
Abstract
Background
Breast cancer (BC) is the most common type of cancer in women and the second cause of cancer-related mortality world-wide. The majority of BC-related deaths is due to metastasis. Bone, lung, brain and liver are the primary target sites of BC metastasis. The clinical implications and mechanisms underlying bone metastasis have been reviewed before. Given the fact that BC lung metastasis (BCLM) usually produces symptoms only after the lungs have been vastly occupied with metastatic tumor masses, it is of paramount importance for diagnostic and prognostic, as well as therapeutic purposes to comprehend the molecular and cellular mechanisms underlying BCLM. Here, we review current insights into the organ-specificity of BC metastasis, including the role of cancer stem cells in triggering BC spread, the traveling of tumor cells in the blood stream and their migration across endothelial barriers, their adaptation to the lung microenvironment and the initiation of metastatic colonization within the lung.
Conclusions
Detailed understanding of the mechanisms underlying BCLM will shed a new light on the identification of novel molecular targets to impede daunting pulmonary metastases in patients with breast cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Breast cancer is the most common type of cancer in women with over 252,000 new diagnoses per year in the USA alone, accounting for about 15% of all new cancer cases. It is the second cause of cancer-related death in women and responsible for more than 40,000 deaths in the USA per year, representing 6.8% of all cancer-related deaths [1]. The majority of breast cancer-related deaths is due to the occurrence of metastases. Up to 50% of the patients with breast cancer will develop metastases, often decades after the primary tumor is diagnosed, which seriously hampers disease management and eradication. Usually, metastatic breast cancer has a poor prognosis, with an overall 5-year survival rate of about 27% [2,3,4,5].
It has been reported that several factors may influence the prognosis of breast cancer. Tumors at higher stages and with higher grades have an unfavorable prognosis and exhibit higher recurrence risks [6]. Tumor size and lymph node status represent other prognostic factors, i.e., tumors with a size ≥ 5 cm or positive axillary or internal mammary lymph nodes are more likely to metastasize to distant organs [6]. It has also been reported that patients under 35 years of age tend to have more aggressive and higher grade tumors and to exhibit higher recurrence risks [6], indicative of a relatively poor overall prognosis. In addition, various histological and molecular characteristics have been found to exhibit prognostic and/or therapeutic relevance. For instance, triple-negative breast cancers (i.e., estrogen receptor (ER)-, progesterone receptor (PR)- and human epidermal growth factor receptor 2 (HER2)-negative) are at the highest risk of metastasis formation and they do not respond to hormone therapy and/or trastuzumab [7]. Conversely, it has been found that ER+/HER2−/Ki67low breast cancer subtypes exhibit relatively high median overall survival rates [8].
Overt metastasis, which contributes to about 90% of cancer-related deaths, is an inevitable manifestation of most high grade human tumors [9]. Metastasis formation is usually parsed into interrelated steps, beginning with local invasion, intravasation and subsequent dissemination via circulatory and/or lymphatic systems [10, 11]. Next, the disseminated tumor cells may extravasate into the parenchyma of distant organs, which finally leads to the formation of micro-metastases and subsequently the formation of full-fledged secondary tumors [12]. Different primary tumors have a proclivity to metastasize to distinct organs. For instance, bones and lungs are the most frequent sites of breast cancer metastasis [13, 14]. Usually, lung metastases elicit little or no symptoms until the lungs are vastly replaced by metastatic tumor masses. This implies that lung metastasis is a substantial problem in patients with breast cancer and, therefore, heralds serious consideration. Current therapeutic options mostly include chemotherapy, radiotherapy and/or surgical resection, but their long-term impact on survival (especially when more than one pulmonary metastasis exists) is still dismal and not effective enough to prevent relapse [15, 16]. It is anticipated that a further in-depth insight into the molecular and cellular mechanisms underlying breast cancer lung metastasis (BCLM) will provide novel leads for the design of (targeted) therapeutic interventions. Here we review known and emerging concepts of BCLM, which may be instrumental for the identification of new molecular targets for impending lung metastases in breast cancer patients.
2 Organ-specific metastasis of breast cancer
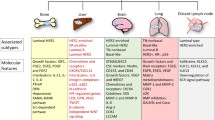
The pattern of cancer spread by which primary tumors tend to metastasize to distinct organs is usually referred to as organ-specific metastasis [17]. Based on organ-selective evolution, metastatic localization does not occur random but rather at preferred sites under the control of a multitude of micro-environmental, cellular and molecular factors. In the past, two prevailing hypotheses have been put forward to explain these metastatic patterns [18]. One hypothesis states that circulation patterns determine which organs are likely to host circulating tumor cells (CTCs), and that these CTCs may be mechanically arrested in the capillary networks they encounter [19]. However, although circulation patterns do affect non-random metastatic organ-specificity, the fact that some organs receive similar blood volumes but show different metastatic patterns indicate that this so-called “mechanical arrest” may not fully explain the organ-specific patterns of metastases that are observed in most human cancers [9, 20,21,22,23]. Along this line, breast cancers have a propensity to metastasize to bone (50–65%), lung (17%), brain (16%) and liver (6%), while metastases to other organs such as spleen, kidney or uterus, are relatively rare [13]. These observations led Stephen Paget to suggest an alternative hypothesis, i.e., the “seed-and-soil hypothesis” [24]. According to this hypothesis, cancer cells or “seeds” shedding from primary tumors and welcoming microenvironments or “soils” are both necessary for organ-specific metastasis formation [18]. This hypothesis implicates that metastasis formation depends on intrinsic properties of distinct cancer types, the architecture of the vascular or lymphatic system, the role of chemo-attractants, and the ability of cancer cells to interact with host cells and/or other micro-environmental factors present within distinct tissues/organs [9]. It appears, however, that the two processes mentioned are not mutually exclusive, but rather act cooperatively to generate organ-specific metastases, i.e., both mechanical processes and molecular characteristics of tumor cells and their interactions with target tissues may determine the propensity of disseminated cancer cells to spread to specific organs [25].
Breast tumors can be classified into basal-like, Her2-amplified, luminal A, luminal B and normal breast-like subtypes based on both their histologic and intrinsic genomic/transcriptomic profiles [26]. Retrospective analyses have indicated that the histologic/molecular characteristics of breast cancers affect their organ-specific metastasis patterns, i.e., lung relapses were found to be most abundant among the basal-like subtype, whereas bone relapses were found to be primarily associated with the luminal subtypes [27, 28]. In recent years, attention has been focused on the molecular determinants that play critical roles in the organ-specific metastasis of breast cancer cells and the cross-talks between various cell types and stromal factors that underlie this organ-specificity. These cross-talks have been found to involve cell-cell and cell-extracellular matrix (ECM) interactions, signaling molecules and adhesion molecules, cytokines and growth factors, as well as their mediators, inhibitors and receptors [4, 29]. Also, various genes have been identified that are specifically and temporally expressed by breast cancer cells and its microenvironmental components and that they, as such, may confer prerequisites for organ-specific metastasis. The genes that underlie the various steps of metastasis have been classified into distinct classes, commonly denoted as metastasis initiation, metastasis progression and metastasis virulence genes (Table 1) [29]. The metastasis initiation genes comprise a subset of genes that initiate tumor outgrowth and angiogenesis and, thereby, facilitate the entrance of tumor cells into the circulation. The metastasis progression genes are those that carry out stimulatory functions in primary tumors and at (pre)metastatic sites. The metastasis virulence genes are those that confer advantages to tumor cells at distant sites rather than at primary sites and, as such, enable metastatic tumor cells to establish macro-metastases. These genes act to promote metastasis formation after locally aggressive tumor cell populations have been established [29, 61]. Although individual genes have been related to organ-specific metastasis, they should be considered holistically, i.e., as components of a complex, dynamic and interactive network that governs complex mechanisms underlying the tissue-tropism of metastatic breast cancer cells [3, 62]. Minn et al. [35] identified a lung metastasis signature in the metastatic breast cancer cell line LM2, consisting of 48 up-regulated and 47 down-regulated genes. Several of these genes were found to be strongly associated with tumor invasiveness and aggressiveness, rather than with functions in the lung microenvironment. In the following sections, recently identified gene expression profiles as well as cellular and molecular mechanisms underlying breast cancer dissemination and metastasis to the lung are described.
3 Metastasis initiating and promoting factors: role of epithelial-mesenchymal transition and cancer stem cells
Metastasis initiating genes are defined as genes that facilitate tumor outgrowth, local invasion, intravasation and dissemination to distant sites. Human anterior gradient 2 (AGR2), one of the many targets of the estrogen receptor (ER), was first discovered as a gene that is over-expressed in ER-positive breast cancer cells [63]. It has subsequently been found that AGR2 may promote BCLM through the (de)regulation of tumor cell adhesion and dissemination. AGR2-induced lung metastases were identified in a rat breast cancer model over-expressing AGR2, apparently through both blood-borne and lymphatic routes, thereby providing in vivo evidence that AGR2 overexpression alone may be sufficient to induce metastases [34]. Next to AGR2, many other genes and molecules have been found to be involved in the initiation of breast cancer metastasis, most of which affect epithelial-mesenchymal transition (EMT) and the formation of cancer stem cells (CSCs). Therefore, in this section, we will focus on cellular and molecular mechanisms underlying EMT in breast cancer and discuss how CSCs may contribute to its metastasis.
3.1 Role of epithelial-mesenchymal transition
EMT is an evolutionary-conserved process that can be activated in cancer cells and is linked to their dissociation from the primary tumor mass and their intravasation into blood vessels [64]. Tumor cells simulate this developmental process to transdifferentiate into mesenchymal-like cells during which they acquire motile and invasive capacities and detach from epithelial cell sheets [65]. As such, EMT represents a critical step in the initiation of metastasis. Several genes are known to be involved in the signaling pathways that stimulate EMT [66], including the Wnt, Notch and TGF-β pathways, of which the TGF-β pathway has been studied most extensively [67,68,69,70,71]. Binding of TGF-β to its receptor results in phosphorylation and activation of Smad2 and Smad3, which can subsequently form trimers with Smad4 and translocate to the nucleus to regulate the expression of TGF-β target genes and, by doing so, affect EMT [72]. Recently, several novel factors have been identified that participate in TGF-β-induced EMT through various mechanisms. Some of these factors have been found to affect the expression of proteins involved in the TGF-β pathway, thereby facilitating EMT. In breast cancer cells it has for instance been found that HOXB7 and SIX1 can directly bind to the promoters of TGF-β2 and the TGF-β type I receptor (TβRI), respectively [30]. It has also been found that SIX1 can shift the TGF-β function in breast cancer cells from pro-metastatic to tumor-suppressive states [73]. Nuclear receptor NR4A1, on the other hand, has been found to stimulate TGF-β signaling through facilitating the degradation of Smad7, whereas inflammation-induced NR4A1 has been found to trigger TGF-β-induced breast cancer cell invasion [74].
It has been frequently reported that the zinc-finger transcription factors Snail1 and Snail2, the zinc finger E-box binding proteins (ZEB)2 and ZEB1, and Twist act as master regulators of EMT [75]. These transcription factors can repress the epithelial marker E-cadherin, which results in a lack of adherent junctions between tumor cells and, thus, their detachment from epithelial cell sheets [76]. Loss of E-cadherin also results in the accumulation and translocation of β-catenin to the nucleus, where it can bind to LEF/TCF and, thereby, activate the expression of mesenchymal markers including N-cadherin, α-SMA and vimentin (Fig. 1) [77, 78]. It has been suggested that microRNA miR-374a, which directly targets multiple regulators of the β-catenin signaling cascade including WIF1 and PTEN, may act as an important stimulator of EMT both in vitro and in vivo [79]. MiR-374a up-regulation in breast cancer has been found to be associated with a poor prognosis and the occurrence of lung metastases, and it has been suggested that its targeting may represent a therapeutic option for metastatic breast cancer [79]. It has also been shown that increased matrix stiffness may induce miR-18a expression which, in turn, may directly or indirectly decrease the expression of PTEN through the targeting of HOXA9 [63]. Metadherin (MTDH) has been found to drive EMT by promoting the expression of TWIST1, a transcription factor that is known to be critical for the induction of cancer cell stemness and metastasis [80]. KLF17 may act as a negative regulator of EMT and down-regulation of KLF17 has been found to promote EMT and metastasis of breast cancer cells to the lung [46]. Transcription factor Fos-related antigen-1 (Fra-1) represents another factor that is required for the motility and invasion of breast cancer cells. Several in vivo and in vitro studies have indicated that Fra-1 may impede the expression and sub-cellular localization of E-cadherin [81], positively regulate the expression of several pro-EMT microRNAs [82] as well as other pro-EMT factors [83] and, thereby, promote the EMT program. These findings suggest that Fra-1 may serve as a tool for the stratification of patients with breast cancer and for predicting disease recurrence. In line with these studies, Desmet et al. [84] assessed the potential of Fra-1 as a novel in vivo therapeutic target for lung metastasis using rodent xenograft models and found that RNAi-induced silencing of Fra-1 strongly suppressed the occurrence of BCLM. Furthermore, by using a synthetic lethal drug screen, they found that pharmacologic blockade of the adenosine receptor A2B (ADORA2B) is toxic to breast tumor cells expressing Fra-1, suggesting that Fra-1 expression predicts responsiveness to targeted breast cancer metastasis therapy [84]. It has recently been reported that human X-box binding protein-1 (XBP1) may act as a novel EMT regulator, i.e., XBP1 over-expression was found to be associated with cancer progression through Snail induction and EMT stimulation [33]. Over-expression and accumulation of lysyl oxidase-like 2 (LOXL2) in the endoplasmic reticulum (ER) results in its interaction with HSPA5, which leads to activation of the inositol requiring enzyme 1α (IRE1)-XBP1 signaling pathway. This activation has been found to lead to up-regulation of several EMT transcription factors, including Snail1, Snail2, ZEB2 and TCF3, all of which are direct transcriptional targets of XBP1 [31].

Molecular mechanisms of EMT and MET underlying BCLM. In the primary tumor environment various signaling pathways, including the TGF-β and Wnt pathways, and different growth factor/receptor tyrosine kinases can induce EMT and generate CSCs. These pathways mainly act through activation of three transcription factors, Snail, ZEB and Twist, which are the major modulators of EMT. Accumulation of β-catenin results in its translocation to the nucleus where it can bind to TCF/LEF and, thereby, induce the expression of mesenchymal factors. CSCs exhibit less adhesion and more motile capacities and, in addition, exhibit resistance to apoptotic signals, which allows them to travel to distant sites. They also abundantly express CXCR4 and CD44 on their surfaces, which enables them to target organs in which their ligands, CXCL12/SDF-1, hyaluronan and OPN, are abundantly present. In the lung parenchyma, TGF-β-dependent up-regulation of ID1 results in MET induction through antagonizing TWIST, which facilitates metastatic colonization
It has also been reported that microRNAs may regulate EMT. As such, miR-374a up-regulation has already been mentioned above as being indicative of a poor prognosis in breast cancer. Also, miR-115 and miR-9 have been found to be up-regulated in invasive breast cancers [85]. Specifically, miR-9 has been found to promote breast cancer progression through targeting CDH1 [86]. MiR-206 has been reported to inhibit Smad2 and Neuropilin-1 (NRP1) in ER-positive breast cancers [87], whereas miR-155 has been found to target CCAAT/enhancer binding protein β (C/EBPβ) and to stimulate TGF-β-mediated EMT in breast cancer cells as well [88]. Mir-122 acts as a tumor suppressor and has been found to inhibit breast cancer development through targeting IGF1R and, by doing so, (de)regulating the PI3K/Akt pathway [89]. It has also been reported that miR-26a may suppress tumor growth and metastasis by down-regulating MTDH in triple-negative breast cancers [90]. Also, miR-320a has been found to inhibit breast cancer-associated lung cancer metastasis by targeting MTDH [91]. The role of miR-200 in breast cancer progression and lung metastasis has so far remained controversial, but some studies indicate that miR-200 can suppress EMT through targeting ZEB1/2 [92]. Others have found that miR-22 may promote the metastatic potential of breast cancer cells through direct targeting of the TET (ten-eleven translocation) family of methylcytosine dioxygenases, thereby inhibiting demethylation of the miR-200 promoter [93]. Other studies indicate, however, that miR-200 expression confers lung-tropic properties to breast cancer cells, i.e., in addition to targeting E-cadherin, miR-200 may promote lung colonization by suppressing SEC23A (Sec23 homolog A) which, in turn, regulates the secretion of IGF binding protein 4 (IGFBP4) and tubule-interstitial nephritis antigen-like 1 (TINAGL1) [94]. The miRNAs that have so far been reported to be involved in BCLM are listed in Table 2.
3.2 Role of cancer stem cells
As stated above, EMT is linked to the acquisition of stem cell-like characteristics [97]. EMT was first proposed as a mechanism through which cancer cells acquire stemness properties and disseminate to form tumors at secondary sites [23, 98]. The cancer stem cell (CSC) hypothesis posits that most primary tumors harbor rare subpopulations of stem-like cells with self-renewal and differentiation properties, from which metastatic cells are derived and seed new tumors at distant sites [99]. According to Al-Hajj et al. [100] and Ginestier et al. [101], the identification of breast cancer stem cells (BCSCs) from pleural effusions, primary breast tumors and breast cancer cell lines is mainly based on ALDH+/CD44+/CD24−/low phenotypes. These BCSCs are postulated to be highly capable of establishing tumors both in vitro and in vivo [102,103,104] and to differentiate into cell populations that comprise the entire tumor and, thus, to recapitulate tumor heterogeneity [105, 106]. It has for instance been shown that as few as 20 BCSCs injected into NOD/SCID mice can give rise to tumors that exhibit phenotypic heterogeneities similar to those of the tumors from which they were derived, while tens of thousands of non stem-like cells from a given tumor were found to fail to give rise to tumors in these mice [101]. Interestingly, a positive correlation has been noted between the proportion of BCSCs present in breast tumors and a poor prognosis of the corresponding patients, confirming a role of BCSCs in the metastatic proficiency of these tumors, as well as their resistance to radiation therapy and chemotherapy [107]. The chemo-resistance observed in CSCs results from the quiescent nature of these cells and their arrest in the G0 phase of the cell cycle, thereby negating the effectiveness of chemotherapeutic agents that target proliferating cells [108]. For BCSCs, it has been reported that a high expression of the breast cancer resistance protein-1 (BCRP1), which is a cell surface efflux pump that expels chemotherapeutic agents out of cells, and a high expression of ALDH, which metabolizes chemotherapeutic agents such as cyclophosphamide and some anti-apoptotic molecules, contribute to chemo-resistance. As a result of chemotherapy, however, new subpopulations of refractory tumor cells may develop through clonal expansion [108,109,110,111].
The contribution of CSCs in organ-specific metastasis is an emerging field in cancer research. Already, several studies have been aimed at resolving the role of BCSCs in the organ tropism of breast cancer. As stated above, bone, lung, brain and liver are the preferred sites of breast cancer metastasis, whereas metastasis to other organs is rarely seen [19]. One possible factor in this organ-specificity is the expression of CD44 on the surface of BCSCs, since hyaluronan and osteopontin (OPN), which are common CD44 ligands, are highly expressed in these organs and have been found to arrest breast CTCs [112]. Moreover, breast cancers have been shown to express high levels of the chemokine receptor CXCR4, which seems to be proportional to the presence of BCSC populations. This expression may help these cells to specifically target bone, lung and brain tissues where the CXCR4 ligand CXCL12/SDF-1 is abundantly present (Fig. 1) [42, 43]. MiR-302a, which has been found to be down-regulated in metastatic breast cancers, is thought to target CXCR4, thereby inhibiting breast cancer metastasis [95]. Thus, besides conferring a high motility and resistance to apoptosis, BCSCs may contribute to organ-specific metastasis through interaction with the microenvironment in distant organs and the induction of pre-metastatic niche formation. In the context of EMT, it has been shown that inhibitor of differentiation 1 (ID1) is commonly expressed in triple-negative breast cancers and, in association with its closely related family member ID3, can facilitate metastatic colonization after infiltration of metastatic tumor cells into the lung parenchyma [113]. ID1 up-regulation is known to be mediated by TGF-β and to result in the induction of mesenchymal-epithelial transition (MET) at metastatic sites through antagonizing Twist1 (Fig. 1), but not at primary sites where the mesenchymal state is maintained by the zinc finger protein Snail1 [32]. In a xenograft model it has been found that ID1 knockdown in metastatic cells prevents MET and, consequently, suppresses lung colonization [32]. EMT also causes stromal matrix degradation, which is mediated mainly through up-regulation of matrix metalloproteinases (MMPs) and plasminogen activator (PA), which confers a more aggressive phenotype to mesenchymal-like BCSCs [114, 115]. In addition, it has been found that EMT contributes to increased angiogenesis through the induction of pro-angiogenic factors, including vascular endothelial growth factor A (VEGF-A), resulting in an excessive vascularization of the primary tumor [116].
In support of the EMT hypothesis, Gupta et al. [117] found that various subpopulations of the human breast cancer-derived cell lines SUM149 and SUM159, purified on basis of their phenotypic states, were able to return to equilibrium proportions of stem-like, basal and luminal cell populations over time. This finding indicates that stochastic processes governing single cell behavior can give rise to a phenotypic equilibrium in a cell population and, thus, that BCSCs arise de novo from non stem-like cells. In addition, it has been found that EMT-induced cells can form 10-fold more mammospheres than non-induced cancer cells, supporting a significant increased tumorigenic capacity of cells that have undergone EMT [98]. Some recent studies are, however, challenging the EMT paradigm by suggesting that EMT may be dispensable for cancer metastasis. Using animal models of pancreatic and breast cancer, Zheng et al. [118, 119] and Fischer et al. [116] showed that EMT may not be exclusively involved in cancer dissemination. So, since EMT may not be the only mechanism underlying CSC formation, there may be additional mechanisms conferring stem-like or aggressive phenotypes to cancer cells facilitating their dissemination to distant organs.
4 Metastasis progression factors: role of the tumor microenvironment, tumor cell circulation and extravasation
Metastasis progression factors are those that carry out several stimulatory functions in both primary tumors and at metastatic sites. Among these functions, the formation of a favorable tumor microenvironment, i.e., an environment for tumor cell proliferation and outgrowth, is of paramount importance. After entering the circulation tumor cells may disperse to various directions, but the subsequent extravasation appears to be organ-specific. In this section, various steps and factors involved in breast cancer metastasis progression will be discussed.
4.1 Role of the tumor microenvironment
Mesenchymal stem cells (MSCs) are emerging as important components of the tumor microenvironment. Recently, it was demonstrated that MSCs may regulate tumor metastasis through crosstalk with TGF-β [120]. It was found that TGF-β can down-regulate CXCL12 expression and, subsequently, restrict tumor progression and metastasis. Simultaneous knock-down of TGFBR2 and CXCL12 expression in MSCs was found to reverse the lung-tropic effect of MSCs that are unresponsive to TGF-β, indicating the significance of the TGF-β-CXCL12 axis in tumor progression and lung metastasis [120]. CXCL12-mediated regulation of metastasis in 4 T1 mouse breast cancer cells was found to be related to its regulation of CXCR7, and CXCR7 blockade was found to suppresses its metastasis. So, MSCs produce CXCL12, which in turn suppresses the metastasis of breast cancer cells through inhibition of CXCR7 expression. TGF-β signaling may abrogate the restriction of MSC-induced CXCL12 expression in tumor cells [120].
Many human cancers undergo (local) hypoxia due to increased cell numbers resulting from deregulated proliferation. Hypoxia activates HIF-1 and HIF-2, which in turn may activate the expression of genes required for cancer progression [121]. The roles of HIFs in the various steps of breast cancer development include the stimulation of lysyl oxidase (LOX) and ECM-remodeling enzymes and the recruitment of bone marrow-derived cells (BMDCs) to pre-metastatic niches (Table 3) [52]. HIF-1 acts as a master regulator of cancer progression [26], regulating HGF to MET and RHOA to ROCK1 signaling to promote cancer cell motility [26]. HIF-1α triggers EMT through modulation of NOTCH and integrin-linked kinase signaling and, indirectly, MMP and urokinase-type plasminogen activator receptor (uPAR) expression, which may affect ECM remodeling [67]. HIF-dependent ECM remodeling may also be brought about through stimulation of collagen cross-linking, which may result from over-expression of procollagen prolyl-4-hydroxylases (P4HA1 and P4HA2), lysyl hydroxylases (PLOD1 and PLOD2) and lysyl oxidases (LOX, LOXL2, and LOXL4), which is required for breast cancer invasion and metastasis [26]. HIF-dependent expression of VEGF has been found to stimulate angiogenesis and to increase vascular permeability, thereby stimulating the extravasation of breast cancer cells [122]. Zhang et al. [123] found that loss of HIF-1 activity in triple-negative breast cancer MDA-MB-231 cells was associated with a decreased primary tumor growth and a dramatic reduction in the metastasis of these cells to the lung. In addition, they found that HIF-1α and HIF-2α may stimulate L1 cell adhesion molecule (L1CAM) expression in hypoxic breast cancer cells, which mediates their interaction with pulmonary endothelial cells and facilitates their extravasation [123]. Ectopic expression or inhibition of miR-18a in an orthotopic metastatic breast cancer xenograft model revealed that this microRNA may inhibit HIF-1α activity and, concomitantly, lung metastasis, thereby underscoring the role of HIF-1α in breast cancer lung metastasis [124].
4.2 Role of circulating tumor cells
The analysis of circulating tumor cells (CTCs) provides a unique opportunity to gain insight into mechanisms of cancer cell dissemination and metastasis [125]. CTCs may travel either as single cells or as clusters of cells (tumor emboli). Recently, it has been found that mesenchymal markers representing EMT may be expressed by CTCs [126]. Using mouse models Aceto et al. [127] revealed that, compared to individual CTCs, CTC clusters are derived from oligoclonal clumps of primary tumor cells and constitute a rare but very metastasis-competent subset of CTCs. These CTC clusters were found to be more resistant to apoptosis than single CTCs following dissemination to the lung. CTC clusters thus exhibit a greater metastatic potential than single CTCs and act as more flourishing seeds to form micro-metastases. The formation and metastatic potential of CTC clusters may be related to their gene expression profiles. RNA sequencing of human breast cancer-derived CTC clusters revealed, for example, that plakoglobin may act as a key mediator of CTC clustering, i.e., plakoglobin expression knockdown resulted in a suppression of CTC cluster formation and a reduction of metastatic spread in mouse models [127]. As such, plakoglobin may serve as a novel prognostic factor and therapeutic target in patients with breast cancer [128]. A recent study on breast cancer CTCs has suggested that EpCAM, CD44, CD47 and MET co-expression may serve as a signature for CTC subsets with an increased metastatic capacity [129]. Also, Chemokine (CXC Motif) Ligand-1 (CXCL1) cytokines have been reported to be involved in the formation of lung metastases the by CTCs [130], and miR-17/20 has been found to suppress breast cancer cell migration and invasion by altering the secretion of CXCL1, IL-8 and CK8 [44].
4.3 Role of extravasation factors
In lung metastasis, one rate-limiting step is the penetration of breast cancer cells through the lung vasculature. Embolus formation (see above) is facilitated by leukocytes and platelets that form complexes with tumor cells through P- and L-selectins [131]. Up-regulation of selectin ligands has been shown to be associated with a poor prognosis and enhanced metastasis formation [132], and inflammation-induced up-regulation of E-selectin has been found to mediate BCLM in mouse models [133]. The local vascular structure is one of the factors determining organ-specificity of human cancers, especially breast cancers [13]. Unlike bone marrow and liver, which have fenestrated vasculatures with lower physical barriers, the pulmonary vasculature is surrounded by a basement membrane and adjacent alveolar cells, which trammel cancer cells to penetrate freely into the lung parenchyma. Therefore, lung-tropic cancer cells require additional specialized properties to breach the pulmonary vasculature and infiltrate into the lung parenchyma [134]. It has been suggested that TGF-β may act as a major cytokine priming the metastasis of breast cancer cells to the lung [40, 135, 136]. TGF-β has been shown to exhibit different roles during breast cancer progression. By inhibiting the proliferation of normal mammary epithelial cells, TGF-β usually suppresses breast cancer initiation, while it enhances the malignancy of late-stage breast cancers [137]. Tian et al. [138] showed that inhibition of the TGF-β downstream effectors Smad2/3 strongly suppressed the metastases of aggressive mammary carcinoma cells to the lung. Dankort et al. [139] used mouse mammary tumor virus (MMTV) transgenic mice expressing oncogenic Neu coupled to the adaptor proteins Grb-2 [Neu(YB)] or Shc [Neu(YD)] to dissect Neu-induced signaling pathways involved in breast cancer metastasis. They found that (MMTV)/Neu(YB) animals developed mammary tumors with a high propensity to metastasize to the lung, while MMTV/Neu(YD) mice exhibited a reduced incidence of pulmonary metastases. Siegel et al. [140] generated Neu-induced MMTV animals expressing activated TGF-β type I receptor (TβRI) or dominant negative TGF-β type II receptor (TβRII) to dissect the role of TGF-β signaling on the metastasis of Neu-induced mammary carcinomas. They found that, when crossed with Neu(YB) or Neu(YD) mice, the Neu-induced carcinomas in activated TβRI mice showed an increased propensity to extravasate to lung parenchyma, while the dominant negative TβRII mice exhibited an impaired Neu-induced tumor growth and a decreased pulmonary metastasis rate. Padua et al. [40] found that TGF-β in the breast tumor microenvironment primes lung metastasis through induction of angiopoietin-like 4 (ANGPTL4) in cancer cells that are about to enter the circulation. ANGPTL4 induction is one of the factors resulting in the disruption of lung vascular endothelial lining and in inducing hyperpermeability of capillaries, leading to the establishment of pulmonary metastases [40]. Huang et al. [37] found that TGF-β-mediated up-regulation of angiopoietin 2 (Angpt2), matrix metalloproteinase (MMP)-3 and MMP-10 in primary B16/F10 tumors destabilized the pulmonary vasculature and facilitated the formation of lung metastases. Using murine TGF-β1-pretreated 4 T1 mammary carcinoma cells injected into mammary fat pads of BALB/c mice, Ye et al. [141] found that during the pre-metastatic phase, TGF-β induced pulmonary vascular hyper-permeability resulting in a pulmonary microenvironment that facilitated extravasation of CTCs and dose-dependently increased the survival and proliferation of metastatic tumor cells. Therefore, TGF-β inhibition may be an efficient approach to treat at least a subset of late-stage breast cancers [142].
It has also been shown that lung-tropic breast cancer cells may express other specific mediators such as secreted protein acidic and cysteine-rich/OPN (SPARC) [50], secreted C-terminal fibrinogen-like domain of angiopoietin-like 4 (cANGPTL4), vascular cell adhesion molecule 1 (VCAM1), MMP-1 and MMP-2, interleukin-13 (IL-13) decoy receptor IL13Rα2, epidermal growth factor (EGF), EGF receptor ligand epiregulin (EREG) and cytochrome c oxidase polypeptide II (COX2), and thereby debilitate cell-cell junctions between pulmonary endothelial cells [35, 36, 40, 143]. It has been found that depletion of IL13Rα2 in metastatic breast cancer cells suppresses lung metastasis formation in vivo and that IL13Rα2 knockdown and IL-13 treatment cooperatively up-regulate the metastasis suppressor tumor protein 63 (TP63) in a STAT6-dependent manner. So, the STAT6-TP63 pathway appears to be involved in impairing metastatic dissemination of breast cancer cells to the lungs [47]. Other factors mediating the infiltration of metastatic breast cancer cells into the lung parenchyma and its homing in metastatic niches include the chemokine receptors CXCR4 and CCR7, which interact with their respective ligands CXCL12/SDF-1 and CCL21 [144], MTDH, which binds to a receptor expressed by the lung endothelium [41] and α6β4 integrin, which interacts with the chloride channel protein CLCA12 on lung vascular endothelial cells [145]. Moreover, it has been reported that in a rat breast cancer model the assembly of fibronectin on the surface of breast cancer cells and their interaction with dipeptidyl peptidase IV (DPP IV) on the pulmonary endothelial cells may facilitate the targeting of the lung vasculature by metastatic breast cancer cells (Fig. 2) [146]. Interestingly, most of the factors mentioned are already up-regulated early in primary breast cancers, indicating that the cellular tools required for metastasis to the lungs are already present at these stages [147] and, thus, that targeting these factors in primary tumors may be instrumental for impeding lung metastasis.

Extravasation of metastatic tumor cells from the pulmonary vasculature. Breast cancer cells in circulation recruit monocytes or macrophages enabling them to evade apoptotic signals. Subsequent interactions of lung-tropic breast cancer cells with pulmonary vascular endothelial cells is followed by embolus formation, which is facilitated by selectins, immune cells and surface molecules. Tumor cells secrete various molecules that directly prime endothelial retardation and extravasation. Tumor cells also secrete CCL2, which may recruit CCR2 expressing inflammatory monocytes. CCL2-CCR2 interactions may enhance the extravasation of metastatic breast cancer cells mainly through the targeted delivery of VEGF which, in turn, promotes vascular permeability
4.4 Role of tumor-associated macrophages
Other key drivers of breast cancer progression and metastasis that have been intensively studied are tumor-associated macrophages (TAMs) [148,149,150]. It has been found that TAMs may induce growth and angiogenesis, as well as migration and invasion of breast tumor cells, and participate in the formation of their metastatic niches [151]. TAMs may direct tumor cells to intravasate and to travel to distant sites including lung and bone [149]. The emerging roles of TAMs in angiogenesis and lymphangiogenesis are becoming increasingly apparent. It has been found that TAMs secrete several pro-angiogenic growth factors including EGF, VEGF, PLGF, MIF, TNF-α, TGF-β, IL-8, IL-1β, thymidine phosphorylase and the chemokines CCL2 and CXCL8 [48]. These factors modulate the vasculature for the dissemination of tumor cells and the balance between vasculature and capillary densities. Hiratsuka et al. [38] reported that primary breast tumors could promote hyper-permeability sites in E0071 and 3LL mice bearing metastatic lung tumors through modulation of the toll-like receptor 4 (TLR4) and its co-receptor MD-2, resulting in the regulation of serum amyloid A3 (SAA3), S100A8, VEGF, CCL2 and TNFα. Metastatic breast cancer cells have also been shown to emit CCL2, which may recruit inflammatory monocytes expressing CCR2 early during pulmonary metastasis. These monocytes can, in turn, differentiate into TAMs that secrete EGF that can bind to the EGFR on breast cancer cells. This feedback loop between TAMs and metastatic breast cancer cells is crucial for the process of breast cancer metastasis. The CCL2-CCR2 interaction enhances extravasation of metastatic cells mainly through targeted delivery of vascular endothelial growth factor (VEGF), which is known to promote extravasation [152]. TAMs within the microenvironment at metastatic sites express receptors different from those interacting with primary breast tumors [149]. The former ones express VEGFR1, CCR2 and CX3CR1, but not surface Tie2 or CXCR4, indicating that these TAMs are different from other pro-angiogenic macrophages [49]. Others have shown that CCR2 can trigger a pro-metastatic chemokine cascade involving the production of CCL3 by TAMs. CCL3 may subsequently signal via CCR1, thereby promoting metastatic progression [39]. In a recently published study Guerriero et al. [153, 154] showed that reprogramming of monocytes and macrophages by TMP195, a novel class IIa histone deacetylase (HDAC) inhibitor, may lead to the inhibition of breast cancer metastasis through anti-tumor macrophages.
5 Metastasis virulence factors: adaptation to the lung microenvironment and metastatic colonization
Recent work has revealed an important role of the pro-metastatic microenvironment in organ-specific metastasis, even before the arrival of tumor cells [59]. After extravasation into the lung parenchyma, metastatic tumor cells confront another rate-limiting step: survival and adaptation to a new microenvironment. This adaptation includes the evasion from apoptotic signals in the new microenvironment and the formation of a de novo niche that favors the proliferation of metastatic tumor cells [12]. Unlike dissemination via circulation, which is a frequent event, only a small percentage of tumor cells can survive at distant sites and form metastases [9]. This inefficiency of metastatic colonization has been documented for various human cancers [147, 155]. It has for example been found that most intravenously injected cancer cells that lodge in the lung will die within 2 days [156], which may be mainly attributed to immune attacks by leukocytes [157]. In addition, it has been found that the early growth of B16F10 tumor cells injected to target mouse lungs was unaffected by the site of extravasation, but that subsequent metastasis formation was enriched along the lung surface and around arterial and venous vessels [158].
5.1 Distant microenvironment creation in the pre-metastatic phase
It has been reported that communication between primary tumors and their prospective metastatic sites already starts before the arrival of circulating tumor cells. This so-called “pre-metastatic phase” represents a crucial step in the establishment of metastases. During the pre-metastatic phase, various primary tumor-derived systemic mediators, including growth factors, cytokines, extracellular matrix (ECM)-remodeling enzymes and exosomes, can modify the pulmonary parenchyma and create a hospitable microenvironment for the seeding, survival and proliferation of tumor cells [159, 160]. Exosomes are small vesicles that are shed from various cell types, including tumor cells [161]. These so called “tumor-derived exosomes’ have the ability to alter tissue microenvironments via fusion with the plasma membranes of target cells and, by doing so, to deliver their cargo. Hoshino et al. [162] found that exosomes derived from lung-tropic breast cancer cells fuse preferentially with lung fibroblasts and epithelial cells, and that this organ-specific exosome uptake “educates” the pre-metastatic niche. They further found that specific exosomal integrins, such as α6β4 and α6β1, may interact with cell-associated ECM components and, thereby, mediate exosome uptake at specific target sites within the lung. This exosomal integrin uptake was subsequently found to activate Src phosphorylation and pro-inflammatory S100 gene expression. Through proteomic profiling, Maji et al. [163] have recently found that the Annexin II levels in exosomes derived from metastatic breast cancer cells were significantly higher than in those derived from normal breast cells. They also found that tumor-derived exo-Anx II promotes tPA-dependent angiogenesis and, at the same time, the establishment of a pre-metastatic niche. Anx II-depleted exosomes were found to reduce the occurrence of brain and lung metastases. After careful evaluation, they found that exo-Anx II increases the secretion of IL-6 and TNFα, as well as macrophage-dependent stimulation of the p38MAPK, NF-κB and STAT3 pathways. It has also been found that breast cancer cell-derived exosomes may recruit myeloid-derived suppressor cells, thereby creating an immunosuppressive and pro-metastatic lung niche. Colony stimulating factor 1-receptor (CSF1R) is a master regulator of myeloid cells, and CSF-1 over-expression has been found to be associated with an increased number of metastatic niches in several cancers, including breast cancer [54].
The lysyl oxidase (LOX) family comprises to a group of factors that are critical to the formation of pre-metastatic niches before the arrival of breast cancer cells [164]. Erler et al. [57] found that hypoxic breast cancer cells secrete LOX, which subsequently accumulates at prospective pre-metastatic sites and cross-links collagen IV in the lung basement membrane, thereby generating a suitable ECM that facilitates the recruitment of CD11b + myeloid cells. These myeloid cells produce MMP-2 which cleaves collagen, thereby enhancing the invasion and recruitment of bone marrow-derived cells (BMDCs) [57]. So, through this process BMDCs are mobilized by the primary tumor and directed to the pulmonary microenvironment prior to the arrival of tumor cells [60]. BMDCs share common markers with hematopoietic progenitor cells (HPCs), including CD34, CD133 and VEGFR1, and interact with fibronectin in the pre-metastatic niche through the expression of VLA-4 (α4β1 integrin). The recruited BMDCs secrete MMPs that digest the ECM and, by doing so, release matrix-bound VEGF [59, 60]. BMDCs also mediate primary tumor-directed systemic instigation of indolent tumor cells and micro-metastases through the secretion of OPN. OPN is required for the activation and incorporation of BMDCs into the stroma of distant indolent tumors. So, indolent metastatic breast cancer cells need OPN-mediated BMDC activation in order to initiate their proliferation [45]. Other components within the lung microenvironment, such as neutrophils, may contribute to the colonization of metastatic breast cancer cells through inflammatory signals. It has recently been reported that neutrophil-derived leukotrienes support lung colonization by selectively enriching the sub-pool of cancer cells that exhibits a high metastatic potential [165]. Inhibition of arachidonate 5-lipoxygenase (Alox5) as a leukotriene-generating enzyme has been found to abrogate the pro-metastatic activity of neutrophils and to reduce the occurrence of pulmonary metastases, which suggests that this enzyme may serve as a potential therapeutic target in the lung microenvironment [165].
5.2 Evasion from apoptosis in the pulmonary microenvironment
To evade apoptosis in a leukocyte-rich microenvironment, such as the lung, metastatic breast cancer cells highly express VCAM1, which tethers macrophages to cancer cells via counter-receptor α4-integrins [53]. In xenograft model systems it has been shown that this interaction triggers the activation of Ezrin, which subsequently activates PI3K-AKT signaling in cancer cells, thereby increasing their survival (Fig. 3) [53]. Using 4 T1 as a highly metastatic breast cancer model, Olkhanud et al. [166] found that only a sub-set of the tumor cells expressing CCR4 can metastasize to the lung. In addition, they found that primary tumor cells can activate the expression of TARC/CCL17 and MDC/CCL22 in lungs. These chemokines act through CCR4 and attract both tumor and immune cells (i.e., CCR4+ regulatory T cells (Tregs)) that can directly kill natural killer (NK) cells using β-galactoside-binding protein, thereby preventing metastatic tumor cells from undergoing apoptosis [166]. So, metastatic breast cancer cells induce the recruitment and expansion of Tregs in order to help them escape from host protective immune cells [166]. Further studies have revealed that a unique subset of regulatory B cells, designated tumor-evoked Bregs (tBregs), can promote pulmonary breast cancer metastasis through TGF-β-dependent conversion of resting CD4+ T cells to Foxp3+ Tregs (Fig. 3) [167]. Targeting tBregs may have therapeutic potential via interrupting breast cancer-induced immunosuppressive events crucial for lung metastasis. It has also been found that metastatic tumor cells have to prevail antagonistic bone morphogenetic protein (BMP)-mediated signals, which have been shown to promote the differentiation of breast cancer cells in the lung in allograft models [168].

Adaptation of metastatic tumor cells to the lung parenchyma. To evade apoptosis in the lung parenchyma, breast cancer cells abundantly express VCAM1, which tethers them to macrophages. This interaction triggers activation of Ezrin which, consequently, activates PI3K-AKT signaling in the breast cancer cells and increases their survival. Another apoptosis-evading mechanism is mediated through CCL17 and CCL22, which are expressed by primary tumor cells and travel to the lung to form a pre-metastatic microenvironment. These chemokines act through CCR4 on the surface of Foxp3+ Tregs and induces them to directly kill NK cells using β-galactoside-binding protein. It has been shown that Foxp3+ Tregs originate from resting CD4+ T cells through a recently recognized subset of regulatory B cells, designated tumor-evoked Bregs (tBregs). These tBregs are thought to originate from B cells mediated by tumor cells. Metastatic tumor cells also prevail antagonistic BMP-mediated signals from lung resident cells by the secretion of Coco. After evading from apoptosis, metastatic tumor cells stimulate lung fibroblasts to express POSTN. POSTN, in turn, may recruit Wnt ligands and stimulate Wnt signaling in breast cancer cells enabling them to form macro-metastases. POSTN also promotes the incorporation of TNC into the ECM to generate an ECM meshwork required for adaptation of the ECM architecture within a mechanical environment. TNC expression by metastatic tumor cells enhances the expression of stemness-maintaining components, including MSI1, which is a positive regulator of NOTCH signaling and, thus, protects this signaling pathway from suppression by STAT5
5.3 De novo formation of pre-metastatic niches
After evasion from apoptosis, metastatic tumor cells “educate” stromal cells in lung parenchyma to facilitate their proliferation and the formation of de novo niches that support the expansion process. In this respect, it has been found that metastatic breast cancer cells can stimulate lung fibroblasts to express the extracellular matrix (ECM) component periostin (POSTN) via the secretion of TGF-β3 [169]. POSTN can, in turn, recruit Wnt ligands and stimulate Wnt signaling in cancer cells, which may help them to maintain their stemness properties and, consequently, to enhance their lung colonizing capacity (Fig. 3) [169]. It has been found that the Wnt signaling inhibitor DKK, which serves as a serological marker of organ-specific breast cancer metastasis, can inhibit the formation of lung metastases. DKK1 can suppresses PTGS2-induced macrophage and neutrophil recruitment in lung metastases by antagonizing non-canonical Wnt/PCP-RAC1-JNK signaling in the cancer cells [170]. POSTN promotes the incorporation of hexameric glycoprotein tenascin-C (TNC) into the ECM and, by doing so, organizes an ECM meshwork needed for adaptation of the ECM architecture to mechanical forces within the microenvironment [171]. Interestingly, TNC over-expression has been found to be associated with the occurrence of lung metastases [58]. Specifically, it was found that TNC expression by metastatic tumor cells may enhance the expression of stemness-maintaining components, including musashi homolog 1 (MSI1), which is a positive regulator of NOTCH signaling and, thus, protect this signaling pathway from suppression by signal transducer and activator of transcription 5 (STAT5) [58]. TNC has also been found to enhance the expression of leucine-rich repeat-containing G protein-coupled receptor 5 (LGR5), which is a target of the Wnt pathway [58]. Ye et al. [141] found that TGF-β can promote the formation of a pre-metastatic microenvironment through the modulation of certain inflammatory chemo-attractants and growth factors, including S100A8, S100A9, Angpt2 and VEGF. It has been shown that S100A8 and S100A9 may contribute to pre-metastatic niche formation through the recruitment of CD11b + myeloid cells to the lung microenvironment (see above) and the activation of toll-like receptor-4 (TLR-4)-dependent nuclear factor-κB (NF-κB), which may result in the survival of metastatic breast cancer cells [172, 173]. NF-κB is a transcription factor that is activated in a variety of human cancers and that acts mainly through inflammatory responses and the protection of transformed cells from apoptosis [174]. TGF-β-mediated NF-κB activation has been reported to be involved in lung metastasis [175]. Huber et al. [176] used an in vitro/in vivo model of mammary carcinogenesis that is dependent on both TGF-β and H-Ras activation. They found that inhibition of NF-κB activity prevented the acquisition of a mesenchymal phenotype and the formation of lung metastases by H-Ras-transformed epithelial cells, whereas TGF-β-dependent activation of the NF-κB pathway induced EMT. Their data suggest that NF-κB plays a crucial role in the progression and metastasis of breast cancer cells and that the lung-seeking role of the H-Ras- and TGF-β-dependent signaling pathways primarily depends on the NF-κB pathway. Therefore, TGF-β inhibition may not only be an attractive strategy to inhibit EMT and the extravasation of breast cancer cells, but also to efficiently impede the survival of metastatic breast cancer cells in the lung parenchyma.
It has been found that up-regulation of miR-122 may serve as another metastasis-promoting mechanism through reprogramming glucose metabolism and, consequently, increasing the nutrient availability at metastatic sites [96]. It has recently been found that up-regulation of tartrate-resistant acid phosphatase (TRACP), which belongs to the family of metalloproteinases, may lead to modulation of breast cancer pre-metastatic niches. TRACP has been found to create pre-metastatic conditions through cell adhesion/angiogenesis signaling alterations and, by doing so, to promote cancer cell invasion and lung metastasis, whereas TRACP knockdown has been found to inhibit these processes. Therefore, TRACP targeting may also serve as a plausible strategy to combat breast cancer metastasis [177].
6 Concluding remarks and future perspectives
Lung metastasis is a pernicious outcome of breast cancer. Current treatment strategies, including chemotherapy, radiotherapy and/or surgical resection, are palliative rather than curative. Therefore, targeted therapies have recently come into the limelight. These therapies are based on the identification of molecules and cellular signaling pathways underlying BCLM. CSC formation is an early event resulting in organ-specific metastasis of breast cancer cells to bones and lungs. Therefore, targeting CSCs and EMT may be considered as pivotal strategies to treat breast cancer metastasis. Interaction of breast cancer cells with the pulmonary vasculature is another critical determinant of BCLM formation. Therefore, molecules involved in the extravasation of breast cancer cells to the lung parenchyma, such as ANGPTL4, may serve as novel targets for impeding BCLM. Of note, TGF-β signaling has been found to be of paramount importance in all steps of BCLM and thus the targeting of this pathway, which results in the impediment of various downstream signaling cascades, may be of particular significance for the treatment of BCLM. Breast cancer cells exhibit sophisticated intrinsic capacities to evade apoptosis and to prosper in lung microenvironments. Blocking of major components within these supportive microenvironments including HIFs and their target genes, may serve as another attractive approach to impede BCLM formation. These approaches may target micro-metastases before their development into overt metastases and, as such, they warrant evaluation in pre-clinical (animal) studies and, ultimately, clinical trials.
References
SEER Stat Fact Sheets: Female Breast Cancer (2017), Available from: http://seer.cancer.gov/statfacts/html/breast.html. Accessed Nov 2017
S.A. Rabbani, A.P. Mazar, Evaluating distant metastases in breast cancer: from biology to outcomes. Cancer Metastasis Rev 26, 663–674 (2007)
S. Valastyan, R.A. Weinberg, Tumor metastasis: molecular insights and evolving paradigms. Cell 147, 275–292 (2011)
R. Sharma, R. Sharma, T.P. Khaket, C. Dutta, B. Chakraborty, T.K. Mukherjee, Breast cancer metastasis: Putative therapeutic role of vascular cell adhesion molecule-1. Cell Oncol 40, 199–208 (2017)
Y. Huang, Q. Jin, M. Su, F. Ji, N. Wang, C. Zhong, Y. Jiang, Y. Liu, Z. Zhang, J. Yang, L. Wei, T. Chen, B. Li, Leptin promotes the migration and invasion of breast cancer cells by upregulating ACAT2. Cell Oncol 40, 537–547 (2017)
C. C. Society, Prognosis and survival for breast cancer Canada: Canadian Cancer Society (2017), Available from: http://www.cancer.ca/en/cancer-information/cancer-type/breast/prognosis-and-survival/?region=bc. Accessed 2017 Nov
L. Ding, M.J. Ellis, S. Li, D.E. Larson, K. Chen, J.W. Wallis, C.C. Harris, M.D. McLellan, R.S. Fulton, L.L. Fulton, R.M. Abbott, J. Hoog, D.J. Dooling, D.C. Koboldt, H. Schmidt, J. Kalicki, Q. Zhang, L. Chen, L. Lin, M.C. Wendl, J.F. McMichael, V.J. Magrini, L. Cook, S.D. McGrath, T.L. Vickery, E. Appelbaum, K. Deschryver, S. Davies, T. Guintoli, L. Lin, R. Crowder, Y. Tao, J.E. Snider, S.M. Smith, A.F. Dukes, G.E. Sanderson, C.S. Pohl, K.D. Delehaunty, C.C. Fronick, K.A. Pape, J.S. Reed, J.S. Robinson, J.S. Hodges, W. Schierding, N.D. Dees, D. Shen, D.P. Locke, M.E. Wiechert, J.M. Eldred, J.B. Peck, B.J. Oberkfell, J.T. Lolofie, F. Du, A.E. Hawkins, M.D. O'Laughlin, K.E. Bernard, M. Cunningham, G. Elliott, M.D. Mason, D.M. Thompson, J.L.I. Jr, P.J. Goodfellow, C.M. Perou, G.M. Weinstock, R. Aft, M. Watson, T.J. Ley, R.K. Wilson, E.R. Mardis, Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 464, 999–1005 (2010)
C.D. Savci-Heijink, H. Halfwerk, G.K. Hooijer, H.M. Horlings, J. Wesseling, M.J. van de Vijver, Retrospective analysis of metastatic behaviour of breast cancer subtypes. Breast Cancer Res Treat 150, 547–557 (2015)
A.C. Obenauf, J. Massague, Surviving at a distance: organ specific metastasis. Trends Cancer 1, 76–91 (2015)
D. Hanahan, R.A. Weinberg, Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011)
R. Paduch, The role of lymphangiogenesis and angiogenesis in tumor metastasis. Cell Oncol 39, 397–410 (2016)
J. Massague, A.C. Obenauf, Metastatic colonization by circulating tumour cells. Nature 529, 298–306 (2016)
X. Lu, Y. Kang, Organotropism of breast cancer metastasis. J Mammary Gland Biol Neoplasia 12, 153–162 (2007)
L.R. Yates, S. Knappskog, D. Wedge, J.H.R. Farmery, S. Gonzalez, I. Martincorena, L.B. Alexandrov, P. Van Loo, H.K. Haugland, P.K. Lilleng, G. Gundem, M. Gerstung, E. Pappaemmanuil, P. Gazinska, S.G. Bhosle, D. Jones, K. Raine, L. Mudie, C. Latimer, E. Sawyer, C. Desmedt, C. Sotiriou, M.R. Stratton, A.M. Sieuwerts, A.G. Lynch, J.W. Martens, A.L. Richardson, A. Tutt, P.E. Lønning, P.J. Campbell, Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 32, 169–84.e7 (2017)
J. Bergh, P.E. Jonsson, B. Glimelius, P. Nygren, A systematic overview of chemotherapy effects in breast cancer. Acta Oncol 40, 253–281 (2001)
C. Ludwig, E. Stoelben, J. Hasse, Disease-free survival after resection of lung metastases in patients with breast cancer. Eur J Surg Oncol 29, 532–535 (2003)
B. Tayyeb, M. Parvin, Pathogenesis of Breast Cancer Metastasis to Brain: a Comprehensive Approach to the Signaling Network. Mol Neurobiol 53, 446–454 (2016)
I.J. Fidler, The pathogenesis of cancer metastasis: the 'seed and soil' hypothesis revisited. Nat Rev Cancer 3, 453–458 (2003)
K.R. Hess, G.R. Varadhachary, S.H. Taylor, W. Wei, M.N. Raber, R. Lenzi, J.L. Abbruzzese, Metastatic patterns in adenocarcinoma. Cancer 106, 1624–1633 (2006)
J. Budczies, M. von Winterfeld, F. Klauschen, M. Bockmayr, J.K. Lennerz, C. Denkert, T. Wolf, A. Warth, M. Dietel, I. Anagnostopoulos, W. Weichert, D. Wittschieber, A. Stenzinger, The landscape of metastatic progression patterns across major human cancers. Oncotarget 6, 570–583 (2015)
I.T. Gavrilovic, J.B. Posner, Brain metastases: epidemiology and pathophysiology. J Neuro-Oncol 75, 5–14 (2005)
A. Mujoomdar, J.H. Austin, R. Malhotra, C.A. Powell, G.D. Pearson, M.C. Shiau, H. Raftopoulos, Clinical predictors of metastatic disease to the brain from non-small cell lung carcinoma: primary tumor size, cell type, and lymph node metastases. Radiology 242, 882–888 (2007)
M. Yousefi, T. Bahrami, A. Salmaninejad, R. Nosrati, P. Ghaffari, S.H. Ghaffari, Lung cancer-associated brain metastasis: Molecular mechanisms and therapeutic options. Cell Oncol 40, 419–441 (2017)
S. Paget, The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev 8, 98–101 (1989)
G. Lorusso, C. Ruegg, New insights into the mechanisms of organ-specific breast cancer metastasis. Semin Cancer Biol 22, 226–233 (2012)
L. Schito, G.L. Semenza, Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2, 758–770 (2016)
M. Smid, Y. Wang, Y. Zhang, A.M. Sieuwerts, J. Yu, J.G. Klijn, J.A. Foekens, J.W. Martens, Subtypes of breast cancer show preferential site of relapse. Cancer Res 68, 3108–3114 (2008)
H. Kennecke, R. Yerushalmi, R. Woods, M.C. Cheang, D. Voduc, C.H. Speers, T.O. Nielsen, K. Gelmon, Metastatic behavior of breast cancer subtypes. J Clin Oncol 28, 3271–3277 (2010)
D.X. Nguyen, J. Massagué, Genetic determinants of cancer metastasis. Nat Rev Genet 8, 341–352 (2007)
K. Jin, T. Li, H. van Dam, F. Zhou, L. Zhang, Molecular insights into tumour metastasis: tracing the dominant events. J Pathol 241(5), 567–577 (2017). https://doi.org/10.1002/path.4871
E.P. Cuevas, P. Eraso, M.J. Mazón, V. Santos, G. Moreno-Bueno, A. Cano, F. Portillo, LOXL2 drives epithelial-mesenchymal transition via activation of IRE1-XBP1 signalling pathway. Sci Rep 7, 44988 (2017)
M. Stankic, S. Pavlovic, Y. Chin, E. Brogi, D. Padua, L. Norton, J. Massague, R. Benezra, TGF-beta-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep 5, 1228–1242 (2013)
H. Li, X. Chen, Y. Gao, J. Wu, F. Zeng, F. Song, XBP1 induces snail expression to promote epithelial- to-mesenchymal transition and invasion of breast cancer cells. Cell Signal 27, 82–89 (2015)
D. Liu, P.S. Rudland, D.R. Sibson, A. Platt-Higgins, R. Barraclough, Human homologue of cement gland protein, a novel metastasis inducer associated with breast carcinomas. Cancer Res 65, 3796–3805 (2005)
A.J. Minn, G.P. Gupta, P.M. Siegel, P.D. Bos, W. Shu, D.D. Giri, A. Viale, A.B. Olshen, W.L. Gerald, J. Massague, Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524 (2005)
R.L. Huang, Z. Teo, H.C. Chong, P. Zhu, M.J. Tan, C.K. Tan, C.R. Lam, M.K. Sng, D.T. Leong, S.M. Tan, S. Kersten, J.L. Ding, H.Y. Li, N.S. Tan, ANGPTL4 modulates vascular junction integrity by integrin signaling and disruption of intercellular VE-cadherin and claudin-5 clusters. Blood 118, 3990–4002 (2011)
Y. Huang, N. Song, Y. Ding, S. Yuan, X. Li, H. Cai, H. Shi, Y. Luo, Pulmonary vascular destabilization in the premetastatic phase facilitates lung metastasis. Cancer Res 69, 7529–7537 (2009)
S. Hiratsuka, S. Ishibashi, T. Tomita, A. Watanabe, S. Akashi-Takamura, M. Murakami, H. Kijima, K. Miyake, H. Aburatani, Y. Maru, Primary tumours modulate innate immune signalling to create pre-metastatic vascular hyperpermeability foci. Nat Commun 4, 1853 (2013)
M.-A.D. Cao, Z. Zhang, A. Gilchrist, CCR1 as a Target for Metastatic Breast Cancer. FASEB J 31, 823.11-.11 (2017)
D. Padua, X.H. Zhang, Q. Wang, C. Nadal, W.L. Gerald, R.R. Gomis, J. Massague, TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 133, 66–77 (2008)
D.M. Brown, E. Ruoslahti, Metadherin, a cell surface protein in breast tumors that mediates lung metastasis. Cancer Cell 5, 365–374 (2004)
M.Z. Dewan, S. Ahmed, Y. Iwasaki, K. Ohba, M. Toi, N. Yamamoto, Stromal cell-derived factor-1 and CXCR4 receptor interaction in tumor growth and metastasis of breast cancer. Biomed Pharmacother 60, 273–276 (2006)
B. Furusato, A. Mohamed, M. Uhlen, J.S. Rhim, CXCR4 and cancer. Pathol Int 60, 497–505 (2010)
Z. Yu, N.E. Willmarth, J. Zhou, S. Katiyar, M. Wang, Y. Liu, P.A. McCue, A.A. Quong, M.P. Lisanti, R.G. Pestell, microRNA 17/20 inhibits cellular invasion and tumor metastasis in breast cancer by heterotypic signaling. Proc Natl Acad Sci USA 107, 8231–8236 (2010)
S.S. McAllister, A.M. Gifford, A.L. Greiner, S.P. Kelleher, M.P. Saelzler, T.A. Ince, F. Reinhardt, L.N. Harris, B.L. Hylander, E.A. Repasky, R.A. Weinberg, Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell 133, 994–1005 (2008)
K. Gumireddy, A. Li, P.A. Gimotty, A.J. Klein-Szanto, L.C. Showe, D. Katsaros, G. Coukos, L. Zhang, Q. Huang, KLF17 is a negative regulator of epithelial–mesenchymal transition and metastasis in breast cancer. Nat Cell Biol 11, 1297–1304 (2009)
P. Papageorgis, S. Ozturk, A.W. Lambert, C.M. Neophytou, A. Tzatsos, C.K. Wong, S. Thiagalingam, A.I. Constantinou, Targeting IL13Ralpha2 activates STAT6-TP63 pathway to suppress breast cancer lung metastasis. Breast Cancer Res 17, 98 (2015)
N. Todorović-Raković, J. Milovanović, Interleukin-8 in breast cancer progression. J Interf Cytokine Res 33, 563–570 (2013)
C. Qian, A. Worrede-Mahdi, R. Kaur, F. Shen, J. Salvino, O. Meucci, A. Fatatis, Targeting CX3CR1 impairs the reseeding of cancer cells recirculating from metastatic tumors. Abstract In: Proceedings of the American Association for Cancer Research Annual Meeting 2017; 2017 Apr 1-5; Washington, DC. Philadelphia (PA): AACR; Cancer Res 2017;77(13 Suppl):Abstract nr 5799. https://doi.org/10.1158/1538-7445.AM2017-5799
J. Lee, G. Kim, M. Park, J. Yoon, Up-regulation of SPARC is associated with breast tumor progression and epithelial SPARC expression is correlated with poor survival and MMP-2 expression in patients with breast carcinoma. Abstract In: Proceedings of the 2016 San Antonio Breast Cancer Symposium; 2016 Dec 6-10; San Antonio, TX. Philadelphia (PA): AACR; Cancer Res 2017;77(4 Suppl):Abstract nr P4–12–11
J. Ma, S. Gao, X. Xie, E. Sun, M. Zhang, Q. Zhou, C. Lu, SPARC inhibits breast cancer bone metastasis and may be a clinical therapeutic target. Oncol Lett 14, 5876–5882 (2017)
C.C.-L. Wong, D.M. Gilkes, H. Zhang, J. Chen, H. Wei, P. Chaturvedi, S.I. Fraley, C.-M. Wong, U.-S. Khoo, I.O.-L. Ng, D. Wirtz, G.L. Semenza, Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc Natl Acad Sci USA 108, 16369–16374 (2011)
Q. Chen, X.H. Zhang, J. Massague, Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell 20, 538–549 (2011)
T.F. Borin, K. Angara, M. Rashid, A. Shankar, A. Iskander, R. Ara, M. Jain, B.R. Achyut, A.S. Arbab, CSF-1R inhibitor prevented pre-metastatic lung niches in metastatic mammary tumor. Abstract In: Proceedings of the American Association for Cancer Research Annual Meeting 2017; 2017 Apr 1-5; Washington, DC. Philadelphia (PA): AACR; Cancer Res 2017;77(13 Suppl):Abstract nr 1043. https://doi.org/10.1158/1538-7445.AM2017-1043
K.E. Luker, G.D. Luker, Functions of CXCL12 and CXCR4 in breast cancer. Cancer Lett 238, 30–41 (2006)
N. Nagarsheth, M.S. Wicha, W. Zou, Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol 17, 559–572 (2017)
J.T. Erler, K.L. Bennewith, T.R. Cox, G. Lang, D. Bird, A. Koong, Q.T. Le, A.J. Giaccia, Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 15, 35–44 (2009)
T. Oskarsson, S. Acharyya, X.H. Zhang, S. Vanharanta, S.F. Tavazoie, P.G. Morris, R.J. Downey, K. Manova-Todorova, E. Brogi, J. Massague, Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med 17, 867–874 (2011)
R.N. Kaplan, B. Psaila, D. Lyden, Bone marrow cells in the 'pre-metastatic niche': within bone and beyond. Cancer Metastasis Rev 25, 521–529 (2006)
R.N. Kaplan, R.D. Riba, S. Zacharoulis, A.H. Bramley, L. Vincent, C. Costa, D.D. MacDonald, D.K. Jin, K. Shido, S.A. Kerns, Z. Zhu, D. Hicklin, Y. Wu, J.L. Port, N. Altorki, E.R. Port, D. Ruggero, S.V. Shmelkov, K.K. Jensen, S. Rafii, D. Lyden, VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438, 820–827 (2005)
D.X. Nguyen, P.D. Bos, J. Massagué, Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer 9, 274–284 (2009)
J.R. Nevins, A. Potti, Mining gene expression profiles: expression signatures as cancer phenotypes. Nat Rev Genet 8, 601–609 (2007)
J.K. Mouw, Y. Yui, L. Damiano, R.O. Bainer, J.N. Lakins, I. Acerbi, G. Ou, A.C. Wijekoon, K.R. Levental, P.M. Gilbert, E.S. Hwang, Y.-Y. Chen, V.M. Weaver, Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nat Med 20, 360 (2014)
J.P. Thiery, H. Acloque, R.Y. Huang, M.A. Nieto, Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890 (2009)
S. Heerboth, G. Housman, M. Leary, M. Longacre, S. Byler, K. Lapinska, A. Willbanks, S. Sarkar, EMT and tumor metastasis. Clin Transl Med 4, 6 (2015)
M.A. Nieto, R.Y. Huang, R.A. Jackson, J.P. Thiery, EMT: 2016. Cell 166, 21–45 (2016)
S.Y. Shin, O. Rath, A. Zebisch, S.M. Choo, W. Kolch, K.H. Cho, Functional roles of multiple feedback loops in extracellular signal-regulated kinase and Wnt signaling pathways that regulate epithelial-mesenchymal transition. Cancer Res 70, 6715–6724 (2010)
A. Eger, A. Stockinger, J. Park, E. Langkopf, M. Mikula, J. Gotzmann, W. Mikulits, H. Beug, R. Foisner, Beta-Catenin and TGFbeta signalling cooperate to maintain a mesenchymal phenotype after FosER-induced epithelial to mesenchymal transition. Oncogene 23, 2672–2680 (2004)
L.A. Timmerman, J. Grego-Bessa, A. Raya, E. Bertran, J.M. Perez-Pomares, J. Diez, S. Aranda, S. Palomo, F. McCormick, J.C. Izpisua-Belmonte, J.L. de la Pompa, Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev 18, 99–115 (2004)
D. Xiao, J. He, Epithelial mesenchymal transition and lung cancer. J Thorac Dis 2, 154–159 (2010)
T.A. DiMeo, K. Anderson, P. Phadke, C. Fan, C.M. Perou, S. Naber, C. Kuperwasser, A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res 69, 5364–5373 (2009)
J. Fuxe, T. Vincent, A. Garcia de Herreros, Transcriptional crosstalk between TGF-beta and stem cell pathways in tumor cell invasion: role of EMT promoting Smad complexes. Cell Cycle 9, 2363–2374 (2010)
D.S. Micalizzi, C.-A. Wang, S.M. Farabaugh, W.P. Schiemann, H.L. Ford, Homeoprotein Six1 increases TGF-β type I receptor and converts TGF-β signaling from suppressive to supportive for tumor growth. Cancer Res 70, 10371–10380 (2010)
F. Zhou, Y. Drabsch, T.J. Dekker, A.G. De Vinuesa, Y. Li, L.J. Hawinkels, K.-A. Sheppard, M.-J. Goumans, R.B. Luwor, C.J. De Vries, Nuclear receptor NR4A1 promotes breast cancer invasion and metastasis by activating TGF-β signalling. Nat Commun 5, 3388 (2014)
B. De Craene, G. Berx, Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 13, 97–110 (2013)
G. Berx, F. van Roy, Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb Perspect Biol 1, a003129 (2009)
K. Polyak, R.A. Weinberg, Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 9, 265–273 (2009)
C. Gilles, M. Polette, M. Mestdagt, B. Nawrocki-Raby, P. Ruggeri, P. Birembaut, J.M. Foidart, Transactivation of vimentin by beta-catenin in human breast cancer cells. Cancer Res 63, 2658–2664 (2003)
J. Cai, H. Guan, L. Fang, Y. Yang, X. Zhu, J. Yuan, J. Wu, M. Li, MicroRNA-374a activates Wnt/beta-catenin signaling to promote breast cancer metastasis. J Clin Invest 123, 566–579 (2013)
Y. Liang, J. Hu, J. Li, Y. Liu, J. Yu, X. Zhuang, L. Mu, X. Kong, D. Hong, Q. Yang, Epigenetic Activation of TWIST1 by MTDH Promotes Cancer Stem–like Cell Traits in Breast Cancer. Cancer Res 75, 3672–3680 (2015)
D.A. Zajchowski, M.F. Bartholdi, Y. Gong, L. Webster, H.L. Liu, A. Munishkin, C. Beauheim, S. Harvey, S.P. Ethier, P.H. Johnson, Identification of gene expression profiles that predict the aggressive behavior of breast cancer cells. Cancer Res 61, 5168–5178 (2001)
S. Stinson, M.R. Lackner, A.T. Adai, N. Yu, H.J. Kim, C. O'Brien, J. Spoerke, S. Jhunjhunwala, Z. Boyd, T. Januario, R.J. Newman, P. Yue, R. Bourgon, Z. Modrusan, H.M. Stern, S. Warming, F.J. de Sauvage, L. Amler, R.F. Yeh, D. Dornan, TRPS1 targeting by miR-221/222 promotes the epithelial-to-mesenchymal transition in breast cancer. Sci Signal 4, ra41 (2011)
H. Chen, G. Zhu, Y. Li, R.N. Padia, Z. Dong, Z.K. Pan, K. Liu, S. Huang, Extracellular signal-regulated kinase signaling pathway regulates breast cancer cell migration by maintaining slug expression. Cancer Res 69, 9228–9235 (2009)
C.J. Desmet, T. Gallenne, A. Prieur, F. Reyal, N.L. Visser, B.S. Wittner, M.A. Smit, T.R. Geiger, J. Laoukili, S. Iskit, B. Rodenko, W. Zwart, B. Evers, H. Horlings, A. Ajouaou, J. Zevenhoven, M. van Vliet, S. Ramaswamy, L.F. Wessels, D.S. Peeper, Identification of a pharmacologically tractable Fra-1/ADORA2B axis promoting breast cancer metastasis. Proc Natl Acad Sci USA 110, 5139–5144 (2013)
Y. Wu, M. Sarkissyan, J.V. Vadgama, Epithelial-mesenchymal transition and breast cancer. J Clin Med 5, 13 (2016)
L. Ma, J. Young, H. Prabhala, E. Pan, P. Mestdagh, D. Muth, J. Teruya-Feldstein, F. Reinhardt, T.T. Onder, S. Valastyan, miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol 12, 247–256 (2010)
K. Yin, W. Yin, Y. Wang, L. Zhou, Y. Liu, G. Yang, J. Wang, J. Lu, MiR-206 suppresses epithelial mesenchymal transition by targeting TGF-β signaling in estrogen receptor positive breast cancer cells. Oncotarget 7, 24537 (2016)
J. Johansson, T. Berg, E. Kurzejamska, M.-F. Pang, V. Tabor, M. Jansson, P. Roswall, K. Pietras, M. Sund, P. Religa, MiR-155-mediated loss of C/EBPβ shifts the TGF-β response from growth inhibition to epithelial-mesenchymal transition, invasion and metastasis in breast cancer. Oncogene 32, 5614–5624 (2013)
B. Wang, H. Wang, Z. Yang, MiR-122 inhibits cell proliferation and tumorigenesis of breast cancer by targeting IGF1R. PLoS One 7, e47053 (2012)
P. Liu, H. Tang, B. Chen, Z. He, M. Deng, M. Wu, X. Liu, L. Yang, F. Ye, X. Xie, miR-26a suppresses tumour proliferation and metastasis by targeting metadherin in triple negative breast cancer. Cancer Lett 357, 384–392 (2015)
J. Yu, J.-G. Wang, L. Zhang, H.-P. Yang, L. Wang, D. Ding, Q. Chen, W.-L. Yang, K.-H. Ren, D.-M. Zhou, Q. Zou, Y.-T. Jin, X.-P. Liu, MicroRNA-320a inhibits breast cancer metastasis by targeting metadherin. Oncotarget 7, 38612–38625 (2016)
M. Korpal, Y. Kang, The emerging role of miR-200 family of microRNAs in epithelial-mesenchymal transition and cancer metastasis. RNA Biol 5, 115–119 (2008)
S.J. Song, L. Poliseno, M.S. Song, U. Ala, K. Webster, C. Ng, G. Beringer, N.J. Brikbak, X. Yuan, L.C. Cantley, A.L. Richardson, P.P. Pandolfi, MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell 154, 311–324 (2013)
M. Korpal, B.J. Ell, F.M. Buffa, T. Ibrahim, M.A. Blanco, T. Celia-Terrassa, L. Mercatali, Z. Khan, H. Goodarzi, Y. Hua, Y. Wei, G. Hu, B.A. Garcia, J. Ragoussis, D. Amadori, A.L. Harris, Y. Kang, Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med 17, 1101–1108 (2011)
Z. Liang, X. Bian, H. Shim, Inhibition of breast cancer metastasis with microRNA-302a by downregulation of CXCR4 expression. Breast Cancer Res Treat 146, 535–542 (2014)
M.Y. Fong, W. Zhou, L. Liu, A.Y. Alontaga, M. Chandra, J. Ashby, A. Chow, S.T.F. O’Connor, S. Li, A.R. Chin, Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol 17, 183–194 (2015)
M.K. Jolly, M. Boareto, B. Huang, D. Jia, M. Lu, E. Ben-Jacob, J.N. Onuchic, H. Levine, Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front Oncol 5, 155 (2015)
S.A. Mani, W. Guo, M.J. Liao, E.N. Eaton, A. Ayyanan, A.Y. Zhou, M. Brooks, F. Reinhard, C.C. Zhang, M. Shipitsin, L.L. Campbell, K. Polyak, C. Brisken, J. Yang, R.A. Weinberg, The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 (2008)
M.S. Wicha, S. Liu, G. Dontu, Cancer stem cells: an old idea--a paradigm shift. Cancer Res 66, 1883–1890; discussion 95-6 (2006)
M. Al-Hajj, M.S. Wicha, A. Benito-Hernandez, S.J. Morrison, M.F. Clarke, Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 100, 3983–3988 (2003)
C. Ginestier, M.H. Hur, E. Charafe-Jauffret, F. Monville, J. Dutcher, M. Brown, J. Jacquemier, P. Viens, C.G. Kleer, S. Liu, A. Schott, D. Hayes, D. Birnbaum, M.S. Wicha, G. Dontu, ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1, 555–567 (2007)
A.K. Croker, D. Goodale, J. Chu, C. Postenka, B.D. Hedley, D.A. Hess, A.L. Allan, High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J Cell Mol Med 13, 2236–2252 (2009)
C. Sheridan, H. Kishimoto, R.K. Fuchs, S. Mehrotra, P. Bhat-Nakshatri, C.H. Turner, R. Goulet Jr., S. Badve, H. Nakshatri, CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res 8, R59 (2006)
E. Charafe-Jauffret, C. Ginestier, F. Iovino, J. Wicinski, N. Cervera, P. Finetti, M.H. Hur, M.E. Diebel, F. Monville, J. Dutcher, M. Brown, P. Viens, L. Xerri, F. Bertucci, G. Stassi, G. Dontu, D. Birnbaum, M.S. Wicha, Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res 69, 1302–1313 (2009)
L.L. Campbell, K. Polyak, Breast tumor heterogeneity: cancer stem cells or clonal evolution? Cell Cycle 6, 2332–2338 (2007)
S. Liu, M.S. Wicha, Targeting breast cancer stem cells. J Clin Oncol 28, 4006–4012 (2010)
M.A. Velasco-Velazquez, N. Homsi, M. De La Fuente, R.G. Pestell, Breast cancer stem cells. Int J Biochem Cell Biol 44, 573–577 (2012)
F. Ishikawa, S. Yoshida, Y. Saito, A. Hijikata, H. Kitamura, S. Tanaka, R. Nakamura, T. Tanaka, H. Tomiyama, N. Saito, M. Fukata, T. Miyamoto, B. Lyons, K. Ohshima, N. Uchida, S. Taniguchi, O. Ohara, K. Akashi, M. Harada, L.D. Shultz, Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol 25, 1315–1321 (2007)
K. Engelmann, H. Shen, O.J. Finn, MCF7 side population cells with characteristics of cancer stem/progenitor cells express the tumor antigen MUC1. Cancer Res 68, 2419–2426 (2008)
C.W. Lee, K. Simin, Q. Liu, J. Plescia, M. Guha, A. Khan, C.C. Hsieh, D.C. Altieri, A functional Notch-survivin gene signature in basal breast cancer. Breast Cancer Res 10, R97 (2008)
Z. Madjd, A.Z. Mehrjerdi, A.M. Sharifi, S. Molanaei, S.Z. Shahzadi, M. Asadi-Lari, CD44+ cancer cells express higher levels of the anti-apoptotic protein Bcl-2 in breast tumours. Cancer Immun 9, 4 (2009)
J.E. Draffin, S. McFarlane, A. Hill, P.G. Johnston, D.J. Waugh, CD44 potentiates the adherence of metastatic prostate and breast cancer cells to bone marrow endothelial cells. Cancer Res 64, 5702–5711 (2004)
G.P. Gupta, J. Perk, S. Acharyya, P. de Candia, V. Mittal, K. Todorova-Manova, W.L. Gerald, E. Brogi, R. Benezra, J. Massague, ID genes mediate tumor reinitiation during breast cancer lung metastasis. Proc Natl Acad Sci USA 104, 19506–19511 (2007)
L.S. Orlichenko, D.C. Radisky, Matrix metalloproteinases stimulate epithelial-mesenchymal transition during tumor development. Clin Exp Metastasis 25, 593–600 (2008)
J.P. Thiery, J.P. Sleeman, Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 7, 131–142 (2006)
B. De Craene, B. Gilbert, C. Stove, E. Bruyneel, F. van Roy, G. Berx, The transcription factor snail induces tumor cell invasion through modulation of the epithelial cell differentiation program. Cancer Res 65, 6237–6244 (2005)
P.B. Gupta, C.M. Fillmore, G. Jiang, S.D. Shapira, K. Tao, C. Kuperwasser, E.S. Lander, Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 146, 633–644 (2011)
K.R. Fischer, A. Durrans, S. Lee, J. Sheng, F. Li, S.T. Wong, H. Choi, T. El Rayes, S. Ryu, J. Troeger, R.F. Schwabe, L.T. Vahdat, N.K. Altorki, V. Mittal, D. Gao, Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527, 472–476 (2015)
X. Zheng, J.L. Carstens, J. Kim, M. Scheible, J. Kaye, H. Sugimoto, C.C. Wu, V.S. LeBleu, R. Kalluri, Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525–530 (2015)
P. Yu, Y. Huang, C. Xu, L. Lin, Y. Han, W. Sun, G. Hu, A. Rabson, Y. Wang, Y. Shi, Downregulation of CXCL12 in mesenchymal stromal cells by TGFβ promotes breast cancer metastasis. Oncogene 36, 840–849 (2017)
G.L. Semenza, Molecular mechanisms mediating metastasis of hypoxic breast cancer cells. Trends Mol Med 18, 534–543 (2012)
R. Sullivan, C.H. Graham, Hypoxia-driven selection of the metastatic phenotype. Cancer Metastasis Rev 26, 319–331 (2007)
H. Zhang, C.C. Wong, H. Wei, D.M. Gilkes, P. Korangath, P. Chaturvedi, L. Schito, J. Chen, B. Krishnamachary, P.T. Winnard Jr., V. Raman, L. Zhen, W.A. Mitzner, S. Sukumar, G.L. Semenza, HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene 31, 1757–1770 (2012)
R. Krutilina, W. Sun, A. Sethuraman, M. Brown, T.N. Seagroves, L.M. Pfeffer, T. Ignatova, M. Fan, MicroRNA-18a inhibits hypoxia-inducible factor 1alpha activity and lung metastasis in basal breast cancers. Breast Cancer Res 16, R78 (2014)
S. Maheswaran, D.A. Haber, Circulating tumor cells: a window into cancer biology and metastasis. Curr Opin Genet Dev 20, 96–99 (2010)
M. Yu, A. Bardia, B.S. Wittner, S.L. Stott, M.E. Smas, D.T. Ting, S.J. Isakoff, J.C. Ciciliano, M.N. Wells, A.M. Shah, Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584 (2013)
N. Aceto, A. Bardia, D.T. Miyamoto, M.C. Donaldson, B.S. Wittner, J.A. Spencer, M. Yu, A. Pely, A. Engstrom, H. Zhu, Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158, 1110–1122 (2014)
W. Goto, S. Kashiwagi, Y. Asano, K. Takada, K. Takahashi, T. Hatano, T. Takashima, S. Tomita, H. Motomura, M. Ohsawa, Circulating tumor cell clusters-associated gene plakoglobin is a significant prognostic predictor in patients with breast cancer. Biomarker Res 5, 19 (2017)
I. Baccelli, A. Schneeweiss, S. Riethdorf, A. Stenzinger, A. Schillert, V. Vogel, C. Klein, M. Saini, T. Bäuerle, M. Wallwiener, Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol 31, 539–544 (2013)
A.M. Dehnavi, M.R. Sehhati, H. Rabbani, Hybrid method for prediction of metastasis in breast cancer patients using gene expression signals. J Med Signals Sens 3(2), 79–86 (2013)
H. Laubli, L. Borsig, Selectins promote tumor metastasis. Semin Cancer Biol 20, 169–177 (2010)
G. Bendas, L. Borsig, Cancer cell adhesion and metastasis: selectins, integrins, and the inhibitory potential of heparins. Int J Cell Biol 2012, 676731 (2012)
M. Jiang, X. Xu, Y. Bi, J. Xu, C. Qin, M. Han, Systemic inflammation promotes lung metastasis via E-selectin upregulation in mouse breast cancer model. Cancer Biol Ther 15, 789–796 (2014)
G.P. Gupta, D.X. Nguyen, A.C. Chiang, P.D. Bos, J.Y. Kim, C. Nadal, R.R. Gomis, K. Manova-Todorova, J. Massague, Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 446, 765–770 (2007)
R.S. Muraoka-Cook, H. Kurokawa, Y. Koh, J.T. Forbes, L.R. Roebuck, M.H. Barcellos-Hoff, S.E. Moody, L.A. Chodosh, C.L. Arteaga, Conditional overexpression of active transforming growth factor beta1 in vivo accelerates metastases of transgenic mammary tumors. Cancer Res 64, 9002–9011 (2004)
R.S. Muraoka, N. Dumont, C.A. Ritter, T.C. Dugger, D.M. Brantley, J. Chen, E. Easterly, L.R. Roebuck, S. Ryan, P.J. Gotwals, V. Koteliansky, C.L. Arteaga, Blockade of TGF-beta inhibits mammary tumor cell viability, migration, and metastases. J Clin Invest 109, 1551–1559 (2002)
Y. Kang, Pro-metastasis function of TGFbeta mediated by the Smad pathway. J Cell Biochem 98, 1380–1390 (2006)
F. Tian, S. DaCosta Byfield, W.T. Parks, S. Yoo, A. Felici, B. Tang, E. Piek, L.M. Wakefield, A.B. Roberts, Reduction in Smad2/3 signaling enhances tumorigenesis but suppresses metastasis of breast cancer cell lines. Cancer Res 63, 8284–8292 (2003)
D. Dankort, B. Maslikowski, N. Warner, N. Kanno, H. Kim, Z. Wang, M.F. Moran, R.G. Oshima, R.D. Cardiff, W.J. Muller, Grb2 and Shc adapter proteins play distinct roles in Neu (ErbB-2)-induced mammary tumorigenesis: implications for human breast cancer. Mol Cell Biol 21, 1540–1551 (2001)
P.M. Siegel, W. Shu, R.D. Cardiff, W.J. Muller, J. Massague, Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci USA 100, 8430–8435 (2003)
Y. Ye, S. Liu, C. Wu, Z. Sun, TGFbeta modulates inflammatory cytokines and growth factors to create premetastatic microenvironment and stimulate lung metastasis. J Mol Histol 46, 365–375 (2015)
J.M. Yingling, K.L. Blanchard, J.S. Sawyer, Development of TGF-beta signalling inhibitors for cancer therapy. Nat Rev Drug Discov 3, 1011–1022 (2004)
M. Tichet, V. Prod'Homme, N. Fenouille, D. Ambrosetti, Tumour-derived SPARC drives vascular permeability and extravasation through endothelial VCAM1 signalling to promote metastasis. Nat Commun 6, 6993 (2015)
A. Muller, B. Homey, H. Soto, N. Ge, D. Catron, M.E. Buchanan, T. McClanahan, E. Murphy, W. Yuan, S.N. Wagner, J.L. Barrera, A. Mohar, E. Verastegui, A. Zlotnik, Involvement of chemokine receptors in breast cancer metastasis. Nature 410, 50–56 (2001)
M. Abdel-Ghany, H.C. Cheng, R.C. Elble, B.U. Pauli, The breast cancer beta 4 integrin and endothelial human CLCA2 mediate lung metastasis. J Biol Chem 276, 25438–25446 (2001)
H.C. Cheng, M. Abdel-Ghany, R.C. Elble, B.U. Pauli, Lung endothelial dipeptidyl peptidase IV promotes adhesion and metastasis of rat breast cancer cells via tumor cell surface-associated fibronectin. J Biol Chem 273, 24207–24215 (1998)
A.J. Minn, Y. Kang, I. Serganova, G.P. Gupta, D.D. Giri, M. Doubrovin, V. Ponomarev, W.L. Gerald, R. Blasberg, J. Massague, Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest 115, 44–55 (2005)
S.R. Nielsen, M.C. Schmid, Macrophages as Key Drivers of Cancer Progression and Metastasis. Mediat Inflamm 2017, 9624760 (2017)
C.B. Williams, E.S. Yeh, A.C. Soloff, Tumor-associated macrophages: unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer 2, 1–12 (2016)
L.M. Nusblat, M.J. Carroll, C.M. Roth, Crosstalk between M2 macrophages and glioma stem cells. Cell Oncol 40, 471–482 (2017)
B.Z. Qian, J.W. Pollard, Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51 (2010)
B.Z. Qian, J. Li, H. Zhang, T. Kitamura, J. Zhang, L.R. Campion, E.A. Kaiser, L.A. Snyder, J.W. Pollard, CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475, 222–225 (2011)
L. Cassetta, J.W. Pollard, Repolarizing macrophages improves breast cancer therapy. Cell Res 27, 963–964 (2017)
J.L. Guerriero, A. Sotayo, H.E. Ponichtera, J.A. Castrillon, A.L. Pourzia, S. Schad, S.F. Johnson, R.D. Carrasco, S. Lazo, R.T. Bronson, S.P. Davis, M. Lobera, M.A. Nolan, A. Letai, Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 543, 428–432 (2017)
A.F. Chambers, A.C. Groom, I.C. MacDonald, Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2, 563–572 (2002)
C.W. Wong, A. Lee, L. Shientag, J. Yu, Y. Dong, G. Kao, A.B. Al-Mehdi, E.J. Bernhard, R.J. Muschel, Apoptosis: an early event in metastatic inefficiency. Cancer Res 61, 333–338 (2001)
K.J. Luzzi, I.C. MacDonald, E.E. Schmidt, N. Kerkvliet, V.L. Morris, A.F. Chambers, A.C. Groom, Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol 153, 865–873 (1998)
M.D. Cameron, E.E. Schmidt, N. Kerkvliet, K.V. Nadkarni, V.L. Morris, A.C. Groom, A.F. Chambers, I.C. MacDonald, Temporal progression of metastasis in lung: cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res 60, 2541–2546 (2000)
S.S. McAllister, R.A. Weinberg, Tumor-host interactions: a far-reaching relationship. J Clin Oncol 28, 4022–4028 (2010)
C. Coghlin, G.I. Murray, Current and emerging concepts in tumour metastasis. J Pathol 222, 1–15 (2010)
K. Pakravan, S. Babashah, M. Sadeghizadeh, S.J. Mowla, M. Mossahebi-Mohammadi, F. Ataei, N. Dana, M. Javan, MicroRNA-100 shuttled by mesenchymal stem cell-derived exosomes suppresses in vitro angiogenesis through modulating the mTOR/HIF-1alpha/VEGF signaling axis in breast cancer cells. Cell Oncol 40, 457–470 (2017)
A. Hoshino, B. Costa-Silva, T.L. Shen, G. Rodrigues, A. Hashimoto, M. Tesic Mark, H. Molina, S. Kohsaka, A. Di Giannatale, S. Ceder, S. Singh, C. Williams, N. Soplop, K. Uryu, L. Pharmer, T. King, L. Bojmar, A.E. Davies, Y. Ararso, T. Zhang, H. Zhang, J. Hernandez, J.M. Weiss, V.D. Dumont-Cole, K. Kramer, L.H. Wexler, A. Narendran, G.K. Schwartz, J.H. Healey, P. Sandstrom, K.J. Labori, E.H. Kure, P.M. Grandgenett, M.A. Hollingsworth, M. de Sousa, S. Kaur, M. Jain, K. Mallya, S.K. Batra, W.R. Jarnagin, M.S. Brady, O. Fodstad, V. Muller, K. Pantel, A.J. Minn, M.J. Bissell, B.A. Garcia, Y. Kang, V.K. Rajasekhar, C.M. Ghajar, I. Matei, H. Peinado, J. Bromberg, D. Lyden, Tumour exosome integrins determine organotropic metastasis. Nature 527, 329–335 (2015)
S. Maji, P. Chaudhary, I. Akopova, P.M. Nguyen, R.J. Hare, I. Gryczynski, J.K. Vishwanatha, Exosomal annexin II promotes angiogenesis and breast cancer metastasis. Mol Cancer Res 15, 93–105 (2017)
F. Salvador, A. Martin, C. López-Menéndez, G. Moreno-Bueno, V. Santos, A. Vázquez-Naharro, P.G. Santamaria, S. Morales, P.R. Dubus, L. Muinelo-Romay, Lysyl Oxidase–like Protein LOXL2 Promotes Lung Metastasis of Breast Cancer. Cancer Res 77, 5846–5859 (2017)
S.K. Wculek, I. Malanchi, Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 528, 413–417 (2015)
P.B. Olkhanud, D. Baatar, M. Bodogai, F. Hakim, R. Gress, R.L. Anderson, J. Deng, M. Xu, S. Briest, A. Biragyn, Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res 69, 5996–6004 (2009)
P.B. Olkhanud, B. Damdinsuren, M. Bodogai, R.E. Gress, R. Sen, K. Wejksza, E. Malchinkhuu, R.P. Wersto, A. Biragyn, Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4(+) T cells to T-regulatory cells. Cancer Res 71, 3505–3515 (2011)
H. Gao, G. Chakraborty, A.P. Lee-Lim, Q. Mo, M. Decker, A. Vonica, R. Shen, E. Brogi, A.H. Brivanlou, F.G. Giancotti, The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 150, 764–779 (2012)
I. Malanchi, A. Santamaria-Martinez, E. Susanto, H. Peng, H.A. Lehr, J.F. Delaloye, J. Huelsken, Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 481, 85–89 (2011)
X. Zhuang, H. Zhang, X. Li, X. Li, M. Cong, F. Peng, J. Yu, X. Zhang, Q. Yang, G. Hu, Differential effects on lung and bone metastasis of breast cancer by Wnt signalling inhibitor DKK1. Nat Cell Biol 19, ncb3613 (2017)
I. Kii, T. Nishiyama, M. Li, K. Matsumoto, M. Saito, N. Amizuka, A. Kudo, Incorporation of tenascin-C into the extracellular matrix by periostin underlies an extracellular meshwork architecture. J Biol Chem 285, 2028–2039 (2010)
S. Hiratsuka, A. Watanabe, H. Aburatani, Y. Maru, Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol 8, 1369–1375 (2006)
S. Rafii, D. Lyden, S100 chemokines mediate bookmarking of premetastatic niches. Nat Cell Biol 8, 1321–1323 (2006)
V. Baud, M. Karin, Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov 8, 33–40 (2009)
S.Y. Yang, A. Miah, A. Pabari, M. Winslet, Growth Factors and their receptors in cancer metastases. Front Biosci (Landmark Ed) 16, 531–538 (2011)
M.A. Huber, N. Azoitei, B. Baumann, S. Grunert, A. Sommer, H. Pehamberger, N. Kraut, H. Beug, T. Wirth, NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest 114, 569–581 (2004)
M. Dai, C. Wu, L. Chen, C. Chen, T. Chao, Tartrate-resistant acid phosphatase (TRACP) modulates breast cancer pre-metastatic niche. Abstract In: Proceedings of the 2016 San Antonio Breast Cancer Symposium; 2016 Dec 6-10; San Antonio, TX. Philadelphia (PA): AACR; Cancer Res 2017;77(4 Suppl):Abstract nr P4-03-17
Acknowledgments
The authors would like to thank the faculty members at Mashhad University of Medical Sciences for their assistance in the preparation of the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Yousefi, M., Nosrati, R., Salmaninejad, A. et al. Organ-specific metastasis of breast cancer: molecular and cellular mechanisms underlying lung metastasis. Cell Oncol. 41, 123–140 (2018). https://doi.org/10.1007/s13402-018-0376-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-018-0376-6