
Abstract
Altered aerobic glycolysis is a well-recognized characteristic of cancer cell energy metabolism, known as the Warburg effect. Even in the presence of abundant oxygen, a majority of tumor cells produce substantial amounts of energy through a high glycolytic metabolism, and breast cancer (BC) is no exception. Breast cancer continues to be the second leading cause of cancer-associated mortality in women worldwide. However, the precise role of aerobic glycolysis in the development of BC remains elusive. Therefore, the present review attempts to address the implication of key enzymes of the aerobic glycolytic pathway including hexokinase (HK), phosphofructokinase (PFK) and pyruvate kinase (PK), glucose transporters (GLUTs), together with related signaling pathways including protein kinase B(PI3K/AKT), mammalian target of rapamycin (mTOR) and adenosine monophosphate-activated protein kinase (AMPK) and transcription factors (c-myc, p53 and HIF-1) in the research of BC. Thus, the review of aerobic glycolysis in BC may evoke novel ideas for the BC treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breast cancer (BC) remains the most frequently diagnosed malignancy amongst women worldwide, contributing to approximately 25% of all cancer incidences [1]. Despite significant advances in the early detection and development of effective therapeutic intervention [2], BC continues to be the second leading cause of cancer-associated mortality in women worldwide [3, 4]. According to the World Health Organization (WHO), an estimated 1.3 million women are newly diagnosed with BC each year [5]. Clinically, it is grouped into three molecular subtypes, including hormone receptor-positive (HR+) [estrogen receptor alpha-positive (ER+) and/or progesterone receptor (PR+)], human epidermal growth factor receptor 2 positive (HER2+), and triple-negative breast cancers (TNBC) [6]. Notably, different BC subtypes exhibit distinct clinicopathological and molecular profiles, and shows heterogeneous responses to different treatments, which includes surgery, radiotherapy, chemotherapeutics, hormonotherapy, and targeted therapy [7, 8]. Accumulating evidence suggest that aerobic glycolysis is a crucial metabolic adaptation of cancer cells. Thus, targeting the function of the glucose metabolism might be a promising therapeutic strategy for BC.
Regardless of abundant oxygen availability, tumor cells predominantly utilize glycolysis for energy production mechanisms and have higher rates of glycolysis. This aerobic glycolysis phenotype is referred to as the Warburg effect, first described by the German biochemist Otto Warburg in 1920 [9]. Aerobic glycolysis is a scientifically recognized hallmark of cancer cell metabolism and targeting it may provide possible drug target to the strategies for cancer therapy [10]. Lapachol, a PKM2 inhibitor, has been reported to block glycolysis in melanoma cells, ultimately reducing ATP levels and inhibiting cell proliferation [11]. Sainan Li et al. also reported that genistein can inhibit aerobic glycolysis of HCC (hepatocellular carcinoma) cells, and inactivate the expression of HIF-1a to down-regulate GLUT1 and HKII [12].Moreover, the mechanisms underlying the Warburg effect are anfractuous and intimately connected, including glycolytic enzymes, transcription factors, and signaling cascades [13]. The Warburg effect has been documented in different types of solid tumors, including BC [10]. Recently, the relationship between BC and aerobic glycolysis has been intensively followed. Here, we review the implication of glycolytic enzymes, together with oncogenic signaling pathways and transcription factors in BC to evoke new possible therapeutic ideas for BC.
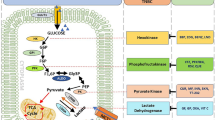
Key enzymes of glycolysis as potential targets for breast cancer
The accelerated rates of glycolysis in tumor cells are predominantly due to the overexpression or enhanced activity of key glycolytic enzymes [14]. Accumulating evidence suggested cancer-specific roles of key glycolytic enzymes as potential anti-tumor therapeutic strategies. The key enzymes of the glycolytic pathway include hexokinase (HK), phosphofructokinase (PFK) and pyruvate kinase (PK) [14] (Fig. 1). Further, we review the relevance of targeting crucial glycolytic pathways enzymes and glucose transporters (GLUTs) as anti-cancer strategies for BC. Besides, important miRNAs regulating the expression of glycolytic genes in BC (Table 1) will also be elaborated and targeting miRNA might provide a means for breast cancer treatment.

Schematic diagram of glycolysis, and three key enzymes: hexokinase(HK2), phosphofructokinase (PFK) and pyruvate kinase (PK)
Hexokinase
Significance of HK
HK is the first rate-limiting enzyme in the glycolytic pathway that catalyzes the phosphorylation of glucose by adenosine triphosphate (ATP) to glucose-6-phosphate [15]. In Mammals, HK exists as four isoenzymes including HKI, HKII, HKIII, and HKIV, encoded by the separate single-copy gene, each located on a different chromosome [16]. In humans, HKIV exhibit tissue-specific expression in the pancreas and liver and is more sensitive to glucose than other hexoses and is designated as glucokinase, while, HKIII is allosterically inhibited by the product, glucose-6-phosphate at physiologic concentrations. HKII is highly expressed in all mammalian tissues, and is recognized as a “housekeeping enzyme”. It is also the predominant HK isoform in many cell types [17]. Previous studies showed that HKII boosts tumor glycolysis, progression and metastasis, and elevated expression levels of HKII have been found in many cancers, including BC [18, 19]. Subsequent studies in BC revealed that HK-specific activity was 13-fold higher in BC tissues compared to normal breast tissues [20].
Effects of HK on breast cancer
Previously, increased hexokinase activity was reported to be correlated with overexpression of HKII in malignant tumor cell including human breast cancers (BCs) [19]. Using immunohistochemistry, Brown et al. demonstrated a significantly higher positive expression of HKII (79%) with untreated primary BCs [21]. A relatively smaller fraction of HKII is induced by HIF-1α and is known to promote proliferation, progression, and clinical recurrence of BC through increased glycolysis. Furthermore, HKII immunoreactivity was significantly associated with the histological grade of BC and the immunoreactivity of HIF-1α and Ki-67LI [22]. Besides, the mitochondrial membrane-bound HKII can promote glycolysis and inhibit cancer cell apoptosis in cancer. The major mechanisms that allow the tumor to continue metabolizing glucose to evade product inhibition and preferentially obtain newly synthesized ATP to phosphorylate glucose [23].
HK, as a therapeutic target for BC, has been extensively studied. Genistein-27 (Gen-27), a newly synthesized isoflavonoid, induces suppression of glycolysis and mitochondrial apoptosis through inhibition of mitochondrial localization and expression of HKII in MDA-MB-231 BC cell. In addition, Gen-27 could reduce the tumor volume by about 35% and exhibit low toxicity in major organs in vivo in BC mice model [24]. Moreover, microRNA is a class of short non-coding RNA sequences that regulates the gene expression at post-transcriptional, and it also represents a critical regulator of aerobic glycolysis in BC [25]. miR-155 inhibits socs1 and activates STAT3 in BC cells, where STAT3 is a transcriptional activator of HKII. Conversely, research evidence also revealed that miR-155 can up-regulate the HKII expression in breast tumor cell through targeting C/EBPb to suppress miR-143, thereby increasing the rate of glycolysis in ZR-75-30 cells [26]. Concurrently, Jiang et al. reported that miR-143 can mediate post-transcriptional regulation of HKII protein expression in BC cells. There was a negative correlation between miR-143 expression and HKII protein expression, and down-regulation of HKII decreases proliferation and survival of MDA-MB-231 cells [26]. Overall, HKII plays a critical role in the glycolytic pathway and is a putative candidate target in the treatment of BC.
Phosphofructokinase
Significance of PFK
6-Phosphate fructose-1-kinase (PFK-1) is the second rate-limiting enzyme in glycolysis, which catalyzes the conversion of fructose 6-phosphate to fructose 1, 6-bisphosphate and adenosine diphosphate (ADP). It is allosterically inhibited by ATP concentrations, phosphoenolpyruvate (PEP) and activated by fructose-2,6-bisphosphate (F26BP) [27]. The 6-phosphofructo 2-kinase/fructose 2 and 6-bisphosphatase (PfKfb) are bifunctional enzymes and are encoded by four different (PfKfb1-4) genes and can stimulate the expression level of F26BP [28]. Interestingly, PFKFB3 is significantly involved in tumor glycolysis, growth, and metastasis, and is closely regulated by HIF-1α, AKT, and PTEN [29].
Effects of PFK on breast cancer
BC cells express elevated expression levels of PFKFB3 [30]. Besides, increased activity of PFKFB3 enzyme is associated with estradiol activation of estrogen receptors, which promote the growth of HER-2-positive BC cells. Consistently, overexpression of PFKFB3 is also correlated with the increased expression of HER2 and poor progression-free survival (PFS) and distant metastatic-free survival (DMFS), and it also consistent with poor overall survival (OS) in patients with BC [30, 31]. Owing to increased kinase activity, PFKFB3 has been recognized as the major contributor to the high glycolytic activity observed in transformed cells; however, it may also translocate to the nucleus to regulate the activity of cyclin-dependent kinase (Cdks) [32]. Moreover, overexpression of PFKFB3 has also been associated with increased expression of VEGFα, which eventually leads to increased angiogenesis and distant metastasis in BC [31].
p27 predominantly inhibits the G1-to-S-phase transition and increases apoptosis. By inhibiting the phosphorylation of P27 through cyclin/cyclin-dependent kinase-1 (cdk1), PFKFB3 inhibition can act as both anti-proliferative and pro-apoptotic in HeLa cells [33]. Similarly, Peng et al. revealed that inhibition of PFKFB3 could also reduce the expression level of pAKT to promote the expression of p27 in BC cells. In addition, down-regulation of PFKFB3 subsequently decreased breast cancer cell (MDA-MB-231 and MDA-MB-468) proliferation, migration and invasion [31]. The miRNAs act as critical tumor suppressors or promoters in different molecular subtypes of BC as confirmed by enormous studies. Ge et al. indicated that miR-206 combined with the 3′-untranslated region (UTR) of PFKFB3 mRNA can reduce PFKFB3 protein expression in MCF-7, T47D, and SUM159 cell lines, and suppress the proliferation and migration of BC cells [34]. Breast carcinoma cells, however, also express PFKFB4 RNA and other isozymes [35]. In a recent study, Dasgupta et al. revealed that PFKFB4 regulates the oncogenic steroid receptor coactivator-3 (SRC-3) transcription through phosphorylation of SRC-3 at serine 857, and then, the activated SRC-3 drives the glucose flux to pentose phosphate pathway (PPP) in BC. The PFKFB–SRC-3 interaction also mediates the purine synthesis, causing BC cells proliferation and metastasis [36, 37]. Taken together, studies suggest that targeting the PFKFB 3 or PFKFB 4 might be a therapeutically valuable strategy in BC.
Pyruvate kinase
Significance of PK
PK is the final and rate-limiting enzyme in the glycolytic pathway and essentially contributes to aerobic glycolysis and provides a selective growth advantage for tumorigenesis. PK catalyzes the conversion of phosphoenolpyruvate (PEP) to pyruvate and ADP to ATP [38]. PK comprises four isoforms including liver-type PK (PKL), red blood cell PK (PKR), and PK muscle isozymes M1 and M2 (PKM1 and PKM2, respectively). PK isoforms are encoded by two genes, PKLR and PKM [39]. PKL is primarily expressed in liver, kidney, intestine, and pancreas, whereas PKR is exclusively expressed in erythrocytes. PKM1 and PKM2 are alternatively spliced products of mutually exclusive exons of the PKM gene [40]. Unlike the constitutively expressed PKM1, isozyme PKM2 is tightly regulated and is up-regulated in many cancer types. The activity of PKM2 is controlled by its oligomerized state, numerous allosteric effectors, and post-translational modifications [41]. Interestingly, tyrosine phosphorylation of PKM2 leads to a paradoxical effect, where phosphorylation reduces its activity, and the reduced activity promotes enhanced glycolytic rate and production of lactate and increased cancer cell proliferation [42].
Effects of PK on breast cancer
In tumor microenvironment, PKM2 is universally expressed in malignant cancer cells [43]. Elevated expression of PKM2 is associated with poor clinical outcomes and prognosis in BC. Accumulating evidence also suggests that PKM2 is an independent predictor for BC and is associated with poor PFS and OS [43]. However, the expression of PKM2 was positively correlated with chemosensitivity of BC cells to chemotherapy drugs including, epirubicin (EPI) and 5-fluorouracil (5-FU) in vitro [43, 44]. Nevertheless, down-regulation of PKM2 expression reduced STAT3 and phospho-STAT3 (pSTAT3) expression, which results in the inhibition of gene transcription and suppression of breast tumor cell proliferation [45].
PKM2 is also diffusely involved in nonmetabolic transcriptional regulation [46]. Recently, in 2016, Huang et al. reported that the tandem zinc-finger protein tristetraprolin (TTP), a well-recognized mRNA decay protein directly interacts with PKM2 to regulate its own transcription. Furthermore, PKM2 inhibits TTP-mediated mRNA decay in invasive BC MDA-MB-231 cells [47]. Recent studies have indicated that miRNAs also target PKM2. Interestingly, it was shown that miRNA let-7a-5p down-regulates Stat3 to modulate aerobic glycolysis and proliferation of BC cells and stat3 can up-regulate hnRNP-A1, which is a crucial regulator of PKM2 transcription. It was also revealed that let‐7a‐5p/stat3/hnRNP‐A1/PKM2 forms a feedback loop to regulate PKM2 expression in BC [48]. Unsurprisingly, a study by Wen et al. suggested that miR-152 inhibits proliferation and angiogenesis in BC via suppression both β-catenin and PKM2. β-catenin, the downstream molecule of IGF-1, has also been implicated to play an important role in the regulation of cell proliferation [49]. Recently, increasing evidence showed that activation of miR-148a/152 contributes to inhibition of PKM2 and NF-κB p56 expression in TNBC cells. NF-κB p56 directly interacts with PKM2 to control the expression of EGR1, which can bind with the miRNA gene promoters at multiple binding sites to modulate the expression of both miR-148a and miR-152 [50]. Collectively, the studies point towards PKM2 as an attractive potential target for BC therapeutic intervention.
Glucose transporters
Importance of GLUTs
The human GLUT family comprises of 14 members, which have varying tissue expression profiles and substrate specificities [51]. Of which, class I facilitative glucose transporters, represented by GLUT 1–4 are the best characterized, and have high relevance to cancer. GLUT 1 is a constitutive glucose transporter and is highly expressed on the membrane of erythrocytes and accounts for 3–5% of total membrane protein [52, 51]. GLUT2 mainly transports glucose across the membrane in hepatocytes, intestinal, and renal epithelial cells [53, 54], while GLUT3 predominantly exhibits high affinity for glucose and transport capacity in neurons [55]. GLUT 4 is an insulin-sensitive glucose transporter expressed in insulin-sensitive tissues such as fat and muscle tissue to equilibrate blood glucose levels and play an important role in systemic glucose balance [56].
Effects of GLUTs on breast cancer
In BC, seven GLUTs including GLUT1–6 and 12 have been reported to be overexpressed [51]. Notably, elevated expression of GLUT-1 has been associated with higher grade and poorly differentiated tumors and correlated with high proliferation rates and aggressiveness in BC [57, 58]. In addition, studies have also demonstrated that GLUT4 mRNA and protein were expressed in tissues that are not considered to be insulin sensitive, including BC [59]. Furthermore, Pablo et al. indicated that inhibition of GLUT4 critically reduces basal glucose uptake and induces metabolic reprogramming in BC cells including MCF7 and MDA-MB-231, eventually inhibiting cell proliferation [60]. Like GLUT-1 and GLUT4, the remaining five GLUTs have been identified to be expressed in BC tissues; however, studies on GLUTs remain limited [51].
Interplay of signaling pathway and breast cancer
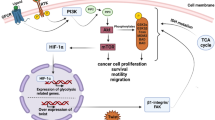
A hallmark difference between cancer cells and healthy counterparts is metabolic reprogramming, while aerobic glycolysis has been regarded as the major metabolic phenotype of cancer [61]. In fact, cancer cells can significantly increase glucose uptake and utilization and aerobic glycolysis can rapidly produce ATP and biomass synthesis, ultimately promoting tumorigenesis and metastasis [61, 62]. Simultaneously, a series of signaling pathways currently studied in cancer include JAK-STAT, PI3K/AKT, mTOR, MAPK, Wnt, AMPK, and Notch. However, PI3K/AKT, mTOR, MAPK, Wnt and AMPK signaling pathways are involved in regulating aerobic glycolysis of cancer cells [63,64,65,66,67,68]. Below we will review the study of these signaling pathways in aerobic glycolysis and BC, including PI3K/AKT, mTOR and AMPK. Figure 2 illustrates that the above three signaling pathways are inextricably interconnected to glycolytic enzymes and BC, and the components of mTOR.

Interaction of PI3K/AKT, mTOR and AMPK in key enzymes of glycolysis and breast cancer, and the components of mTOR
PI3K/AKT pathway
Given the relationship between glycolytic signaling pathways and cancer, studies related to glycolytic pathways in BC have recently gained much attention. In this context, the PI3K/AKT/mTOR signaling pathway has been known to play a significant role in inducing glucose metabolism in cancer cells. Numerous studies demonstrated that this pathway is altered in common cancers and regulate a variety of important cellular functions, including proliferation and apoptosis, glucose homeostasis, angiogenesis, and invasion and metastasis [64, 69]. The phosphatidylinositol-3-kinases (PI3Ks) are a family of signaling enzymes and these enzymes comprise three major classes of lipid kinases, I–III (Class I is further subcategorized into Ia and Ib), and a distantly related Class IV [70]. Akt, known as protein kinase B, is an essential serine/threonine protein kinase that is directly activated by PI3K and is essential for multiple cellular processes including cellular growth, metabolism, and survival [71]. Although Akt is the primary effector of PI3K, Akt-independent pathways were also activated by PI3K, including the TEC families of non-receptor tyrosine kinases, the Bruton tyrosine kinase (BTK), serum- and glucocorticoid-regulated kinases (SGKs), and regulators of small GTPases [72, 73].
PI3K-dependent AKT directly phosphorylates and activates phosphofructokinase 2 (PFK2) and enhances fructose-2, 6-diphosphate production, eventually activating the glycolytic rate-limiting enzyme phosphofructokinase1 (PFK1) [74, 75]. Recently, Melstrom et al. reported that up-regulation of the PI3K/Akt signaling pathway increases the expression of GLUT-1 [76]. Further studies showed the translocation of the GLUT-1 from the cytoplasm to the plasma membrane of other endocrine organs through the PI3K/Akt pathway [77]. GLUT1 is the major glucose transporter and is overexpressed in many tumor types, including BC [78]. Cuesta et al. also revealed that 17-estradiol activates Akt through phosphorylation of Ser473, which leads to the translocation of GLUT4 to the plasma membrane in an estrogen receptor (ER)-dependent manner, promoting ER-positive MCF-7 BC cells to increase glucose uptake [79]. Moreover, the lipid metabolism gene, FTO was reported to be overexpressed in BC, and contribute to up-regulating the activity of PK and HK and promote glycolysis. The underlying mechanism behind these effects may be attributed to the PI3K/AKT signaling pathway [80]. Thus, PI3K/AKT signaling pathway can up-regulate some enzyme expression in the glycolytic pathway.
Furthermore, activation of the PI3K/Akt pathway plays a critical role in multiple cellular functions including cell proliferation, differentiation, and intracellular trafficking, all of which are involved in cancer development [81]. Oncogenic mutations in PIK3CA, loss of PTEN activity, and mutations in AKT1 are the most common genetic alterations associated with abnormal activation of this pathway in BC and mammary tumorigenesis and angiogenesis [82]. PIK3CA is the most frequently altered in BC, and the somatic mutation rate is more than 10% and is significantly more frequently mutated than PTEN and AKT1 [83]. Although PIK3CA activating mutations are commonly identified in ER-positive and HER2-positive BC, they are rarely detected in basal cells of the breast. Activating mutations in PIK3CA confer resistance to BC HER2-targeted therapy through pathologic complete response (pCR) measurement. The pCR rate of trastuzumab and lapatinib combination therapy in HER2-positive BC patients with PIK3CA activating mutation decreased to 28.6%, compared with the wild-type PIK3CA (53.1%) [84]. Furthermore, in BC tissues and cells, aberrant expression of miR-106b and miR-93 facilitate BC development and progression. Mechanistically, miR-106b and miR-93 regulate the PI3K/Akt pathway through down-regulation of PTEN. Meanwhile, it has also been reported that microRNA-130b-3p exhibits characteristics similar to the above-mentioned miRNAs [85, 86]. HSPC159, a galectin-related protein, is aberrantly expressed in BC cells. Further analysis by Zheng et al. indicated that HSPC159 contributed to BC cell proliferation, migration, and invasion by activating the PI3K/AKT pathway, as well as epithelial–mesenchymal transition (EMT) and the F-actin polymerization process. Clinically, OS and disease-free survival (DFS) were significantly lower in patients with high expression of HSPC159 as compared to patients with low expression of HSPC159 [87]. Literature suggested that the PI3K pathway has a critical role in cell cycle G2/M transition, and potent PI3K inhibitor HS-106 can induce cell cycle arrest and apoptosis in BC cells [88]. Nevertheless, the PI3K/AKT pathway plays a prominent role in cancer cell cycle. Chun Wang et al. also demonstrated that beta-naphthoflavone (BNF, an agonist of aryl hydrocarbon receptor) inhibits PI3K/AKT signaling and induces G0/G1 cell cycle arrest and senescence, thereby contributing to inactivating cyclin D1/D3 and CDK4, as a promising anticancer drug for ER-positive MCF-7 BC cell [89]. Overall, the down-regulation of PI3K/AKT pathway may result in the inhibition of BC proliferation and migration.
mTOR pathway
The mammalian target of rapamycin (TOR) (mTOR) pathway regulates cell growth and organismal homeostasis by orchestrating both anabolic and catabolic processes with nutrient, energy, and oxygen availability and growth factors [90]. As a downstream effector of AKT, mTOR is encoded by the mTOR gene and interacts with protein binding partners to form the catalytic subunit of two distinct multi-subunit complexes: mTOR complex 1 (mTORC1) and 2 (mTORC2) [91,92,93].
mTORC1, as a multi-protein complex, is composed of the mTOR catalytic subunit and three associated proteins: Raptor, mLST8 (previously known as GβL) and PRAS40. mTORC1 regulates anabolic growth and proliferation by transforming glucose oxidative phosphorylation into glycolysis and increases the translation of transcription factor HIF1α; eventually, HIF1α drives the expression of several glycolytic enzymes, including PFK [94]. Meanwhile, mTORC1 also regulates the PPP, autophagy, mRNA translation, and lipid synthesis [95, 96]. The PPP remains an essential pathway for glucose metabolism in AML cells, exhibiting the high activity of mTORC1. Recent findings suggested that glucose-6-phosphate dehydrogenase (G6PD) is a major enzyme of PPP; however, higher mTORC1 activity induces the susceptibility to G6PD inhibition [97].
mTORC2 contains five subunits: mTOR, Rictor, mSin1, mLST8, and PRR5; subsequently, it can up-regulate the downstream targets of AGC kinase family, including Akt, serum/glucocorticoid-regulated kinase (SGK) and protein kinase C (PKC), via which mTORC2 promotes cell survival, glucose uptake, glycolysis, and lipogenesis [98,99,100,101,102]. c-myc is a crucial regulator of the Warburg effect, and mTORC2 makes glioblastoma (GBM) cells more dependent on glucose concentration by regulating intracellular c-myc levels [103, 104]. Recent studies demonstrate an intriguing Akt-independent role of mTORC2 in inducing metabolic reprogramming in GBM [105]. Moreover, Beg et al. reported that mTORC2 enhances Glut1-mediated glucose uptake and induces glycolytic metabolism by phosphorylated hydrophobic motif (HM) domain [106]. As one of the essential elements of mTORC2, Sin1 is associated with the development of CD4-CD8-double-negative (DN) stage thymocytes, and it is destroyed by the deficiency of Sin1 [107, 108]. Overexpression of PKM2 by Sin1-mTORC2 leads to a proliferation of DN thymocytes [108]. Increased PKM2 transcription is believed to be due to the activation of PPAR-γ, a known transcription activator of PKM2 [109]. Hence, mTORC1 and mTORC2 play key roles in up-regulating key enzymes in glycolysis.
mTOR signaling is often up-regulated in BC, and inhibition of mTOR is an effective strategy for the treatment of BC, including slowing down tumor growth and limiting the spread of cancer. Multiple mechanisms are attributed for the activation of mTOR, including up-regulation of the ErbB family receptors or alterations of PI3K signaling, and mutations and alterations in mTOR itself [110]. Moreover, clinical studies have revealed that the activation of mTOR signaling is associated with resistance to drug therapy in BC. The resistance to tamoxifen, an estrogen receptor targeting drug, has been correlated with the mTOR pathway through phosphorylation of ERa in Ser118, particularly in BC. A higher expression of HER2 (more than 15–20%) in BCs contributes to the overactivation of mTOR signaling, which has been implicated in conferring resistance to therapies that target HER2 in BC [111,112,113]. The non-carcinogenic susceptibility of HER2 + BC is significantly associated with the endoplasmic reticulum (ER)-associated degradation (ERAD) pathway. However, inhibition of ERAD has a critical role in impairing HER2+ cells through development of protein toxicity of ER induced by activated HER2–mTOR signaling [114]. Suppression of triple-negative breast cancers (TNBCs), which represent 10–20% of all BCs, have also been reported to be associated with mTOR inhibitors. For example, DHM25, a novel selective and covalent inhibitor of mTOR, strikingly suppresses the growth and metastasis in TNBC cells [115]. Consistently, Haiyu Zhang et al. also showed similar findings on mTOR inhibitors in TNBC; this mTOR inhibitor shows 77–99% inhibition of growth in the xenograft model of TNBC [116]. Thus, mTOR is a crucial anti-cancer target for BC.
AMPK pathway
The AMP-activated protein kinase (AMPK) is a highly conserved Ser/Thr kinase heterotrimeric complex, consisting of catalytic α and regulatory β, and γ subunits, that functions as a cellular energy homeostasis regulator and a sensor of cellular energy status [117, 118]. Under metabolic stress, AMPK works by conserving ATPs mechanisms through activation of catabolic pathways generating ATPs such as autophagy, as well as inhibiting ATP-consuming processes, including lipid biosynthesis, cell proliferation, and mTORC1-dependent protein biosynthesis [119,120,121].
It has been also reported previously that under hypoxia, AMPK mediates the activation of 6-phosphate fructose-2-kinase (PFK-2) to enhance glycolysis in myocardia. In addition, AMPK is also known to induce the translocation of the glucose transporter, GLUT4, in skeletal and cardiac muscle [122]. Thus, AMPK activation increases GLUT-4 translocation to improve myocardial glucose uptake in ischemia-induced hearts, which is independent of the PI3K pathway [123]. Moreover, AMPK has also been indicated to increase GLUT1 levels through multiple mechanisms [124]. Recently, Castro et al. revealed that overexpression of occludin (known as an NADH oxidase) significantly enhances the expression level of GLUT1 and GLUT4 in the blood–brain barrier (BBB) in an AMPK-mediated manner, thus, contributing to an impact on glucose uptake and ATP concentration [125]. Interestingly, the AMPK signal pathway is critical for tumor glucose metabolism and it might be beneficial to target AMPK activation as a therapeutic strategy.
As described earlier, the AMPK is a crucial integrator of the metabolism and signaling pathway, which can regulate the upstream kinase LKB1 with tumor suppressor roles [126]. Furthermore, the AMPK-related drug targets have also been studied in vitro and in vivo, to explore the association with the tumorigenesis and malignancy [127]. In TNBC, a significantly higher aberrant expression of AMPK was observed than non-triple-negative breast cancer (NTNBC) and AMPK expression also showed a potential relationship between the patients with BC and clinicopathologic characteristics including TNM stage and distant metastasis. Besides, positive expression of AMPK was consistent with shorter OS and DFS [128]. Moreover, the AMPK was confirmed to accelerate Skp2 S256 phosphorylation and promote cancer progression in BC mouse model, which was correlated with Akt activation and anti-EGF therapy responsiveness in patients with BC [129].
AMPK and Akt were identified to exhibit a reversible, double-negative feedback loop between matrix-attached and matrix-deprived conditions [130]. The Akt is known to be an upstream kinase for mTORC1 activation [91], and the AMPK-mediated mTORC1 inactivation may also alter the response to the growth factor, EGF and suppress the protein synthesis [131]. AMPK activation is positively correlated with TNBCs through targeted inhibition of the Akt/mTOR pathway [132]. Thus, AMPK-mediated oncogenesis and drug sensitivity may drive BC. The narciclasine, isolated from Narcissus L. bulbs, was identified to inhibit the cancer cells proliferation [133]. Evidently, the Narciclasine was confirmed to induce the tumor cell death in vitro and in vivo through AMPK–ULK1 pathways in TNBC, which predominantly stimulate the expression and phosphorylation of AMPK [134]. The lower phosphorylation of AMPK is most commonly associated with tumor cell growth and survival during metastasis of BC cell lines, MDA-MB-231 and MCF7 [130]. Furthermore, metformin was believed to exert its anticancer effect through activation of intracellular target AMPK in BC cells. Thus, a possible implicated underlying mechanism might be that by suppressing the expression of mTOR and pS6K, phosphorylation of AMPK inhibits the cell proliferation [135]. Moreover, inhibition of hexokinase-2 down-regulated the activity of Akt/mTOR/ HIF-1α, subsequently, reduced the phosphorylated of AMPK. Interestingly, overactivation of HIF-1α drives aerobic glycolysis to treat tamoxifen resistance in BC by regulating Akt/mTOR and/or AMPK signaling cascades [136]. Collectively, these researches provide further evidence that AMPK is most likely to influence BC progression and can be utilized as target therapy in the near future.
Transcription factors and breast cancer
Transcription factors play an important role in regulating the Warburg effect. Activated oncogenes (c-myc) can transactivate the glycolytic genes and increase aerobic glycolysis. On the contrary, tumor suppressor genes (p53) can hamper the transcription of glycolytic genes, inducing a down-regulation in aerobic glycolysis [10]. Simultaneously, the hypoxic environment in the tumor can up-regulate the glucose transporter and glycolytic enzymes by increasing HIF-1 levels [61]. Taken together, c-myc, p53, and HIF-1 significantly modulated the glycolytic enzymes in Table 2, and they have also been well studied in BC. Figure 3 indicated that P53, HIF-1 and c-myc regulate glycolysis in tumor.

P53, HIF-1 and c-myc regulate glycolysis in tumor
c-myc
c-myc is a short-lived oncogenic protein that is rapidly degraded by the ubiquitin (Ub)–proteasome system in nontransformed cells [137]. The protein product of the c-myc gene is an oncogenic pleiotropic transcription factor, a c-myc oncoprotein, which is involved in ribosome biogenesis, cellular metabolism, growth, proliferation, and apoptosis [138]. c-myc enhances ribosome biogenesis in nucleoli, to effectively promote cell growth and tumorigenesis [139]. In tumor cells, mutation of the c-myc gene itself or the induction of c-myc expression through upstream carcinogenic pathway can increase the function of c-myc [140]. c-myc enhances aerobic glycolysis by directly up-regulating the transcription of glycolytic genes and the expression of GLUTs, HK, phosphoglucose isomerase, PFK, glyceraldehyde-3-phosphate dehydrogenase, phosphoglycerate kinase (PGK), enolase, and LDHA [141]. c-myc can also up-regulate the expression of glutamine synthetase (GS), which converts glutamine into glutamate in the tricyclic acid(TCA) cycle [142].
A majority of patients with BCs are estrogen receptor (ER)-positive BC, and increased expression of c-myc is one of the earliest transcriptional responses to estrogen. Recently, Alison J Butt, identified HSPC111, as an estrogen-responsive c-myc target gene, that is predominantly localized in the nucleus and is correlated with an adverse outcome in patients with BC [143]. Furthermore, a study by Jain showed that inhibition of c-myc regulated translation and transcription of the glucose transporter GLUT1 inhibits cell growth in ER-negative mammary tumors [144]. Hence, targeting c-myc may become a therapeutic strategy for BC.
p53
p53 is the most extensively studied transcription factor, directly involved in metabolic reprogramming during malignant transformation and regulating crucial cellular functions including DNA repair, cell proliferation, autophagy, apoptosis, and senescence. TIGAR (TP53-induced glycolytic and apoptosis regulators) acts as a novel p53-inducible gene that alters the way cells utilize glucose. TIGAR shows similarity in the functional sequence shared by the functional protein, including the bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase (PFK-2/FBPase-2). TIGAR reduces the level of fructose 2,6-diphosphate (FRU-2, 6-P2) in cells similar to FBPase-2, thereby inhibiting the glycolytic pathway in tumor cells and promoting the glucose flux to pentose phosphate [145, 146]. Moreover, increased pentose phosphate shunt and enhanced NADPH production increase GSH levels required to clear ROS. And the p53 response elements located in the promoters of the HKII and phosphoglycerate mutase (PGM) genes demonstrated that p53 directly regulates the transcription of glycolytic genes. However, the expression of PGM and the glucose transporters (GLUT) 1, 3, and 4 was down-regulated by p53 [147, 148].
Through up-regulation of HKI or GLUT1, the activity of p53 can be significantly suppressed by increased glucose metabolism. p53 is the most extensively studied tumor suppressor gene that has been implicated in tumorigenesis [149]. Interestingly, studies indicated that p53-mediated inhibition of proliferation is crucial in cancer. Furthermore, p53 directly inhibit cell growth by up-regulating the expression of AMPK, TsC2, and sestrins, the members of the AMPK pathway [150]. The previous study also provides evidence that p53-dependent inactivation of mTOR and the p53 target of DRAM (damage-regulated autophagy modulator) promote autophagy [151, 152].
Noticeably, activation of p53 is significantly associated with tumor suppression, including BC. Previous studies have also suggested that binding of ERα to p53 inhibits transcriptional function, thereby inhibiting p53-mediated cell cycle arrest and apoptosis in ER-positive human BC cells [153, 154]. ERα can block inhibition of expression of p53 and its downstream targets, MDM2 and p21, thus activating the tumor suppressor and promoting the cancer cells to possibly undergo apoptosis [155]. Besides, MDM2 is induced by activation of p53 and MDM2 increases p53 poly-ubiquitination and degradation. SHARPIN (Shank-Interacting protein-like 1, SIPL1) can connect with MDM2 and promote its stability, thus, contributing to the inhibition of p53 and facilitating BC proliferation [156]. Moreover, miRNAs are also key players in the p53 signaling pathway, and p53 inhibits miR-191-5p, which can target SOX4 in BC. Inhibition of miR-191-5p expression leads to apoptosis in BC cell lines (MCF7 and ZR-75). Interestingly, the p53-miR-191-SOX4 axis is the regulator of apoptosis and drug resistance in BC [157]. P53 mutations occur in more than 50% of human cancers, with no exception to BC. Compared with many other cancer types, p53 mutations occur most frequently in 20–30% of all breast malignancies. Interestingly, mutant p53 consistently occur in ERα-negative or TNBC (approximately 80%). P53 mutations have also been correlated with worse OS and DFS in BC [158,159,160]. Furthermore, prolyl isomerase (Pin1) induces mutant p53 transcriptional program to promote invasiveness and enhances the suppression of the anti-metastatic factor p63 in BC [161]. Prior studies have suggested that wild-type p53 conceives an aspiring effect on the treatment response and prognosis in patients with BC [162]. Growing evidence suggests that wild-type p53 may also have a role in the regulation of tumor cell migration and invasion. Further studies indicated that wild-type p53 protein binds to a specific response element within the epithelial cell adhesion molecule (EpCAM) gene and down-regulates EpCAM (known as a transmembrane glycoprotein) expression, and this negative repression contributes to p53 control of invasion of BC [163]. Overall, p53, tumor suppressor plays a crucial role in cellular processes in BC.
HIF-1
HIF-1 is a heterodimeric transcription factor, comprised of two subunits, the constitutively expressed HIF-1β and the rate-limiting HIF-1α. The two important domain oxygen-dependent degradation (ODD) and transactivation domains (N-TAD and C-TAD) are located within HIF-1α protein molecule [164]. HIF-1α is a key regulator of glycolytic metabolism [10]. Expression of HIF-1α is induced in a hypoxic environment. Hypoxia is the most common characteristic of many solid tumors, and activation of HIF-1 transcription factors is the most recognized pathway-acquired mechanism by hypoxic cells in these tumors [165].
Activated HIF-1 regulates the transcription of multiple target genes. These genes are majorly involved in vital biological processes such as glucose metabolism, cell proliferation, metastasis, angiogenesis, and chemotherapy and radiation resistance [166]. By inducing the glycolytic pathway enzymes including HKII, PFK1, LDHA, aldolase, and GLUT-1 and-3, HIF-1 switches the glucose metabolism of hypoxic tumor cells to the glycolytic pathway and this metabolic switch causes a shift in energy production [166]. Furthermore, down-regulation of mitochondrial function can be achieved by transactivating genes such as pyruvate dehydrogenase kinase 1 (PDK1) and MAX interaction 1 (MXI1) through HIF [166]. MXI1, the negative regulator, is a member of the myc family, which can inactivate myc [167].
Various clinical studies have demonstrated the relationship between HIF-1α expression and survival in patients with BC. However, using immunohistochemistry, no significant high level of HIF-1a protein expression was observed in normal BC epithelial tissues [168]. HIF-1α was indicated to be an independent prognostic factor for DMFS, DFS and OS in patients with BC [169, 170]. Further, Yan et al. suggested that positive HIF-1α is associated with shorter recurrence-free survival (RFS) in patients with BC [171]. Particularly, as a direct transcriptional target of ERα, HIF-1α can compensate for the loss of ERα function; and its expression is significantly correlated with lower survival rates for endocrine therapy with ERα + BC [172]. Moreover, activation of HIF-1a promotes c-erbB2-mediated BC invasiveness, angiogenesis, and migration pathways [173]. Furthermore, Zhang indicated that HIF-1 directly activates HIF-dependent CD47 transcription, resulting in reduced phagocytosis of bone marrow-derived macrophages in BC cells, which eventually promotes cancer progression [174]. Hence, HIF-1 might be an excellent metabolic target for BC treatment.
Perspective
The present review addresses the function of glycolysis enzymes, together with associated signaling pathways, and transcription factors, which are essential for the energy metabolism in BC cells. At present, there are still many problems in targeting aerobic glycolysis as a cancer treatment. For instance, targeting key glycolysis enzymes for cellular metabolism has the mutation risk and targeting transcription factors could have many side effects, both in tumor and normal cells. Therefore, what kind of treatments are pursued by researchers may focus on exploiting these relevant targets of aerobic glycolysis in clinical practice for treatment of cancers and its related risk.
Conclusion
Hyperactivity of glycolysis is a hallmark of cancer metabolic reprogramming, known as the Warburg Effect, which is distinctively perceived by cancer cells [9]. Stable genetic or epigenetic alterations may constitutively up-regulate the aerobic glycolysis in malignant tumors like BC. Thus, this metabolic reprogramming provides a significant excuse for tumor proliferation and migration in cancer cells [175]. Collectively, the key enzymes of the aerobic glycolytic pathway (HK, PFK and PK), GLUTs, together with associated signaling pathways, PI3K/AKT, mTOR and AMP and transcription factors (c-myc, p53 and HIF-1) all play critical roles in tumor energy metabolism. Moreover, BC proliferation and cell survival are prudently regulated by aerobic glycolysis. Thus, the review of aerobic glycolysis in BC may evoke novel ideas for the BC treatment.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. https://doi.org/10.3322/caac.21262.
Yang ZY, Di MY, Yuan JQ, Shen WX, Zheng DY, Chen JZ, et al. The prognostic value of phosphorylated Akt in breast cancer: a systematic review. Sci Rep. 2015;5:7758. https://doi.org/10.1038/srep07758.
Li W, Hou JZ, Niu J, Xi ZQ, Ma C, Sun H, et al. Akt1 inhibition promotes breast cancer metastasis through EGFR-mediated beta-catenin nuclear accumulation. Cell Commun Signal. 2018;16(1):82. https://doi.org/10.1186/s12964-018-0295-1.
Yousefi M, Nosrati R, Salmaninejad A, Dehghani S, Shahryari A, Saberi A. Organ-specific metastasis of breast cancer: molecular and cellular mechanisms underlying lung metastasis. Cell Oncol (Dordr). 2018;41(2):123–40. https://doi.org/10.1007/s13402-018-0376-6.
Prager GW, Braga S, Bystricky B, Qvortrup C, Criscitiello C, Esin E, et al. Global cancer control: responding to the growing burden, rising costs and inequalities in access. ESMO Open. 2018;3(2):e000285. https://doi.org/10.1136/esmoopen-2017-000285.
Dey N, De P, Leyland-Jones B. PI3K-AKT-mTOR inhibitors in breast cancers: From tumor cell signaling to clinical trials. Pharmacol Ther. 2017;175:91–106. https://doi.org/10.1016/j.pharmthera.2017.02.037.
Tang Y, Wang Y, Kiani MF, Wang B. Classification, treatment strategy, and associated drug resistance in breast cancer. Clin Breast Cancer. 2016;16(5):335–43. https://doi.org/10.1016/j.clbc.2016.05.012.
Wu L, Yang X. Targeting the hippo pathway for breast cancer therapy. Cancers. 2018. https://doi.org/10.3390/cancers10110422.
Ganapathy-Kanniappan S. Taming tumor glycolysis and potential implications for immunotherapy. Front Oncol. 2017;7:36. https://doi.org/10.3389/fonc.2017.00036.
Li L, Liang Y, Kang L, Liu Y, Gao S, Chen S, et al. Transcriptional regulation of the Warburg effect in cancer by SIX1. Cancer Cell. 2018;33(3):368–85.e7. https://doi.org/10.1016/j.ccell.2018.01.010.
Shankar Babu M, Mahanta S, Lakhter AJ, Hato T, Paul S, Naidu SR. Lapachol inhibits glycolysis in cancer cells by targeting pyruvate kinase M2. PLoS ONE. 2018;13(2):e0191419. https://doi.org/10.1371/journal.pone.0191419.
Li S, Li J, Dai W, Zhang Q, Feng J, Wu L, et al. Genistein suppresses aerobic glycolysis and induces hepatocellular carcinoma cell death. Br J Cancer. 2017;117(10):1518–28. https://doi.org/10.1038/bjc.2017.323.
Garber K. Energy deregulation: licensing tumors to grow. Science. 2006;312(5777):1158–9. https://doi.org/10.1126/science.312.5777.1158.
Li XB, Gu JD, Zhou QH. Review of aerobic glycolysis and its key enzymes—new targets for lung cancer therapy. Thorac Cancer. 2015;6(1):17–24. https://doi.org/10.1111/1759-7714.12148.
Tan VP, Miyamoto S. HK2/hexokinase-II integrates glycolysis and autophagy to confer cellular protection. Autophagy. 2015;11(6):963–4. https://doi.org/10.1080/15548627.2015.1042195.
Criss WE. A review of isozymes in cancer. Can Res. 1971;31(11):1523–42.
Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, et al. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014;8(5):1461–74. https://doi.org/10.1016/j.celrep.2014.07.053.
Chen J, Yu Y, Li H, Hu Q, Chen X, He Y, et al. Long non-coding RNA PVT1 promotes tumor progression by regulating the miR-143/HK2 axis in gallbladder cancer. Mol Cancer. 2019;18(1):33. https://doi.org/10.1186/s12943-019-0947-9.
Yang T, Ren C, Qiao P, Han X, Wang L, Lv S, et al. PIM2-mediated phosphorylation of hexokinase 2 is critical for tumor growth and paclitaxel resistance in breast cancer. Oncogene. 2018;37(45):5997–6009. https://doi.org/10.1038/s41388-018-0386-x.
Hennipman A, Smits J, van Oirschot B, van Houwelingen JC, Rijksen G, Neyt JP, et al. Glycolytic enzymes in breast cancer, benign breast disease and normal breast tissue. Tumour Biol. 1987;8(5):251–63. https://doi.org/10.1159/000217529.
Brown RS, Goodman TM, Zasadny KR, Greenson JK, Wahl RL. Expression of hexokinase II and Glut-1 in untreated human breast cancer. Nucl Med Biol. 2002;29(4):443–53.
Sato-Tadano A, Suzuki T, Amari M, Takagi K, Miki Y, Tamaki K, et al. Hexokinase II in breast carcinoma: a potent prognostic factor associated with hypoxia-inducible factor-1alpha and Ki-67. Cancer Sci. 2013;104(10):1380–8. https://doi.org/10.1111/cas.12238.
Mathupala SP, Ko YH, Pedersen PL. Hexokinase-2 bound to mitochondria: cancer's stygian link to the "Warburg Effect" and a pivotal target for effective therapy. Semin Cancer Biol. 2009;19(1):17–24. https://doi.org/10.1016/j.semcancer.2008.11.006.
Tao L, Wei L, Liu Y, Ding Y, Liu X, Zhang X, et al. Gen-27, a newly synthesized flavonoid, inhibits glycolysis and induces cell apoptosis via suppression of hexokinase II in human breast cancer cells. Biochem Pharmacol. 2017;125:12–25. https://doi.org/10.1016/j.bcp.2016.11.001.
Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Can Res. 2006;66(17):8319–26. https://doi.org/10.1158/0008-5472.can-06-0410.
Jiang S, Zhang LF, Zhang HW, Hu S, Lu MH, Liang S, et al. A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J. 2012;31(8):1985–98. https://doi.org/10.1038/emboj.2012.45.
Okar DA, Manzano A, Navarro-Sabate A, Riera L, Bartrons R, Lange AJ. PFK-2/FBPase-2: maker and breaker of the essential biofactor fructose-2,6-bisphosphate. Trends Biochem Sci. 2001;26(1):30–5.
Kim SG, Manes NP, El-Maghrabi MR, Lee YH. Crystal structure of the hypoxia-inducible form of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB3): a possible new target for cancer therapy. J Biol Chem. 2006;281(5):2939–44. https://doi.org/10.1074/jbc.M511019200.
Panigrahy D, Singer S, Shen LQ, Butterfield CE, Freedman DA, Chen EJ, et al. PPARgamma ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J Clin Investig. 2002;110(7):923–32. https://doi.org/10.1172/jci15634.
O'Neal J, Clem A, Reynolds L, Dougherty S, Imbert-Fernandez Y, Telang S, et al. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) suppresses glucose metabolism and the growth of HER2+ breast cancer. Breast Cancer Res Treat. 2016;160(1):29–40. https://doi.org/10.1007/s10549-016-3968-8.
Peng F, Li Q, Sun JY, Luo Y, Chen M, Bao Y. PFKFB3 is involved in breast cancer proliferation, migration, invasion and angiogenesis. Int J Oncol. 2018;52(3):945–54. https://doi.org/10.3892/ijo.2018.4257.
Yalcin A, Clem BF, Imbert-Fernandez Y, Ozcan SC, Peker S, O'Neal J, et al. 6-Phosphofructo-2-kinase (PFKFB3) promotes cell cycle progression and suppresses apoptosis via Cdk1-mediated phosphorylation of p27. Cell Death Dis. 2014;5:e1337. https://doi.org/10.1038/cddis.2014.292.
Yalcin A, Clem BF, Simmons A, Lane A, Nelson K, Clem AL, et al. Nuclear targeting of 6-phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases. J Biol Chem. 2009;284(36):24223–32. https://doi.org/10.1074/jbc.M109.016816.
Ge X, Lyu P, Cao Z, Li J, Guo G, Xia W, et al. Overexpression of miR-206 suppresses glycolysis, proliferation and migration in breast cancer cells via PFKFB3 targeting. Biochem Biophys Res Commun. 2015;463(4):1115–21. https://doi.org/10.1016/j.bbrc.2015.06.068.
Minchenko OH, Ochiai A, Opentanova IL, Ogura T, Minchenko DO, Caro J, et al. Overexpression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-4 in the human breast and colon malignant tumors. Biochimie. 2005;87(11):1005–100. https://doi.org/10.1016/j.biochi.2005.04.007.
Dasgupta S, Rajapakshe K, Zhu B, Nikolai BC, Yi P, Putluri N, et al. Metabolic enzyme PFKFB4 activates transcriptional coactivator SRC-3 to drive breast cancer. Nature. 2018;556(7700):249–54. https://doi.org/10.1038/s41586-018-0018-1.
Goncalves MD, Cantley LC. A glycolysis outsider steps into the cancer spotlight. Cell Metab. 2018;28(1):3–4. https://doi.org/10.1016/j.cmet.2018.06.017.
Harris I, McCracken S, Mak TW. PKM2: a gatekeeper between growth and survival. Cell Res. 2012;22(3):447–9. https://doi.org/10.1038/cr.2011.203.
Dayton TL, Jacks T, Vander Heiden MG. PKM2, cancer metabolism, and the road ahead. EMBO Rep. 2016;17(12):1721–30. https://doi.org/10.15252/embr.201643300.
Mendez-Lucas A, Li X, Hu J, Che L, Song X, Jia J, et al. Glucose catabolism in liver tumors induced by c-MYC can be sustained by various PKM1/PKM2 ratios and pyruvate kinase activities. Can Res. 2017;77(16):4355–64. https://doi.org/10.1158/0008-5472.can-17-0498.
Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci USA. 2010;107(5):1894–9. https://doi.org/10.1073/pnas.0914845107.
Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008;452(7184):181–6. https://doi.org/10.1038/nature06667.
Lin Y, Lv F, Liu F, Guo X, Fan Y, Gu F, et al. High expression of pyruvate kinase M2 is associated with chemosensitivity to epirubicin and 5-fluorouracil in breast cancer. J Cancer. 2015;6(11):1130–9. https://doi.org/10.7150/jca.12719.
Benesch C, Schneider C, Voelker HU, Kapp M, Caffier H, Krockenberger M, et al. The clinicopathological and prognostic relevance of pyruvate kinase M2 and pAkt expression in breast cancer. Anticancer Res. 2010;30(5):1689–94.
Guan M, Tong Y, Guan M, Liu X, Wang M, Niu R, et al. Lapatinib inhibits breast cancer cell proliferation by influencing PKM2 expression. Technol Cancer Res Treat. 2018;17:1533034617749418. https://doi.org/10.1177/1533034617749418.
Lee J, Kim HK, Han YM, Kim J. Pyruvate kinase isozyme type M2 (PKM2) interacts and cooperates with Oct-4 in regulating transcription. Int J Biochem Cell Biol. 2008;40(5):1043–54. https://doi.org/10.1016/j.biocel.2007.11.009.
Huang L, Yu Z, Zhang Z, Ma W, Song S, Huang G. Interaction with pyruvate kinase M2 destabilizes tristetraprolin by proteasome degradation and regulates cell proliferation in breast cancer. Sci Rep. 2016;6:22449. https://doi.org/10.1038/srep22449.
Yao A, Xiang Y, Si YR, Fan LJ, Li JP, Li H, et al. PKM2 promotes glucose metabolism through a let-7a-5p/Stat3/hnRNP-A1 regulatory feedback loop in breast cancer cells. J Cell Biochem. 2019;120(4):6542–54. https://doi.org/10.1002/jcb.27947.
Wen YY, Liu WT, Sun HR, Ge X, Shi ZM, Wang M, et al. IGF-1-mediated PKM2/beta-catenin/miR-152 regulatory circuit in breast cancer. Sci Rep. 2017;7(1):15897. https://doi.org/10.1038/s41598-017-15607-y.
Xu Q, Liu LZ, Yin Y, He J, Li Q, Qian X, et al. Regulatory circuit of PKM2/NF-kappaB/miR-148a/152-modulated tumor angiogenesis and cancer progression. Oncogene. 2015;34(43):5482–93. https://doi.org/10.1038/onc.2015.6.
Barron CC, Bilan PJ, Tsakiridis T, Tsiani E. Facilitative glucose transporters: Implications for cancer detection, prognosis and treatment. Metabolism. 2016;65(2):124–39. https://doi.org/10.1016/j.metabol.2015.10.007.
Olson AL, Pessin JE. Structure, function, and regulation of the mammalian facilitative glucose transporter gene family. Annu Rev Nutr. 1996;16:235–56. https://doi.org/10.1146/annurev.nu.16.070196.001315.
Fukumoto H, Seino S, Imura H, Seino Y, Eddy RL, Fukushima Y, et al. Sequence, tissue distribution, and chromosomal localization of mRNA encoding a human glucose transporter-like protein. Proc Natl Acad Sci USA. 1988;85(15):5434–8. https://doi.org/10.1073/pnas.85.15.5434.
Thorens B, Cheng ZQ, Brown D, Lodish HF. Liver glucose transporter: a basolateral protein in hepatocytes and intestine and kidney cells. Am J Physiol. 1990;259(6 Pt 1):C279–C285285. https://doi.org/10.1152/ajpcell.1990.259.2.C279.
Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med. 2013;34(2–3):121–38. https://doi.org/10.1016/j.mam.2012.07.001.
Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab. 2007;5(4):237–52. https://doi.org/10.1016/j.cmet.2007.03.006.
Younes M, Brown RW, Mody DR, Fernandez L, Laucirica R. GLUT1 expression in human breast carcinoma: correlation with known prognostic markers. Anticancer Res. 1995;15(6b):2895–8.
Brown RS, Wahl RL. Overexpression of Glut-1 glucose transporter in human breast cancer. An immunohistochemical study. Cancer. 1993;72(10):2979–85. https://doi.org/10.1002/1097-0142(19931115)72:10%3c2979:aid-cncr2820721020%3e3.0.co;2-x.
Godoy A, Ulloa V, Rodriguez F, Reinicke K, Yanez AJ, Garcia Mde L, et al. Differential subcellular distribution of glucose transporters GLUT1-6 and GLUT9 in human cancer: ultrastructural localization of GLUT1 and GLUT5 in breast tumor tissues. J Cell Physiol. 2006;207(3):614–27. https://doi.org/10.1002/jcp.20606.
Garrido P, Osorio FG, Moran J, Cabello E, Alonso A, Freije JM, et al. Loss of GLUT4 induces metabolic reprogramming and impairs viability of breast cancer cells. J Cell Physiol. 2015;230(1):191–8. https://doi.org/10.1002/jcp.24698.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. https://doi.org/10.1016/j.cell.2011.02.013.
Jia D, Lu M, Jung KH, Park JH, Yu L, Onuchic JN, et al. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc Natl Acad Sci USA. 2019;116(9):3909–18. https://doi.org/10.1073/pnas.1816391116.
Irey EA, Lassiter CM, Brady NJ, Chuntova P, Wang Y, Knutson TP, et al. JAK/STAT inhibition in macrophages promotes therapeutic resistance by inducing expression of protumorigenic factors. Proc Natl Acad Sci USA. 2019;116(25):12442–51. https://doi.org/10.1073/pnas.1816410116.
Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–19. https://doi.org/10.1038/nrg1879.
Yoshida GJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res. 2015;34:111. https://doi.org/10.1186/s13046-015-0221-y.
Cai CF, Ye GD, Shen DY, Zhang W, Chen ML, Chen XX, et al. Chibby suppresses aerobic glycolysis and proliferation of nasopharyngeal carcinoma via the Wnt/beta-catenin-Lin28/let7-PDK1 cascade. J Exp Clin Cancer Res. 2018;37(1):104. https://doi.org/10.1186/s13046-018-0769-4.
Han J, Zhang L, Guo H, Wysham WZ, Roque DR, Willson AK, et al. Glucose promotes cell proliferation, glucose uptake and invasion in endometrial cancer cells via AMPK/mTOR/S6 and MAPK signaling. Gynecol Oncol. 2015;138(3):668–75. https://doi.org/10.1016/j.ygyno.2015.06.036.
Hibdon ES, Razumilava N, Keeley TM, Wong G, Solanki S, Shah YM, et al. Notch and mTOR signaling pathways promote human gastric cancer cell proliferation. Neoplasia. 2019;21(7):702–12. https://doi.org/10.1016/j.neo.2019.05.002.
Guerrero-Zotano A, Mayer IA, Arteaga CL. PI3K/AKT/mTOR: role in breast cancer progression, drug resistance, and treatment. Cancer Metastasis Rev. 2016;35(4):515–24. https://doi.org/10.1007/s10555-016-9637-x.
Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–62. https://doi.org/10.1038/nrc2664.
Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14(5):381–95.
Qiu Y, Kung HJ. Signaling network of the Btk family kinases. Oncogene. 2000;19(49):5651–61. https://doi.org/10.1038/sj.onc.1203958.
Cain RJ, Ridley AJ. Phosphoinositide 3-kinases in cell migration. Biol Cell. 2009;101(1):13–29. https://doi.org/10.1042/bc20080079.
Lee JH, Liu R, Li J, Wang Y, Tan L, Li XJ, et al. EGFR-phosphorylated platelet isoform of phosphofructokinase 1 promotes PI3K activation. Mol Cell. 2018;70(2):197–210.e7. https://doi.org/10.1016/j.molcel.2018.03.018.
Novellasdemunt L, Tato I, Navarro-Sabate A, Ruiz-Meana M, Mendez-Lucas A, Perales JC, et al. Akt-dependent activation of the heart 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB2) isoenzyme by amino acids. J Biol Chem. 2013;288(15):10640–51. https://doi.org/10.1074/jbc.M113.455998.
Melstrom LG, Salabat MR, Ding XZ, Milam BM, Strouch M, Pelling JC, et al. Apigenin inhibits the GLUT-1 glucose transporter and the phosphoinositide 3-kinase/Akt pathway in human pancreatic cancer cells. Pancreas. 2008;37(4):426–31. https://doi.org/10.1097/MPA.0b013e3181735ccb.
Samih N, Hovsepian S, Aouani A, Lombardo D, Fayet G. Glut-1 translocation in FRTL-5 thyroid cells: role of phosphatidylinositol 3-kinase and N-glycosylation. Endocrinology. 2000;141(11):4146–55. https://doi.org/10.1210/endo.141.11.7793.
Kang SS, Chun YK, Hur MH, Lee HK, Kim YJ, Hong SR, et al. Clinical significance of glucose transporter 1 (GLUT1) expression in human breast carcinoma. Jpn J Cancer Res. 2002;93(10):1123–8. https://doi.org/10.1111/j.1349-7006.2002.tb01214.x.
Garrido P, Moran J, Alonso A, Gonzalez S, Gonzalez C. 17beta-estradiol activates glucose uptake via GLUT4 translocation and PI3K/Akt signaling pathway in MCF-7 cells. Endocrinology. 2013;154(6):1979–89. https://doi.org/10.1210/en.2012-1558.
Liu Y, Wang R, Zhang L, Li J, Lou K, Shi B. The lipid metabolism gene FTO influences breast cancer cell energy metabolism via the PI3K/AKT signaling pathway. Oncol Lett. 2017;13(6):4685–90. https://doi.org/10.3892/ol.2017.6038.
Lopez-Knowles E, O'Toole SA, McNeil CM, Millar EK, Qiu MR, Crea P, et al. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int J Cancer. 2010;126(5):1121–31. https://doi.org/10.1002/ijc.24831.
Castaneda CA, Cortes-Funes H, Gomez HL, Ciruelos EM. The phosphatidyl inositol 3-kinase/AKT signaling pathway in breast cancer. Cancer Metastasis Rev. 2010;29(4):751–9. https://doi.org/10.1007/s10555-010-9261-0.
Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. https://doi.org/10.1038/nature11412.
Majewski IJ, Nuciforo P, Mittempergher L, Bosma AJ, Eidtmann H, Holmes E, et al. PIK3CA mutations are associated with decreased benefit to neoadjuvant human epidermal growth factor receptor 2-targeted therapies in breast cancer. J Clin Oncol. 2015;33(12):1334–9. https://doi.org/10.1200/jco.2014.55.2158.
Li N, Miao Y, Shan Y, Liu B, Li Y, Zhao L, et al. MiR-106b and miR-93 regulate cell progression by suppression of PTEN via PI3K/Akt pathway in breast cancer. Cell Death Dis. 2017;8(5):e2796. https://doi.org/10.1038/cddis.2017.119.
Miao Y, Zheng W, Li N, Su Z, Zhao L, Zhou H, et al. MicroRNA-130b targets PTEN to mediate drug resistance and proliferation of breast cancer cells via the PI3K/Akt signaling pathway. Sci Rep. 2017;7:41942. https://doi.org/10.1038/srep41942.
Zheng J, Zhang M, Zhang L, Ding X, Li W, Lu S. HSPC159 promotes proliferation and metastasis by inducing epithelial-mesenchymal transition and activating the PI3K/Akt pathway in breast cancer. Cancer Sci. 2018;109(7):2153–63. https://doi.org/10.1111/cas.13631.
Li GY, Jung KH, Lee H, Son MK, Seo J, Hong SW, et al. A novel imidazopyridine derivative, HS-106, induces apoptosis of breast cancer cells and represses angiogenesis by targeting the PI3K/mTOR pathway. Cancer Lett. 2013;329(1):59–67. https://doi.org/10.1016/j.canlet.2012.10.013.
Wang C, Xu CX, Bu Y, Bottum KM, Tischkau SA. Beta-naphthoflavone (DB06732) mediates estrogen receptor-positive breast cancer cell cycle arrest through AhR-dependent regulation of PI3K/AKT and MAPK/ERK signaling. Carcinogenesis. 2014;35(3):703–13. https://doi.org/10.1093/carcin/bgt356.
Leung EY, Kim JE, Askarian-Amiri M, Joseph WR, McKeage MJ, Baguley BC. Hormone resistance in two MCF-7 breast cancer cell Lines is associated with reduced mTOR signaling, decreased glycolysis, and increased sensitivity to cytotoxic drugs. Front Oncol. 2014;4:221. https://doi.org/10.3389/fonc.2014.00221.
Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. https://doi.org/10.1038/nrc839.
Baretic D, Williams RL. The structural basis for mTOR function. Semin Cell Dev Biol. 2014;36:91–101. https://doi.org/10.1016/j.semcdb.2014.09.024.
Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110(2):177–89.
Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39(2):171–83. https://doi.org/10.1016/j.molcel.2010.06.022.
Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol. 2013;15(6):555–64. https://doi.org/10.1038/ncb2763.
Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. 2014;15(3):155–62. https://doi.org/10.1038/nrm3757.
Poulain L, Sujobert P, Zylbersztejn F, Barreau S, Stuani L, Lambert M, et al. High mTORC1 activity drives glycolysis addiction and sensitivity to G6PD inhibition in acute myeloid leukemia cells. Leukemia. 2017;31(11):2326–35. https://doi.org/10.1038/leu.2017.81.
Cybulski N, Hall MN. TOR complex 2: a signaling pathway of its own. Trends Biochem Sci. 2009;34(12):620–7. https://doi.org/10.1016/j.tibs.2009.09.004.
Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem J. 2008;416(3):375–85. https://doi.org/10.1042/bj20081668.
Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008;27(14):1919–31. https://doi.org/10.1038/emboj.2008.119.
Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14(14):1296–302. https://doi.org/10.1016/j.cub.2004.06.054.
Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–74. https://doi.org/10.1016/j.cell.2007.06.009.
Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009;15(21):6479–83. https://doi.org/10.1158/1078-0432.ccr-09-0889.
Masui K, Cavenee WK, Mischel PS. mTORC2 dictates Warburg effect and drug resistance. Cell Cycle. 2014;13(7):1053–4. https://doi.org/10.4161/cc.28377.
Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013;18(5):726–39. https://doi.org/10.1016/j.cmet.2013.09.013.
Beg M, Abdullah N, Thowfeik FS, Altorki NK, McGraw TE. Distinct Akt phosphorylation states are required for insulin regulated Glut4 and Glut1-mediated glucose uptake. eLife. 2017. https://doi.og/10.7554/eLife.26896.
Liu P, Gan W, Inuzuka H, Lazorchak AS, Gao D, Arojo O, et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat Cell Biol. 2013;15(11):1340–50. https://doi.org/10.1038/ncb2860.
Ouyang X, Han Y, Qu G, Li M, Wu N, Liu H, et al. Metabolic regulation of T cell development by Sin1-mTORC2 is mediated by pyruvate kinase M2. J Mol Cell Biol. 2019;11(2):93–106. https://doi.org/10.1093/jmcb/mjy065.
Panasyuk G, Espeillac C, Chauvin C, Pradelli LA, Horie Y, Suzuki A, et al. PPARgamma contributes to PKM2 and HK2 expression in fatty liver. Nat Commun. 2012;3:672. https://doi.org/10.1038/ncomms1667.
Hare SH, Harvey AJ. mTOR function and therapeutic targeting in breast cancer. Am J Cancer Res. 2017;7(3):383–404.
Viedma-Rodriguez R, Baiza-Gutman L, Salamanca-Gomez F, Diaz-Zaragoza M, Martinez-Hernandez G, Ruiz Esparza-Garrido R, et al. Mechanisms associated with resistance to tamoxifen in estrogen receptor-positive breast cancer (review). Oncol Rep. 2014;32(1):3–15. https://doi.org/10.3892/or.2014.3190.
Schettini F, Buono G, Cardalesi C, Desideri I, De Placido S, Del Mastro L. Hormone receptor/human epidermal growth factor receptor 2-positive breast cancer: Where we are now and where we are going. Cancer Treat Rev. 2016;46:20–6. https://doi.org/10.1016/j.ctrv.2016.03.012.
Margariti N, Fox SB, Bottini A, Generali D. "Overcoming breast cancer drug resistance with mTOR inhibitors". Could it be a myth or a real possibility in the short-term future? Breast Cancer Res Treat. 2011;128(3):599-606. https://doi.org/10.1007/s10549-010-0986-9.
Singh N, Joshi R, Komurov K. HER2-mTOR signaling-driven breast cancer cells require ER-associated degradation to survive. Sci Signal. 2015;8(378):ra52. https://doi.org/10.1126/scisignal.aaa6922.
Fouque A, Delalande O, Jean M, Castellano R, Josselin E, Malleter M, et al. A novel covalent mTOR inhibitor, DHM25, shows in vivo antitumor activity against triple-negative breast cancer cells. J Med Chem. 2015;58(16):6559–733. https://doi.org/10.1021/acs.jmedchem.5b00991.
Zhang H, Cohen AL, Krishnakumar S, Wapnir IL, Veeriah S, Deng G, et al. Patient-derived xenografts of triple-negative breast cancer reproduce molecular features of patient tumors and respond to mTOR inhibition. Breast Cancer Res. 2014;16(2):R36. https://doi.org/10.1186/bcr3640.
Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–62. https://doi.org/10.1038/nrm3311.
Hardie DG. AMPK–sensing energy while talking to other signaling pathways. Cell Metab. 2014;20(6):939–52. https://doi.org/10.1016/j.cmet.2014.09.013.
Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–61. https://doi.org/10.1126/science.1196371.
Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18(3):283–93. https://doi.org/10.1016/j.molcel.2005.03.027.
Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–90.
Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, et al. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000;10(20):1247–55.
Russell RR 3rd, Bergeron R, Shulman GI, Young LH. Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am J Physiol. 1999;277(2):H643–H649649. https://doi.org/10.1152/ajpheart.1999.277.2.H643.
Barnes K, Ingram JC, Porras OH, Barros LF, Hudson ER, Fryer LG, et al. Activation of GLUT1 by metabolic and osmotic stress: potential involvement of AMP-activated protein kinase (AMPK). J Cell Sci. 2002;115(Pt 11):2433–42.
Castro V, Skowronska M, Lombardi J, He J, Seth N, Velichkovska M, et al. Occludin regulates glucose uptake and ATP production in pericytes by influencing AMP-activated protein kinase activity. J Cereb Blood Flow Metab. 2018;38(2):317–32. https://doi.org/10.1177/0271678x17720816.
Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004;101(10):3329–35. https://doi.org/10.1073/pnas.0308061100.
Cao W, Li J, Hao Q, Vadgama JV, Wu Y. AMP-activated protein kinase: a potential therapeutic target for triple-negative breast cancer. Breast Cancer Res. 2019;21(1):29. https://doi.org/10.1186/s13058-019-1107-2.
Huang X, Li X, Xie X, Ye F, Chen B, Song C, et al. High expressions of LDHA and AMPK as prognostic biomarkers for breast cancer. Breast. 2016;30:39–46. https://doi.org/10.1016/j.breast.2016.08.014.
Han F, Li CF, Cai Z, Zhang X, Jin G, Zhang WN, et al. The critical role of AMPK in driving Akt activation under stress, tumorigenesis and drug resistance. Nat Commun. 2018;9(1):4728. https://doi.org/10.1038/s41467-018-07188-9.
Saha M, Kumar S, Bukhari S, Balaji SA, Kumar P, Hindupur SK, et al. AMPK-Akt double-negative feedback loop in breast cancer cells regulates their adaptation to matrix deprivation. Can Res. 2018;78(6):1497–510. https://doi.org/10.1158/0008-5472.can-17-2090.
Ng TL, Leprivier G, Robertson MD, Chow C, Martin MJ, Laderoute KR, et al. The AMPK stress response pathway mediates anoikis resistance through inhibition of mTOR and suppression of protein synthesis. Cell Death Differ. 2012;19(3):501–10. https://doi.org/10.1038/cdd.2011.119.
Montero JC, Esparis-Ogando A, Re-Louhau MF, Seoane S, Abad M, Calero R, et al. Active kinase profiling, genetic and pharmacological data define mTOR as an important common target in triple-negative breast cancer. Oncogene. 2014;33(2):148–56. https://doi.org/10.1038/onc.2012.572.
Furst R. Narciclasine—an amaryllidaceae alkaloid with potent antitumor and anti-inflammatory properties. Planta Med. 2016;82(16):1389–94. https://doi.org/10.1055/s-0042-115034.
Cao C, Huang W, Zhang N, Wu F, Xu T, Pan X, et al. Narciclasine induces autophagy-dependent apoptosis in triple-negative breast cancer cells by regulating the AMPK-ULK1 axis. Cell Prolif. 2018;51(6):e12518. https://doi.org/10.1111/cpr.12518.
Cai H, Zhang Y, Han TK, Everett RS, Thakker DR. Cation-selective transporters are critical to the AMPK-mediated antiproliferative effects of metformin in human breast cancer cells. Int J Cancer. 2016;138(9):2281–92. https://doi.org/10.1002/ijc.29965.
Woo YM, Shin Y, Lee EJ, Lee S, Jeong SH, Kong HK, et al. Inhibition of aerobic glycolysis represses Akt/mTOR/HIF-1alpha axis and restores tamoxifen sensitivity in antiestrogen-resistant breast cancer cells. PLoS ONE. 2015;10(7):e0132285. https://doi.org/10.1371/journal.pone.0132285.
Farrell AS, Sears RC. MYC degradation. Cold Spring Harbor perspectives in medicine. 2014. https://doi.org/10.1101/cshperspect.a014365.
Sun XX, He X, Yin L, Komada M, Sears RC, Dai MS. The nucleolar ubiquitin-specific protease USP36 deubiquitinates and stabilizes c-Myc. Proc Natl Acad Sci USA. 2015;112(12):3734–9. https://doi.org/10.1073/pnas.1411713112.
van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer. 2010;10(4):301–9. https://doi.org/10.1038/nrc2819.
Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002;2(10):764–76. https://doi.org/10.1038/nrc904.
Hsieh AL, Walton ZE, Altman BJ, Stine ZE, Dang CV. MYC and metabolism on the path to cancer. Semin Cell Dev Biol. 2015;43:11–21. https://doi.org/10.1016/j.semcdb.2015.08.003.
Bott AJ, Peng IC, Fan Y, Faubert B, Zhao L, Li J, et al. Oncogenic Myc induces expression of glutamine synthetase through promoter demethylation. Cell Metab. 2015;22(6):1068–77. https://doi.org/10.1016/j.cmet.2015.09.025.
Butt AJ, Sergio CM, Inman CK, Anderson LR, McNeil CM, Russell AJ, et al. The estrogen and c-Myc target gene HSPC111 is over-expressed in breast cancer and associated with poor patient outcome. Breast Cancer Res. 2008;10(2):R28. https://doi.org/10.1186/bcr1985.
Jain S, Wang X, Chang CC, Ibarra-Drendall C, Wang H, Zhang Q, et al. Src inhibition blocks c-Myc translation and glucose metabolism to prevent the development of breast cancer. Can Res. 2015;75(22):4863–75. https://doi.org/10.1158/0008-5472.can-14-2345.
Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8(4):275–83. https://doi.org/10.1038/nrm2147.
Green DR, Chipuk JE. p53 and metabolism: inside the TIGAR. Cell. 2006;126(1):30–2. https://doi.org/10.1016/j.cell.2006.06.032.
Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9(10):691–700. https://doi.org/10.1038/nrc2715.
Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008;10(5):611–8. https://doi.org/10.1038/ncb1724.
Zhao Y, Coloff JL, Ferguson EC, Jacobs SR, Cui K, Rathmell JC. Glucose metabolism attenuates p53 and Puma-dependent cell death upon growth factor deprivation. J Biol Chem. 2008;283(52):36344–53. https://doi.org/10.1074/jbc.M803580200.
Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134(3):451–60. https://doi.org/10.1016/j.cell.2008.06.028.
Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA. 2005;102(23):8204–9. https://doi.org/10.1073/pnas.0502857102.
Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126(1):121–34. https://doi.org/10.1016/j.cell.2006.05.034.
Liu W, Ip MM, Podgorsak MB, Das GM. Disruption of estrogen receptor alpha-p53 interaction in breast tumors: a novel mechanism underlying the anti-tumor effect of radiation therapy. Breast Cancer Res Treat. 2009;115(1):43–50. https://doi.org/10.1007/s10549-008-0044-z.
Liu W, Konduri SD, Bansal S, Nayak BK, Rajasekaran SA, Karuppayil SM, et al. Estrogen receptor-alpha binds p53 tumor suppressor protein directly and represses its function. J Biol Chem. 2006;281(15):9837–40. https://doi.org/10.1074/jbc.C600001200.
Berger CE, Qian Y, Liu G, Chen H, Chen X. p53, a target of estrogen receptor (ER) alpha, modulates DNA damage-induced growth suppression in ER-positive breast cancer cells. J Biol Chem. 2012;287(36):30117–27. https://doi.org/10.1074/jbc.M112.367326.
Yang H, Yu S, Wang W, Li X, Hou Y, Liu Z, et al. SHARPIN facilitates p53 degradation in breast cancer cells. Neoplasia. 2017;19(2):84–92. https://doi.org/10.1016/j.neo.2016.12.002.
Sharma S, Nagpal N, Ghosh PC, Kulshreshtha R. P53-miR-191-SOX4 regulatory loop affects apoptosis in breast cancer. RNA. 2017;23(8):1237–46. https://doi.org/10.1261/rna.060657.117.
Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA. 2005;102(38):13550–5. https://doi.org/10.1073/pnas.0506230102.
Bailey ST, Shin H, Westerling T, Liu XS, Brown M. Estrogen receptor prevents p53-dependent apoptosis in breast cancer. Proc Natl Acad Sci USA. 2012;109(44):18060–5. https://doi.org/10.1073/pnas.1018858109.
Synnott NC, Bauer MR, Madden S, Murray A, Klinger R, O'Donovan N, et al. Mutant p53 as a therapeutic target for the treatment of triple-negative breast cancer: preclinical investigation with the anti-p53 drug, PK11007. Cancer Lett. 2018;414:99–106. https://doi.org/10.1016/j.canlet.2017.09.053.
Girardini JE, Napoli M, Piazza S, Rustighi A, Marotta C, Radaelli E, et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell. 2011;20(1):79–91. https://doi.org/10.1016/j.ccr.2011.06.004.
Bergh J, Norberg T, Sjogren S, Lindgren A, Holmberg L. Complete sequencing of the p53 gene provides prognostic information in breast cancer patients, particularly in relation to adjuvant systemic therapy and radiotherapy. Nat Med. 1995;1(10):1029–34.
Sankpal NV, Willman MW, Fleming TP, Mayfield JD, Gillanders WE. Transcriptional repression of epithelial cell adhesion molecule contributes to p53 control of breast cancer invasion. Can Res. 2009;69(3):753–7. https://doi.org/10.1158/0008-5472.can-08-2708.
Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92(12):5510–4. https://doi.org/10.1073/pnas.92.12.5510.
Masoud GN, Li W. HIF-1alpha pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin Bs. 2015;5(5):378–89. https://doi.org/10.1016/j.apsb.2015.05.007.
Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8(9):705–13. https://doi.org/10.1038/nrc2468.
Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11(5):407–20. https://doi.org/10.1016/j.ccr.2007.04.001.
Kimbro KS, Simons JW. Hypoxia-inducible factor-1 in human breast and prostate cancer. Endocr Relat Cancer. 2006;13(3):739–49. https://doi.org/10.1677/erc.1.00728.
Trastour C, Benizri E, Ettore F, Ramaioli A, Chamorey E, Pouyssegur J, et al. HIF-1alpha and CA IX staining in invasive breast carcinomas: prognosis and treatment outcome. Int J Cancer. 2007;120(7):1451–8. https://doi.org/10.1002/ijc.22436.
Schindl M, Schoppmann SF, Samonigg H, Hausmaninger H, Kwasny W, Gnant M, et al. Overexpression of hypoxia-inducible factor 1alpha is associated with an unfavorable prognosis in lymph node-positive breast cancer. Clin Cancer Res. 2002;8(6):1831–7.
Yan M, Rayoo M, Takano EA, Fox SB. BRCA1 tumours correlate with a HIF-1alpha phenotype and have a poor prognosis through modulation of hydroxylase enzyme profile expression. Br J Cancer. 2009;101(7):1168–74. https://doi.org/10.1038/sj.bjc.6605287.
Yang J, AlTahan A, Jones DT, Buffa FM, Bridges E, Interiano RB, et al. Estrogen receptor-alpha directly regulates the hypoxia-inducible factor 1 pathway associated with antiestrogen response in breast cancer. Proc Natl Acad Sci USA. 2015;112(49):15172–7. https://doi.org/10.1073/pnas.1422015112.
Konecny GE, Meng YG, Untch M, Wang HJ, Bauerfeind I, Epstein M, et al. Association between HER-2/neu and vascular endothelial growth factor expression predicts clinical outcome in primary breast cancer patients. Clin Cancer Res. 2004;10(5):1706–16.
Zhang H, Lu H, Xiang L, Bullen JW, Zhang C, Samanta D, et al. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc Natl Acad Sci USA. 2015;112(45):E6215–E62236223. https://doi.org/10.1073/pnas.1520032112.
Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):891–9. https://doi.org/10.1038/nrc1478.
Funding
This work was supported by grants from the Union Project of Luzhou City and the Southwest Medical University (Nos. 14JC0144, 2013LZLY-J40).
Author information
Authors and Affiliations
Contributions
JBW, SZF and JJ were responsible for review of the literature. ZXW wrote the manuscript. QJZ drew the figures. JBW, SZF and JJ designed the study and contributed to the valuable discussion and revision of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
For this type of study, informed consent is not required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wu, Z., Wu, J., Zhao, Q. et al. Emerging roles of aerobic glycolysis in breast cancer. Clin Transl Oncol 22, 631–646 (2020). https://doi.org/10.1007/s12094-019-02187-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-019-02187-8