165, Glu168, Glu169, Leu173, Thr203, Leu206, Arg207, His345 and Phe346. Compounds ZINC95100383, ZINC95100382, ZINC95100378, ZINC95100381, ZINC70707332, ZINC70705594 and ZINC8791059 have shown nearly a similar pattern of interaction except some difference. Compounds ZINC95100383, ZINC95100382, ZINC95100378, ZINC95100381, ZINC70707332, ZINC70705594 and ZINC8791059 utilize a set of residues for binding which includes Leu140, Lys144, Tyr148, Lys165, Glu168, Leu173, Thr203, Leu206, Arg207, His345 and Phe346. Two residues of PfSAHH, i.e., Leu140 and Arg207 are observed common in interaction with compounds ZINC95100383, ZINC95100382, ZINC95100378, ZINC95100381, ZINC70707332, ZINC70705594 and ZINC8791059. Compounds ZINC68583591, ZINC8917968 and ZINC2095149 have shown a nearly common set of residues in interaction. Common interacting residues for compounds ZINC68583591, ZINC8917968 and ZINC2095149 are Asp134, Thr201, Lys230, His345, and Phe346.
ZINC2095149 has shown a strong affinity for the target enzyme PfSAHH and it has shown interactions with Asp134, Thr201, Thr202, Thr203, Asn225, Lys230, Asn235, Gly266, Val268, Cys319, Leu389, Gly344, Phe346, His345, His398 residues. It forms a favorable hydrogen bonding interaction (09) with Thr201, Thr203, Asn225, Lys230, Val268 and Gly344 residues with the highest H bond scores (− 12.39 Kcal/mol). BBB and HIA values for this compound were predicted good with a positive outcome and it was also predicted non-carcinogenic and non-toxic with a low probability of Cytochrome450 inhibitory effect. The Molecular weight and Xlopp of ZINC2095149 are 513.598 (gm/mol) and 2.88, respectively. Hydrogen bond donor, acceptor and rotatable bonds for this compound are 3, 9 and 6, respectively. This compound is very close to the Lipinski rule of five for a drug than other compounds in the study set. AutoDock has also evaluated the good binding energy of complex (− 10.32 Kcal/mol) for ZINC2095149 with PFSAHH. Similarly, ZINC8917968 has shown the formation of a better energy complex (− 11.33 Kcal/mol) with PfSAHH. Most of the top hits obtained from biogenic subset have a higher molecular weight (> 500). There are many known drugs that do not satisfy the Lipinski rule but it is better to choose the compounds satisfying most of the parameters of Lipinski rule. Molecular weight, Xlopp, hydrogen bond donor, acceptor and rotatable bonds properties for the ZINC8917968 are 502.522 (g/mol), 7.32, 0, 6 and 7, respectively. This compound is not satisfying some parameters of Lipinski rule but still, it can serve as a drug. The BBB (+/0.85), HIA (+/0.1) and caco (+ 0.56) values are very favorable for the ZINC8917968, but this compound was predicted as an inhibitor of glycoprotein with a probability value of 0.74. ZINC8917968 was predicted as non-carcinogenic and non-toxic.
Here, we can see that all compounds may have a little or some negative outcome in ADMET testing or in vivo treatment. It is also important to mention that even recommended and well-tested drugs were predicted to have some negative outcome related to ADMET. Therefore, these compounds need to be synthesized and tested to investigate their real effect on the patient. Top scoring inhibitors screened from this study can be optimized to enhance their affinity and binding interaction with the PfSAHH target. The absorption, distribution, metabolism and toxicity properties of these inhibitors can also be optimized by performing different chemical modifications in the base structure to achieve better pharmacokinetics response. In vitro inhibition assay of some screened compounds can be carried out in Plasmodium culture to evaluate the efficacy of PfSAHH inhibition. The ADMET and biological activity of these compounds can be optimized if some critical issues observed in the future. ADMET are very important parameters for a compound to qualify as a drug. Absorption of the drug should be higher so that an effective concentration can enter in the systemic circulation. Absorption of drug depends on the lipophilicity. High lipophilicity of a drug increases the risk of binding with plasma proteins, self-aggregation, and off-target interaction. Distribution of a drug depends on its lipophilicity, molecular size, the degree of ionization, protein binding and affinity to other molecules. A drug should be easily metabolized by the liver enzymes to avoid the toxicity. Metabolic product of a drug should not be toxic. The rate of metabolism affects the oral bioavailability and elimination of drug in human. The rate of metabolism for a drug can be altered as per requirement by chemical modifications. Toxicity of a compound can be reduced by removing toxic substructure or by altering the group causing off-target recognition.
4 Conclusion
Targeting pfSAHH can be one of the best approaches to inhibit the growth of Plasmodium parasite inside the host. Computational docking and ADMET studies have suggested some potential compounds that can be synthesized and their inhibitory effects can be tested by in vitro inhibition assays. Compounds such as ZINC95100383, ZINC95100382, ZINC95100378, ZINC95100381, ZINC8214433, ZINC85867831, ZINC2119660, ZINC2095149, and ZINC8917968 have shown good binding interactions with the PfSAHH target but some of their ADMET properties were not observed favorable. ADMET properties of these compounds can be optimized to improve their pharmacokinetic response. Different strategies used for the searching and screening of top scoring inhibitors based on binding affinity and ADMET parameters can be very helpful in finding a therapeutic solution to disease. These compounds satisfy most of the ADMET parameters and their ADMET as well specificity, selectivity and binding properties can also be optimized in future if required.
References
Beck HP, Wampfler R, Carter N, Koh G, Osorio L, Rueangweerayut R (2016) Estimation of tafenoquine anti-relapse efficacy using Plasmodium vivax genotyping. J Infect Dis 213:794–799
Burrows JN, Duparc S, Gutteridge WE, Hooft van Huijsduijnen R, Kaszubska W, Macintyre F, Mazzuri S, Möhrle JJ, Wells TNC (2017) New developments in anti-malarial target candidate and product profiles. Malar J 16(1):26
Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, Lee PW, Tang Y (2012) admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model 52:3099–3105
Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Overington JP (2012) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res 40(Database issue):D1100–D1107
Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG (2012) ZINC: a free tool to discover chemistry for biology. J Chem Inf Model 52:1757–1768
Kitade Y, Kojima H, Zulfiqur F, Kim HS, Wataya Y (2003) Synthesis of 2-fluoronoraristeromycin and its inhibitory activity against Plasmodium falciparum S-adenosyl-l-homocysteine hydrolase. Bioorg Med Chem Lett 13:3963–3965
Koes DR, Camacho CJ (2012) ZINCPharmer: pharmacophore search of the ZINC database. Nucleic Acids Res 40:W409–W414
Kojima H, Yamaguchi T, Kozaki A, Nakanishi M, Ueno Y, Kitade Y (2002) Synthesis of noraristeromycin analogues possessing SAH hydrolase inhibitory activity for the development of antimalaria agents. Nucleic Acids Res Suppl 2:141–142
Kusakabe Y, Ishihara M, Umeda T, Kuroda D, Nakanishi M, Kitade Y, Gouda H, Nakamura KT, Tanaka N (2015) Structural insights into the reaction mechanism of S-adenosyl-l-homocysteine hydrolase. Sci Rep 5:16641
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791
Phillips T (2008) The role of methylation in gene expression. Nat Educ 1:116
Reddy MC, Kuppan G, Shetty ND, Owen JL, Ioerger TR, Sacchettini JC (2008) Crystal structures of Mycobacterium tuberculosis S-adenosyl-l-homocysteine hydrolase in ternary complex with substrate and inhibitors. Protein Sci 17:2134–2144
Rosling JEO, Ridgway MC, Summers RL, Kirk K, Lehane AM (2018) Biochemical characterization and chemical inhibition of PfATP4-associated Na(+)-ATPase activity in Plasmodium falciparum membranes. J Biol Chem 293:13327–13337
Singh DB (2018) Natural lead compounds and strategies for optimization. In: Ul-Haq Z, Wilson AK (eds) Frontiers in computational chemistry, vol 4. Bentham science, pp 1–47
Singh DB, Dwivedi S (2016a) Structural insight into binding mode of inhibitor with SAHH of Plasmodium and human: interaction of curcumin with anti-malarial drug targets. J Chem Biol 9:107–120
Singh DB, Dwivedi S (2016b) Docking and molecular dynamics simulation study of inhibitor 2-Fluoroaristeromycin with anti-malarial drug target PfSAHH. Netw Model Anal Health Inf Bioinf 5:16
Singh DB, Gupta MK, Singh DV, Singh SK, Misra K (2013) Docking and in silico ADMET studies of noraristeromycin, curcumin and its derivatives with Plasmodium falciparum SAH hydrolase: a molecular drug target against malaria. Interdiscip Sci 5:1–12
Talsania N, Modi K, Chaudhary P (2012) A Study of investigation report on death audit due to malaria in New Civil Hospital, Ahmedabad City, Gujarat, India. Healthline 3:26–29
Thomsen R, Christensen MH (2006) MolDock: a new technique for high-accuracy molecular docking. J Med Chem 49:3315–3321
Trott O, Olson AJ (2010) AutoDock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 31:455–461
Van de Waterbeemd H, Gifford E (2003) ADMET in silico modelling: towards prediction paradise? Nat Rev Drug Discov 2:192–204
Verma P, Singh DB, Gupta AK (2016) Designing and virtual screening of potential inhibitors of PFHGPRT against malaria. J Chem Pharm Res 8:635–643
Wells TNC, Hooft van Huijsduijnen R, Van Voorhis WC (2015) Malaria medicines: a glass half full? Nat Rev Drug Discov 14:424–442
White NJ, Pukrittayakamee S, Phyo AP, Rueangweerayut R, Nosten F, Jittamala P (2014) Spiroindolone KAE609 for falciparum and vivax malaria. N Engl J Med 371:403–410
Yang SY (2010) Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov Today 15:444–450
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Singh, D.B., Dwivedi, S. Computational screening and ADMET-based study for targeting Plasmodium S-adenosyl-l-homocysteine hydrolase: top scoring inhibitors. Netw Model Anal Health Inform Bioinforma 8, 4 (2019). https://doi.org/10.1007/s13721-019-0183-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13721-019-0183-7