
Abstract
Blood-borne parasite, Plasmodium vivax and Plasmodium falciparum are the most severe malaria causing organisms. Drug resistance against the present antimalarial drugs increased malaria-related morbidity and mortality. Inter-conversion of lactate and pyruvate in the glycolytic pathway requires a metabolic enzyme called lactate dehydrogenase A (LDHA). In recent years, anti-malarial therapy included several small molecules which are LDHA inhibitor in nature. Parthenin is the most active phytochemical of Parthenium hysteropohorus (Asteraceae). Maestro 9.6 software package was used for docking of parthenin like compounds (n = 85) against P. vivax, P. falciparum and the Homo sapiens lactate dehydrogenase proteins (PDB 2A92, 2A94, and 4R68 respectively) to appraise the interaction pattern of target protein and selected ligands. Docking analysis explored some ligands having excellent binding affinity against P. vivax (CID72786361, 78178433, and 11552273), P. falciparum (CID296217, 3482907, 77977597, 78178433). These lead compounds does not showed interaction with the mammalian LDH protein. Glide docking score of selected compounds and standard inhibitor of the target proteins ranged from −5.58 to −8.6 to −6.8 to −7.38 respectively. Structural investigation of selected ligands with P. vivax, P. falciparum, and mammalian LDH complexes revealed the involvement of strong hydrophobic interactions and hydrogen bonding pattern. The QikProp module of Maestro 9.6 was used to predict ADME/T (Absorption, Distribution, Metabolism, Excretion and Toxicity) properties of the lead compounds. In conclusion, CID72786361, 78178433, 11552273 and CID296217, 3482907, 77977597, 78178433 may serve as lead LDHA inhibitor compounds to target P. vivax and P. falciparum malarial parasite respectively. Further in vitro and in vivo studies are required to assess the anti-malarial drug discovery potential of the parthenin like compounds.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
Malaria is a life-threatening disease caused by Plasmodium parasites conveyed to people through malaria vectors. About three million people die and five million have been reported to be infected with malaria annually worldwide (WHO 2011). The dearth of effectual anti-malarial vaccines, chemotherapy assumes a critical part in control of the illness, but unfortunately, drug-resistant strains of Plasmodium (P. falciparum and P. vivax) have shown up against a large portion of antimalarials present till date. Thus, expanded endeavors are instantly desired for antimalarial drug discovery. The objective must be advanced for safe and inexpensive new medications to slug the spread of malaria parasites that are impervious to existing medications. The malarial parasite lives mainly in the host erythrocytes, where they use cell component as a nourishment for their life-cycle growth. These parasites destroyed the hemoglobin fractions of the infected erythrocyte, which leads to severe ailments such as anemia exclusively in both pregnant women and children (Qidwai et al. 2014; Chen 2014). Malarial parasite present in the erythrocyte fundamentally depends on the glycolysis pathway for their energy production. Cytoplasmic pyruvate fermentation and/or mitochondrial electron transport chain are the mechanism by which the consumed NAD+ during glycolysis is regenerated. In Plasmodia, lactate is the end-product of the glycolytic pathway because pyruvate does not enter the citric acid cycle. In the presence of NADH, reduction reaction converts pyruvate into lactate in the presence of lactate dehydrogenase (LDH) enzyme. Here, Plasmodium pyruvate is not able to work as substrate inhibitor, which allows fast energy production, mandatory in the fast-growing malarial parasite (Ramya et al. 2002). Plasmodial LDH differs from its human counterpart by the presence of a five amino acid insertion at the pyruvate binding site. Thus LDH has been used for testing the novel Plasmodium LDH inhibitors versus human LDH. The malarial LDH also has a vast cleft at its active site which can accommodate consilient colossal inhibitors like gossypol derivatives. Based on the above argument LDH may act as a latent target for malaria. Parthenium hysterophorus is a proverbial weed found throughout the world. P. hysterophorus have numerous medical advantages such as medication for rheumatic aching, urinary tract infections, neuralgia, diarrhea, dysentery and skin inflammation. Parthenin is an important ingredient of P. hysterophorus stem, blossom, leaf, and root (Kumar et al. 2013). Because of this, we used different parthenin like compounds to target P. falciparum and P. vivax LDH enzyme in the present study.
In-silico screening approach is the foremost strategy for introductory identification of new inhibitors for target proteins and evaluating their interactive mode. The larger parts of known anti-malarial drugs are small molecules intended to interact and temper the biological mode of action of the distinctive pathogen proteins. Molecular docking comprises three distinctive objectives sequentially posture prediction, virtual screening, and binding affinity calculation. Computer-aided drug designing are able to discriminate hits, pick leads and rationalize leads to convert naturally active compounds into decent medications by upgrading their physicochemical, pharmaceutical and ADME\T (absorption, distribution, metabolism, excretion, and toxicity) properties. Thus, in-silico approach is utilized impressively to reduce the risk, time and asset necessities for both in-vitro and in-vivo chemical synthesis and biological testing (Ekins et al. 2007). In the present study, we employed Maestro 9.6 software for docking studies of 85 parthenin like compounds. Furthermore, Maestro 9.6 QikProp module was used to evaluate the ADME/T properties of the best-docked parthenin like compounds.
2 Materials and Methods
2.1 In-silico Methodology
2.1.1 Selection of Protein and Ligand Molecules
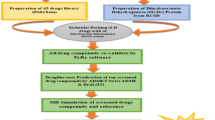
Protocol for the GLIDE based molecular docking took from our earlier published article with a few modifications (Singh et al. 2018) (Fig. 31.1). The present work is the extension of previously published in-silico antimalarial drug discovery from our laboratory (Singh et al. 2018). Same ligands reported in the published article were used to check their multi-targeted antimalarial potential. List of ligands and their structural and physiochemical parameters are reported in previously published article (Singh et al. 2018).

Workflow of study design
Ligprep wizard module of Maestro 9.6 Schrodinger Inc. was used for the ligand preparation. The module accomplishes several modifications in ligands such as hydrogen atom addition, two dimensional to three-dimensional conversion, bond angles and their length corrections, ring conformation, stereochemistry and low energy structure. Ionization was not changed and tautomers were not generated for the ligands. The X-ray crystal structure of test proteins (LDH, PDB: 2A92, LDH PDB: 2A94 and LDH PDB: 4R68) were retrieved from protein data bank (Chaikuad et al. 2005; Labadie et al. 2015). PDB raw structure was modified by using protein preparation wizard module.
2.1.2 Molecular Docking
Maestro 9.6 Schrodinger Inc. software package was used for molecular docking studies of selected ligand molecules (Friesner et al. 2004). Ligands were docked against target protein molecules. Ligand dataset and target proteins were prepared by using appropriate modules available in Maestro 9.6 package. The optimized potential for liquid simulations (OPLS_2005) force field was applied for energy minimization and geometry optimization (Shivakumar et al. 2010; Jorgensen and Tirado-Rives 1988; Jorgensen et al. 1996). The one ligand-one conformation was opted for molecular docking protocol. The receptor-grid file was generated to implement the partial atomic charge of 0.25 and 1 Å Van der Waal radii for the receptor atom. After that, ligand-receptor molecular docking was performed. Based on the minimum glide score, ten compounds were selected for the prediction of type of interactions, optimal energy value, bonding potential, and conformations.
2.1.3 ADME/T Properties Studies
The ADME/T (Absorption, Distribution, Metabolism, Excretion and Toxicity) properties of lead ligands were evaluated by using the Maestro 9.6 QikProp module. Different pharmacological parameters such as overall CNS activity, log BB, MDCK and Caco-2 cell permeability and logKhsa (human serum albumin binding), etc. of the lead ligand molecules were assessed by QikProp module (Jorgensen and Duffy 2002; Lu et al. 2004).
3 Results and Discussion
3.1 Analysis of Docking Results of Promising Compounds for Plasmodium vivax LDH
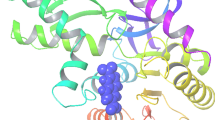
Crystal structure of P. vivax LDH in complex with inhibitor deliver information about position and conformation of enzyme binding site (Kongsaeree et al. 2005). X-ray structure of P. vivax LDH (PDB:2A92) was used for docking study. Anti-malarial combinatorial possessions of artemisinin derivatives (ACT) with quinoline compounds (amodiaquine and mefloquine) are known for their widespread high resistance. Our selected lactate dehydrogenase of Plasmodium vivax structural insights suggest a general approach for developing new generations of antimalarial LDH inhibitors that accommodate the only substrate for their active site, would retain a binding affinity with the mutant enzymes (Kongsaeree et al. 2005). Molecular docking was achieved by using XP mode of GLIDE. Our result highlighted that; CID72786361, 78178433, and 11552273 yielded a pre-eminent dock score for with proteins Plasmodium vivax LDH −8.6, −7.73, −7.55, Kcal/mol respectively (Table 31.1). Most of the interactions made by compounds with residues in the active site of LDH (P. vivax) seem to be hydrophobic in nature. Protein-ligand interactions of 2A92 with compounds showed that amino acids Met30, Ile31, Leu112, Val54, Phe100, Ala98, Val138, Pro250, Leu163, Leu167 and Pro246 appeared in the hydrophobic interactions. Furthermore, amino acid amino acids Asn140, Ser245, Ile31 and Gly99 involved in back-bone hydrogen bonding of protein-ligand interactions (Fig. 31.2).

(a) Ribbon presentation of LDH (PDB, 2A92) protein molecule with CID 72786361 (b) Protein-ligand interactions profile of 2A92 with CID72786361 (c) Protein-ligand interactions profile of 2A92 with CID78178433 (d) Protein-ligand interactions profile of 2A92 with CID11552273
3.2 Interaction Modes Between the Parthenin Like Compounds and Human LDH
Molecular docking of Plasmodium vivax LDH, Plasmodium falciparum LDH and human LDH against natural compounds has been carried out. In the present investigation, our result highlighted that; CID 3482907, 70498184, 73199557, 77977597 yielded a pre-eminent dock score for with proteins human LDH −5.72, −5.58, −6.59, −7.64 Kcal/mol respectively (Table 31.2).
Protein-ligand interactions outline emphasized that the hydrogen bonding, lipophilic, and π-π stacking interactions at the active site plays a vital role in protein-ligand interactions. Molecular docking steps recognize the docking free energy value (Gscore) against the target protein biomolecules. Protein-ligands interactions tinted the electrostatic, lipophilic, and hydrogen bond interactions are a key contributor in protein-ligand interactions. The binding modes of parthenin like compounds in the interactive site of human LDH were identified using intermolecular docking simulations by means of Maestro 9.6 program. All the selected compounds were docked into the human LDH active site, using the same procedure. Figure 31.3 represents the binding pattern of the parthenin like compounds in the LDH binding pocket. Human LDH active site comprises of mostly hydrophobic amino acids as Pro246, Pro250, Leu167, Ile254, Val138, Leu163, Ile31, Tyr247, Met30, Phe100, and Ala98. These amino acid residues are involved in strong hydrophobic interactions with the parthenin like compounds. As anticipated, inhibitors used in this study interact with the same site like the earlier bounded ligand in the crystallographic complex.

(a) Ribbon presentation of LDH (PDB, 2A94) protein molecule with CID 3482907 (b) Protein- ligand interactions profile of 2A94 with CID 3482907 (c) Protein- ligand interactions profile of 2A94 with CID78178433 (d) Protein- ligand interactions profile of 2A94 with CID 77977597 (e) Protein- ligand interactions profile of 2A94 with CID296217
3.3 Interaction Modes Between the Parthenin Like Compounds and Plasmodium falciparum LDH
We used X-ray structure of Plasmodium falciparum LDH in complex inhibitor (PDB code 2A94) for the molecular docking study using GLIDE XP module. Our result highlighted that; CID296217, 3482907, 77977597, and 78178433 yielded a pre-eminent dock score for with proteins Plasmodium falciparum LDH −6.59, −7.84, −6.73, −6.76 Kcal/mol respectively (Table 31.3). Most of the interactions made by compounds with residues in the active site of Plasmodium falciparum LDH seem to be hydrophobic in nature. Protein-ligand interactions of 2A94 with compounds showed that amino acids Leu164, Ile241, Tyr238, Ala237, Val234, and Pro138 appeared in the hydrophobic interactions. Furthermore, amino acid amino acids Arg168, Asn137 and Hid192 involved in back-bone hydrogen bonding of protein-ligand interactions (Fig. 31.4).

(a) Ribbon presentation of LDH (PDB, 4R68) protein molecule with CID77977597 (b) Protein-ligand interactions profile of 4R68 with CID77977597 (c) Protein-ligand interactions profile of 4R68 with CID3482907 (d) Protein-ligand interactions profile of 4R68 with CID73199557 (e) Protein-ligand interactions profile of 4R68 with CID70498184
3.4 ADME/T Properties of Leads Molecules
ADME/T properties of lead compounds were reviewed by Qikprop module of Maestro 9.6 (Kerns and Di 2010). Compound EGCG was found to be promising based on their docking free energy score and binding mode. A most fascinating aspect of CID73199557, 3482907, 73199557, 72786361, 77977597 are their admirable, Qplogpo/w, QplogHERG, QplogBB, QPP MDCK, Qplogkhsa, and proportion of human oral value which justify the Lipinski’s rule of five (Table 31.4). Moreover, high oral bioavailability, polar surface area, H-bond acceptors and donors being imperious criteria for the therapeutic agent’s development. It has been suggested that compounds having a polar surface area equal to or less than 140 angstrom and 10 or less rotatable bonds (or 12 or fewer H-bond donors and acceptors) may have a high possibility for best oral bioavailability in-vivo (Veber et al. 2002). Furthermore, it is also reported that the polar surface area is inversely proportional to permeation rate (Becker et al. 1998). These compounds have batter SASA values that are claimed to be suitable for therapeutic agents. These results designate that these compounds will have a better penetration rate.
4 Conclusion
The present study utilized in-silico approach to search novel Plasmodium spp. lactate dehydrogenase inhibitors using parthenin like compounds as a scaffold. Pharmacological and drug-likeness properties of the selected test compounds showed anti-LDH potential. Beside various lead compounds, CID 78178433 showed potentially against both P. vivax and P. falciparum LDH enzyme. Strong hydrophobic and H-bonding interaction between phytochemicals and pathogen protein was found as revealed by high Dock score. We hereby suggest the in-vitro and in-vivo validation of these lead antimalarial compounds which might provide cost-effective and safer natural antimalarial drug.
References
Becker S, Groner B, Müller CW. Three-dimensional structure of the Stat3β homodimer bound to DNA. Nature. 1998;394(6689):145–51.
Chaikuad A, Fairweather V, Conners R, Joseph-Horne T, Turgut-Balik D, Leo R. Brady structure of lactate dehydrogenase from Plasmodium Vivax: complexes with NADH and APADH†. Biochemistry. 2005;44:16221–8.
Chen C. Development of antimalarial drugs and their application in China: a historical review. Infect Dis Poverty. 2014;3(1):1–10.
Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: applications to targets and beyond. Br J Pharmacol. 2007;152(1):21–37.
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–49.
Jorgensen WL, Duffy EM. Prediction of drug solubility from structure. Adv Drug Deliv Rev. 2002;54(3):355–66.
Jorgensen WL, Tirado-Rives J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J Am Chem Soc. 1988;110(6):1657–66.
Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118(45):11225–36.
Kerns E, Di L. Drug-like properties: concepts, structure design and methods: from ADME to toxicity optimization. Amsterdam: Academic Press; 2010.
Kongsaeree P, Khongsuk P, Leartsakulpanich U, Chitnumsub P, Tarnchompoo B, Walkinshaw MD, Yuthavong Y. Crystal structure of dihydrofolate reductase from Plasmodium vivax: pyrimethamine displacement linked with mutation-induced resistance. Proc Natl Acad Sci U S A. 2005;102(37):13046–51.
Kumar S, Mishra A, Pandey AK. Antioxidant mediated protective effect of Parthenium hysterophorus against oxidative damage using in vitro models. BMC Complement Altern Med. 2013;120:1–9.
Labadie S, Dragovich PS, Chen J, Fauber BP, Boggs J, Corson LB. Optimization of 5-(2, 6-dichlorophenyl)-3-hydroxy-2-mercaptocyclohex-2-enones as potent inhibitors of human lactate dehydrogenase. Bioorg Med Chem Lett. 2015;25(1):75–82.
Lu JJ, Crimin K, Goodwin JT, Crivori P, Orrenius C, Xing L, Tandler PJ, Vidmar TJ, Amore BM, Wilson AG, Stouten PF. Influence of molecular flexibility and polar surface area metrics on oral bioavailability in the rat. J Med Chem. 2004;47(24):6104–7.
Qidwai T, Jamal F, Khan MY, Sharma B. Exploring drug targets in isoprenoid biosynthetic pathway for Plasmodium falciparum. Biochem Res Int. 2014;2014:1–12.
Ramya TNC, Surolia N, Surolia A. Survival strategies of the malarial parasite Plasmodium falciparum. Curr Sci. 2002;83(7):818–25.
Shivakumar D, Williams J, Wu Y, Damm W, Shelley J, Sherman W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J Chem Theory Comput. 2010;6(5):1509–19.
Singh P, Kushwaha PP, Kumar S. Novel potent inhibitors of Plasmodium vivax dihydrofolate reductase: an in silico antimalarial drug discovery. Indian J Pharm Educ Res. 2018;52:1–14.
Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615–23.
WHO. World malaria report 2011. Geneva: World Health Organ; 2011.
Acknowledgments
PS and PPK acknowledge Indian Council of Medical Research (ICMR), India and UGC-CSIR, India respectively for providing the financial assistance in the form of Postdoc and Senior Research Fellowship. SK acknowledges the Central University of Punjab for providing infrastructure facilities.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Singh, P., Kushwaha, P.P., Kumar, S. (2019). Parthenin and Its Similar Structure as Potential Lead Inhibitors of Plasmodium vivax and Plasmodium falciparum Lactate Dehydrogenase. In: Kumar, S., Egbuna, C. (eds) Phytochemistry: An in-silico and in-vitro Update. Springer, Singapore. https://doi.org/10.1007/978-981-13-6920-9_31
Download citation
DOI: https://doi.org/10.1007/978-981-13-6920-9_31
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-6919-3
Online ISBN: 978-981-13-6920-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)