
Abstract
Hypoxia refers to the decrease in oxygen tension in the tissues, and the central effector of the hypoxic response is the transcription factor Hypoxia-Inducible Factor α (HIF1-α). Transient hypoxia in acute events, such as exercising or regeneration after damage, play an important role in skeletal muscle physiology and homeostasis. However, sustained activation of hypoxic signaling is a feature of skeletal muscle injury and disease, which can be a consequence of chronic damage but can also increase the severity of the pathology and worsen its outcome. Here, we review evidence that supports the idea that hypoxia and HIF-1α can contribute to the establishment of fibrosis in skeletal muscle through its crosstalk with other profibrotic factors, such as Transforming growth factor β (TGF-β), the induction of profibrotic cytokines expression, as is the case of Connective Tissue Growth Factor (CTGF/CCN2), or being the target of the Renin-angiotensin system (RAS).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Hypoxic signaling
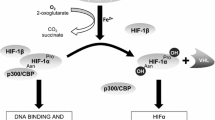
Hypoxia refers to the decrease in oxygen tension in the tissue. The central effector of the hypoxic response is the transcription factor Hypoxia-Inducible Factor α (HIF-α) (Koh et al. 2009). Under normoxic conditions, HIF-α is constitutively expressed and continuously degraded via the ubiquitin-proteasome pathway following its hydroxylation by HIF-prolyl hydroxylases domain-containing enzymes (PHD) (Ivan et al. 2001). The Von Hippel-Lindau E3 ubiquitin-ligase recognizes the hydroxylated form of HIF-α for ubiquitination and degradation. By contrast, during hypoxia, low oxygen pressure inhibits PHD, allowing HIF-α stabilization and translocation to the nucleus where it promotes the expression of target genes that are necessary for cell survival and adaptation under hypoxic conditions. Among the target genes, we found: the glucose transporter 1, which favors glycolic anaerobic metabolism to allow cell survival; Vascular Endothelial Growth Factor (VEGF), which promotes angiogenesis to reestablish the vascular network; and Erythropoietin, which promotes production of erythrocytes to increase oxygen transport through the bloodstream (Ameln et al. 2005; Ke and Costa 2006). The discovery of this oxygen sensing and response pathway (Nobel Prize in Physiology or Medicine 2019), has helped to understand many physiopathological processes in several types of tissues and organs (Leu et al. 2019; Zhang et al. 2019). Moreover, due to the hypoxic environment present in some tumors, hypoxic signaling has been widely studied in the context of different types of cancer and used as a target for reducing tumor vascularization and growth (Akanji et al. 2019).
The role of HIF-1α in skeletal muscle
The physiological role of HIF-1α in skeletal muscle is related to transient hypoxia, which might occur when oxygen pressure drops in response to acute dynamic exercise (Richardson et al. 1995). In this scenario, metabolic oxygen consumption by muscle cells during contraction could exceed oxygen availability from capillaries. As a response to transient hypoxia, HIF-1α stabilizes and in response, VEGF expression is induced (Ameln et al. 2005). HIF-1α has a pro-angiogenic function in skeletal muscle since the overexpression of HIF-1α and HIF-2α induces VEGF expression and the development of capillaries that express CD-31 in vivo (Niemi et al. 2014). Consequently, VEGF triggers angiogenesis in muscle subjected to exercise but is also essential for angiogenesis in sedentary muscle (Olfert et al. 2010; Tang et al. 2004). Moreover, skeletal muscle contraction by itself also induces vasodilation in microvasculature through nitric oxide (NO)-dependent and independent mechanisms in order to try to recover oxygen supply for its metabolic demands (Hong and Kim 2017).
Hypoxic signaling does not seem to be involved in muscle formation since neither HIF-1α nor HIF-2α are necessary for skeletal muscle development. Double knockout experiments for HIF-1α and HIF-2α show that they are dispensable for skeletal muscle development (Yang et al. 2017). Similar results have been reported using myogenic progenitors that do not express HIF-1α (Majmundar et al. 2015). Thus, HIF-mediated hypoxic signaling seems to be more associated with muscle homeostasis than muscle formation.
After an injury, the skeletal muscle niche becomes hypoxic. It has been reported that acute damage induced by eccentric contraction, cardiotoxin (CTX), notexin (NTX) or barium chloride injection trigger a marked disruption in the muscle’s microvasculature (Hardy et al. 2016; Hotta et al. 2018). In fact, HIF-1α signaling has been detected in injured skeletal muscle, increasing from day 1 to day 7 after CTX injection (Drouin et al. 2019). In parallel, after eccentric contraction injury the expression of HIF target genes, such as VEGF, are increased during regeneration. This increment in gene expression occurs in a temporal window that correlates with microvasculature permeability (Hotta et al. 2018). Functional experiments demonstrated that VEGF expression stimulates muscle regeneration, supporting that HIF downstream signaling through VEGF plays a pro-angiogenic role during regeneration (Arsic et al. 2004; Borselli et al. 2010; Mason et al. 2004; Messina et al. 2007). Moreover, VEGF and angiogenesis can improve the outcome and increase regeneration in chronic skeletal muscle diseases (Ennen et al. 2013; Shimizu-Motohashi and Asakura 2014).
Another axis of HIF-mediated hypoxic signaling is related to skeletal muscle adult stem cells (satellite cells). However, HIF function in muscle progenitors is very complex. In vitro experiments in myoblasts have shown that myogenesis is impaired under hypoxic conditions concomitantly with HIF-1α activation (Gustafsson et al. 2005; Majmundar et al. 2012). On the other hand, myogenesis is also inhibited in the absence of HIF-1α (Ono et al. 2006). Therefore, HIF-1α signaling in myogenesis is complex and reports seem to be contradictory however, the data unveils that its function might be restricted to a very bounded threshold.
In vivo, satellite cells reside in a hypoxic niche and they express HIF-2α at basal levels (Xie et al. 2018). In vivo experiments have shown that muscle regeneration is delayed under environmental hypoxic conditions, resulting in decreased expression of myogenic markers, such as myogenin or MyoD, and loss of muscle mass (Chaillou et al. 2014). However, the role of HIF-mediated hypoxic signaling on satellite cells can be very specific. Some reports show that HIF-1α deletion in satellite cells accelerates regeneration and myogenesis in ischemia experiments, suggesting that HIF-1α negatively regulates regeneration through satellite cell proliferation (Majmundar et al. 2015). On the other hand, HIF-1α and HIF-2α double knockout experiments in satellite cells show delayed regeneration after injury and self-renewal inhibition of satellite cells under hypoxic conditions, evidencing the active role of HIF-2α (Yang et al. 2017). Interestingly, transient inhibition of HIF-2α in satellite cells contributed to a better understanding of the mechanism, showing accelerated muscle regeneration and myoblast proliferation in the short term. Nevertheless, long term HIF-2α ablation produces depletion of satellite cells and regenerative failure, indicating that HIF-2α is responsible for stemness maintenance and self-renewal of satellite cells (Xie et al. 2018). Therefore, HIF-mediated signaling seems to be implicated in muscle stem cell quiescence and activation, having an impact on skeletal muscle regeneration.
Myeloid hypoxic signaling also plays an important role in skeletal muscle homeostasis. Experiments in knockout mice for HIF-1α in myeloid cells demonstrate that hypoxic signaling in these cells is essential for skeletal muscle regeneration after injury. In these knockout mice, macrophage invasion is delayed and there are fewer myogenic MyoD positive cells recruited to damaged muscles (Scheerer et al. 2013). Accordingly, activation of HIF-mediated signaling through PHD inhibition protects muscle from eccentric injury through myeloid HIF-1α-mediated iNOS activity (Billin et al. 2018). Besides, NO has been related to satellite cell activation (Anderson 2000; Buono et al. 2012; Rigamonti et al. 2013). Therefore, the physiological role of HIF-1α in myeloid cells affects skeletal muscle homeostasis through macrophage activation and NO signaling during regeneration.
Recently, we showed that different models of skeletal muscle damage and fibrosis (hindlimb denervation, Amyotrophic lateral sclerosis (ALS) murine model, and mdx dystrophic skeletal muscle) have decreased capillary density and activated hypoxia signaling (Valle-Tenney et al. 2019). This evidence supports the idea that HIF-mediated hypoxic signaling activation occurs and has a role during acute and chronic events of damage in skeletal muscle.
Skeletal muscle fiber type-specific hypoxic response
It has been hypothesized that regulation of the oxygen homeostasis system in skeletal muscle depends on fiber type. The basal expression level of HIF-1α is different in each type of fiber and is tightly related to the metabolic profile of each one: fast-twitch oxidative glycolytic (FOG) fibers express higher basal levels of HIF-1α than slow-twitch oxidative (SO) fibers (Mounier et al. 2010; Pisani and Dechesne 2005).
In agreement with this hypothesis, the reports describe the effect of hypoxia signaling activation as a slow-to-fast fiber type switch. HIF-1α overexpression in C2C12 myoblasts that are then differentiated to myotubes, induces the expression of fast MyHC isoforms. Moreover, HIF-1α overexpression in EDL and soleus muscles in vivo displays a slow-to-fast fiber type switch (Lunde et al. 2011). Similarly, in hypoxic muscle, the proportion of fiber types changes to adapt to oxygen deprivation in several physiological and pathological conditions, tending to increase FOG fibers over SO fibers (Chaillou 2018).
On the other hand, reports using different genetic approaches do not support this hypothesis. PHD1−/− mice show more tolerance to hypoxia, partly through metabolic reprogramming leading to a shift from oxidative toward anaerobic/glycolytic metabolism in skeletal muscle without causing a switch in myofiber type (Aragones et al. 2008). Genetic ablation of HIF-2α showed a reduction in slow-twitch fibers and an up-regulation of fast-twitch fibers (Rasbach et al. 2010). Besides, HIF-1α deletion in the skeletal muscle caused an endurance training-like adaptation in resting mice without significant changes in fiber type composition. Nevertheless, following six-weeks of endurance training, knockout mice for HIF-1α do not improve toward an oxidative metabolic profile and there is no fast-to-slow fiber-type shift as shown in wt mice (Mason and Johnson 2007). A similar effect has been reported in high-altitude trekkers. Mountaineers show an increase in slow fibers accompanied by a decrease in fast muscle fibers after an expedition to the Himalaya where they spend 23 days over 5000 m a.s.l. (Doria et al. 2011).
Thus, the contribution of hypoxia-mediated signaling to muscle fiber type composition seems to be more related to the overall effect of hypoxia and is highly influenced by training conditions as recapitulated in Chaillou 2018. Furthermore, the involvement of HIF-1α or HIF-2α signaling pathways on fiber type switch needs to be fully understood. Despite this, the contribution of the hypoxic response in pathological states such as muscle fibrosis could be differentially regulated according to fiber type. Specifically, FOG fibers that express more HIF-1α could be more prone to exert hypoxic HIF-1α-dependent signaling over pro-fibrotic effectors eliciting the fibrotic response.
Skeletal muscle fibrosis
Skeletal muscle fibrosis, in which functional and contractile fibers are replaced by rigid scar-like tissue, underlies several pathological conditions. Many muscle pathologies including dystrophies, motor-neuron diseases and pathological states associated with chronic myotrauma cause the development of fibrosis in the skeletal muscle tissue (Gonzalez et al. 2017; Pessina et al. 2014; Smith and Barton 2018).
The fibrotic environment in skeletal muscle is characterized by the elevated deposition of extracellular matrix (ECM) proteins, overexpression of pro-fibrotic factors, myofibroblast over-activation and persistent inflammation (Mahdy 2018; Mann et al. 2011; Wynn and Ramalingam 2012). Among the ECM proteins that show increased levels in muscle fibrosis, we found fibronectin, collagens, and proteoglycans (Alvarez et al. 2002; Caceres et al. 2000; Pessina et al. 2014; Serrano and Munoz-Canoves 2017).
The mononuclear cells that infiltrate fibrotic skeletal muscle include fibroblasts, immune, myogenic, and vascular cells. These different cell populations proliferate transiently during acute damage and regeneration in a synchronized and coordinated fashion (Bentzinger et al. 2013). However, the overlapping degeneration and regeneration cycles that occur in muscular dystrophies and chronic damage from other pathologies lead to the asynchronous remodeling of the microenvironment that finally progresses to fibrosis (Dadgar et al. 2014).
Vasculature in skeletal muscle fibrosis
The vascular hypothesis of muscular dystrophy has been studied for several years but little is known for other skeletal muscle pathologies. This hypothesis explains part of the pathological state of the disease through muscle fiber apoptosis via functional ischemia due to reduced vasculature and mislocalization of the muscle-specific isoform of neuronal nitric oxide synthase (nNOSμ) (Gargioli et al. 2008; Thomas 2013; Thomas et al. 2003). Nitric oxide (NO) produced by sarcolemmal nNOSμ acts as a local paracrine signal that inhibits vasoconstriction in active muscles (Chavoshan et al. 2002; Jendzjowsky and DeLorey 2013). Further research has shown that the deteriorated vascular component is part of the fibrotic disease and can be manipulated to improve muscle condition. Vascular therapies including vaso-relaxation drugs and pro-angiogenic treatments have been successful in improving the dystrophic phenotype in Duchenne muscular dystrophy (DMD) patients or mdx mice (Ennen et al. 2013; Matsakas et al. 2013; Podkalicka et al. 2019; Shimizu-Motohashi and Asakura 2014). Nevertheless, even when there is considerable evidence that links vascular damage to skeletal muscle pathologies associated with fibrosis, such as muscular dystrophies (DMD, mdx and Sgcd −/−) and denervation (Borisov et al. 2000; Gargioli et al. 2008; Hudlicka 1967; Matsakas et al. 2013), the role of the damage induced by decreased oxygen availability and consequent hypoxic signaling in skeletal muscle fibrosis is not fully understood.
Relationship between hypoxia and skeletal muscle fibrosis
Experimental data obtained in tissues other than skeletal muscle pointed to a relationship between hypoxia and fibrosis (Darby and Hewitson 2016; Norman et al. 2000). It has been described that hypoxia by itself is able to exacerbate the expression of ECM proteins in epidermal fibroblasts from systemic sclerosis patients (Distler et al. 2007), human renal fibroblasts (Norman et al. 2000), proximal tube cells (Orphanides et al. 1997), tubular epithelial cells (Kimura et al. 2008) and epithelial kidney cells (Rana et al. 2015). Nevertheless, the relationship between hypoxia and fibrosis has not been fully addressed in skeletal muscle. In vivo experiments have shown that muscle regeneration is delayed under ambient hypoxic conditions (Chaillou et al. 2014). In this line, there is evidence showing that altered skeletal muscle regeneration results in muscle fibrosis: the overlapping degeneration and regeneration cycles that occur in muscular dystrophies lead to an asynchronous remodeling of the microenvironment that finally develops fibrosis (Dadgar et al. 2014; Serrano et al. 2011). Chronic damage induced by six consecutive rounds of intramuscular injections of barium chloride has the same fibrotic effect (Contreras et al. 2016; Pessina et al. 2014; Riquelme et al. 2014). The histological analysis made by Chaillou et al. 2014 shows that muscles damaged with an intramuscular injection of NTX under hypoxic conditions for 28 days presented delayed regeneration. Moreover, although not the focus of the same work, the published images suggest an increment of the interstitial area at the end of the treatment that could be related to exacerbated connective tissue deposition. Furthermore, HIF-1α activated signaling has been found in different models of skeletal muscle damage, all of them associated with fibrosis (Valle-Tenney et al. 2019).
Furthermore, although VEGF plays a pro-angiogenic role as a downstream effector of the HIF-mediated hypoxic signaling, it could have pro-fibrotic effects. VEGF induces stress fiber formation in fibroblasts isolated from dystrophic muscle, a marker of differentiation to myofibroblasts, which are the main producers of ECM (Gutpell and Hoffman 2015). Therefore, sustained/chronic hypoxic signaling activation in dystrophic muscle due to vasculature damage could be one of the mechanisms contributing to fibroblast-overactivation, as a result of VEGF overexpression, which finally elicits fibrosis.
Interestingly, data we obtained using a prolyl-hydroxylase inhibitor, dimethyloxalylglycine (DMOG), administered daily in a chronic type approach to sustainedly activate HIF-hypoxic signaling shows induction of ECM proteins in skeletal muscle that are characteristic of a fibrotic phenotype. Administration of DMOG leads to a transient increase of HIF-1α, with a peak around 4 h post-dose, inducing the expression of the HIF-1α target gene VEGF in skeletal muscle (Fig. 1a). To obtain sustained pharmacological HIF-1α stabilization, the drug was administered daily for a total of 17 days I.P. to 10-month-old wt C57BL10 mice (Fig. 1b). In homogenates obtained from the diaphragm, we observed that sustained HIF-1α pharmacological stabilization induces the expression of collagen and fibronectin, and increases the expression of profibrotic factor Connective Tissue Growth Factor (CTGF/CCN2), see below), suggesting a potential relationship between the chronic hypoxia response and muscle fibrosis (Fig. 2c,d).

Sustained pharmacological HIF activation leads to a fibrotic muscle phenotype. a. 10-month-old wt C57BL10 mice were treated with DMOG (I.P. 150 mg/Kg) and euthanized 2, 4 and 6 h after administration. Western blot of total homogenates from hindlimb gastrocnemius muscle of treated mice shows that DMOG treatment can induce transient HIF-1α stabilization, which peaks 4 h post-administration, and also the expression of the HIF-1α downstream target gene VEGF. b. Scheme of the sustained DMOG treatment protocol: 10-month-old wt C57BL10 mice were treated daily with DMOG (I.P. 150 mg/Kg/day) or vehicle for 17 days. 4 h after the last DMOG injection, mice were euthanized, and muscle samples were collected. c. Collagen (Sirius red) staining and fibronectin immunofluorescence in diaphragm cryosections of DMOG or vehicle-treated mice. d. Western blot from diaphragm homogenates. HIF pharmacological stabilization induces the overexpression of collagen, fibronectin, IgG, and CCN2

HIF-Hypoxia signaling in skeletal muscle physiology and fibrosis. In normoxia, blood flows through vessels and capillaries (5 per muscle fiber on average) (Valle-Tenney, 2019) allowing oxygen to reach the cells in the muscle tissue. In this condition, the HIF-α transcription factor is continuously hydroxylated by HIF-prolyl hydroxylases domain-containing enzymes (PHD). This mark is recognized by Von Hippel-Lindau E3 ubiquitin-ligases and ubiquitinated for degradation via the ubiquitin-proteasome pathway. During exercise, oxygen consumption could exceed oxygen availability, in addition to vasoconstriction, decreased oxygen diffusion generates a hypoxic state where HIF-signaling is activated. In the hypoxic state, HIF-α is no longer hydroxylated and it accumulates and translocates to the nucleus where it binds to HIF-β to exert its function as a transcription factor. The same effect has been described for muscle regeneration where the skeletal muscle cell niche becomes hypoxic after injury by extravasation of capillaries and impaired blood flow through the tissue. In this context, HIF-signaling plays multiple roles including angiogenesis, myeloid cell activation and myogenesis by satellite cell proliferation. In situations of exercise and regeneration, normoxia is reestablished after a transient period of hypoxia. However, in chronic skeletal muscle pathologies the hypoxic state, in addition to repetitive damage and pro-fibrotic factors overexpression, elicits the establishment of fibrosis. In this condition, sustained HIF-activation in crosstalk with other pro-fibrotic pathways (TGF-β or Renin-Angiotensin system) could promote the overexpression of key profibrotic cytokines such as CCN2
Acute, intermittent or short-term pharmacological inhibition of PHD, and subsequent HIF-1α stabilization, has been shown to be protective in several conditions: cerebral ischemic damage (Reischl et al. 2014), cisplatin-induced kidney injury (Yang et al. 2018), and ischemic cardiac protection (Olenchock et al. 2016). PHD inhibition also accelerates blood recovery after severe irradiation (Forristal et al. 2013). Moreover, HIF controls the production of erythropoietin production by the kidney and the liver coordinating erythropoiesis with iron metabolism (Joharapurkar et al. 2018). Currently, several prolyl hydrolase inhibitors are now tested under different clinical trials. Of them, three oral inhibitors have now advanced to global phase III clinical trial for the treatment of anemia (Joharapurkar et al. 2018; Sanghani and Haase 2019). Nevertheless, to the light of our results (Valle-Tenney et al. 2019) and Fig. 2, caution should be taken evaluating the potential effect of these inhibitors in long-term sustained treatments in order to avoid undesired consequences such as a muscle fibrotic response.
Regulation of skeletal muscle CCN2/CTGF expression
Transforming growth factor β (TGF-β) signaling has been described as the master fibrotic driver in several organs and tissues including skeletal muscle, where is up-regulated in fibrosis-related pathologies of diverse etiology (Cabello-Verrugio et al. 2012b; Ismaeel et al. 2019; Leask and Abraham 2004). TGF-β signaling occurs through Smad-dependent or Smad-independent pathways driving the expression of target genes such as collagens, fibronectin, and CCN2, among others. CCN2, a member of the CCN family of matricellular proteins, has risen as a crucial pro-fibrotic factor in skeletal muscle, and it is considered one of the downstream effectors of fibrotic TGF-β signaling (Biernacka et al. 2011; Duncan et al. 1999; Vial et al. 2008). CCN2 is overexpressed in biopsies of fibrotic skeletal muscle from DMD (Sun et al. 2008), and in the mdx mouse model for DMD (Au et al. 2011; Morales et al. 2013b), under conditions of repetitive damage (Pessina et al. 2014), the transgenic mouse model for ALS(tg hSOD1G93A) (Gonzalez et al. 2017; Gonzalez et al. 2018), and after denervation by sciatic nerve transection (Rebolledo et al. 2019). Accordingly, CCN2 reduction or blockage attenuates skeletal muscle fibrosis in these pathological models (Gonzalez et al. 2018; Morales et al. 2013b; Rebolledo et al. 2019) evidencing its critical role in fibrosis progression. Therefore, the understanding of how CCN2 is regulated results particularly relevant in fibrotic diseases.
The CCN2 promoter region has several transcription factor response elements, including Smad Binding Elements, Activator Protein 1 (AP-1), Specific protein 1, Nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells, SOX9, and also Hypoxia Response Elements (HRE) (Cordova et al. 2015; Chaqour et al. 2006; Higgins et al. 2004; Leask et al. 2001; Oh et al. 2016). Therefore, CCN2 can be regulated by hypoxia even though the role of HIF-1α on CCN2 expression is intriguing and appears to be highly cell-type specific. Several reports show that hypoxia induces CCN2 expression in skin fibroblasts (Hong et al. 2006; Mingyuan et al. 2018) and in mouse tubular cells (Higgins et al. 2004). On the other hand, the activation of HIF-1α downregulates CCN2 expression in chondrocytes (Tran et al. 2010) and in human renal tubular cells (Preisser et al. 2016). We recently showed that hypoxia, through HIF-1α, induces CCN2 expression in skeletal muscle fibers in vitro, and in vivo (Valle-Tenney et al. 2019).
In the context of fibrosis, CCN2 expression could be regulated by HIF-1α in conjunction with other transcription factors related to non-canonical TGF-β signaling pathways. For example, it has been described that the promoter region of the CCN2 gene has an AP-1 site that is responsive to GLI proteins, which are downstream of the MEK/ERK pathway (Cheng et al. 2016). Also, CCN1 gene expression (another member of the CCN protein family) is induced by hypoxia and HIF-1 interaction with c-Jun/AP-1 in melanoma cells (Kunz and Ibrahim 2003). Therefore, several transcription factors, including TGF-β-activated transcription factors, could be involved in HIF-1α-dependent regulation of CCN2 expression in skeletal muscle fibrosis. When hypoxia or HIF-1α stabilization occurs in an environment enriched in TGF-β, a synergistic overexpression of CCN2 is observed, which is greater than the response for each separate factor. Interestingly, this effect is specific for skeletal muscle myotubes/fibers and not for other skeletal muscle resident cell types, supporting the idea that the regulation of CCN2 by HIF-1α is highly cell-specific (Valle-Tenney et al. 2019). It has been reported that Smads can interact with several transcription factors in some cases serving as a scaffold for a large transcription complex that regulates a variety of genes (Euler-Taimor and Heger 2006). Among them, the SP1/SMAD3/HIF-1α multiprotein complex regulates endoglin (Sanchez-Elsner et al. 2002), and the YAP-TEAD4–Smad3-p300 complex regulates CCN2 in cancer cells (Fujii et al. 2012). Thus, HIF-1α signaling could exert a pro-fibrotic effect in skeletal muscle through the expression of the pro-fibrotic factor CCN2 in sustained hypoxic conditions. However, more research is needed in order to understand the transcriptional regulation of CCN2 by HIF and cofactors in skeletal muscle fibrosis.
Relationship between muscle fibrosis, hypoxia, and the renin-angiotensin system
The renin-angiotensin system (RAS) is involved in numerous physiological functions that regulate vasoconstriction, fluid volume regulation, cardiac output, cell growth, vascular wall integrity and fibrosis (Arendse et al. 2019; Santos et al. 2018). The RAS has two axes with opposite functions. The classical axis is composed of the octapeptide Angiotensin II (Ang-II), the enzyme that synthesizes it, Angiotensin Converting Enzyme (ACE) and Angiotensin Type I receptor (AT1). Ang-II via the AT1 receptor produces vasoconstriction, sodium retention, water reabsorption, vascular remodeling, inflammation and fibrosis (Arendse et al. 2019). Since Ang-II induces vasoconstriction it suggests that it could contribute to a hypoxic niche and could be another way to induce damage. The alternative RAS axis is the non-classical, composed of Angiotensin-(1–7) (Ang-1-7), its receptor Mas and ACE2 (Santos et al. 2019; Santos et al. 2018). Ang-(1–7), through the activation of Mas, has the opposite functions of the classic RAS: induces vasodilation, reduces inflammation, reduces oxidative damage and fibrosis. In cardiac tissue, Ang-(1–7) counteracts the effects of Ang-II, and trough Mas induces activation of eNOS that results in increased NO levels, which reduce oxidative damage and produce an increase in blood flow in the heart by vasodilating the coronary arteries and blood vessels (Costa et al. 2010; Dias-Peixoto et al. 2008; Ferreira et al. 2019; Sampaio et al. 2007), suggesting that one of the beneficial effects of Ang-(1–7) could be a reduction of the hypoxic state.
In skeletal muscle Ang-II is involved in inflammation and fibrosis progression associated with pathologies. It is also involved in the loss of muscle mass by increasing levels in oxidative stress and inflammatory mediators (Brink et al. 2001; Delafontaine and Yoshida 2016; Semprun-Prieto et al. 2011; Sukhanov et al. 2011). In models of muscular dystrophy, the treatment with AT1 antagonists (ARBs) or ACE inhibitors (ACEi) leads to a reduction of skeletal muscle fibrosis, reduced TGF-β signaling, reduction of CCN2 levels, inflammation and oxidative stress (Cabello-Verrugio et al. 2012a; Cohn et al. 2007; Cozzoli et al. 2011; Morales et al. 2013a). These results support a contribution of Ang-II to skeletal muscle damage in these pathologies. The inhibition of ACE activity has been used as a therapeutic treatment in mdx mice and in patients with Becker and DMD to improve the cardiomyopathy associated with dystrophy (Bauer et al. 2009; Russo et al. 2018; Silva et al. 2017).
On the other hand, the non-classical RAS pathway plays a protective role in skeletal muscle diseases. In mdx mice, Ang-(1–7) infusion or oral administration ameliorate skeletal muscle architecture, decrease local fibrosis, and improve muscle function by reducing TGF-β signaling (Acuña et al. 2014) and decreasing CCN2 levels (Acuña et al. 2019). Similar effects in improving the fibrotic phenotype were also observed in the sarcoglycan delta null mice, a different model of skeletal muscular dystrophy (Sabharwal and Chapleau 2014). Moreover, the overexpression of ACE2 reduces fibrosis in mdx mice and in a model of fibrosis caused by chronic injury (Riquelme et al. 2014).
Even though it seems that Ang-(1–7) effects are caused by the attenuation of fibrotic pathways, there is also evidence that in vascular tissue it can induce NOS activity, specifically eNOS in endothelial cells. This events suggests the induction of local vasodilation, which leads to reduced fibrosis, vascular remodeling and inflammation (Santos et al. 2018), Moreover, Ang-(1–7) effects are reduced by treatment with L-NAME or Guanylyl cyclase inhibitors that block NO signaling (Ferreira et al. 2019; Gomes et al. 2010; Ren et al. 2002). Taking this data together, we suggest that the beneficial effect of the Ang-(1–7) axis of the RAS system in muscle fibrosis is tightly linked to NO production and vasodilation, and also inhibition of pro-fibrotic cytokines, leaving open the question if such activity could contribute to decreasing the chronic activation of hypoxia signaling in skeletal muscle.
RAS and HIF-1α in skeletal muscle dystrophies
There is evidence showing that Ang-II induces HIF-1α in the kidney, vascular smooth muscle cells (VSMC), and cardiomyocytes (Huang et al. 2019; Qi et al. 2019; Richard et al. 2000; Yan et al. 2020; Zhu et al. 2011). Interestingly, selective HIF-1α knock out in VSMC indicates that Ang-II-induced vascular remodeling is mediated by HIF-1α. These knockout mice show reduced inflammation and decreased blood pressure in an Ang-II-induced hypertension model (Qi et al. 2019). In another report, Ang-II induced fibrosis and inflammation in podocytes was reduced when HIF-1α expression was knockdown by siRNA (Huang et al. 2019). Similarly, kidney injury caused by Ang-II infusion was reduced by HIF-1α silencing in rats (Zhu et al. 2011). Even though there is no evidence of Ang-II induction of HIF-1α in skeletal muscle, it remains a relevant question since Ang-II induces TGF-β and CCN2 in skeletal muscle and could be acting upstream of these profibrotic factors. Also, Ang-II in VSMC induces HIF-1α by a mechanism independent of hypoxia (Richard et al. 2000). Therefore, it would be interesting to evaluate if ARBs or ACEi treated mdx muscles have reduced levels of HIF-1α, and if HIF-1α could also be a mediator of the Ang-II induced fibrotic response in this model or in other models of skeletal muscle fibrosis.
There is also evidence that links the other RAS axis to HIF-1α. Ang-(1–7) treatment reduces inflammation and damage, by a mechanism involving the induction of NO and reduction of HIF-1α, in a model of reflux esophagitis (Pawlik et al. 2014). In kidney damage by diabetic nephropathy, Ang-(1–7) reduced the induction of HIF-1α (Giani et al. 2012). In vitro, Ang-(1–7) treatment also reduced Hif-1α expression in vascular sarcoma cells (Petty et al. 2012) and attenuated long-term hypoxia-stimulated apoptosis by inhibiting HIF-1α nuclear translocation, via Mas receptor, in cardiomyocytes (Chang et al. 2016). Therefore, the non-classical arm of RAS is a novel target regarding hypoxia and HIF-1α signaling.
In skeletal muscle, RAS plays a role in the progression of fibrosis, whether Ang-II induces HIF-1α and Ang-(1–7) can reduce HIF-1α signaling in skeletal muscle pathologies is still an open question and needs further investigation.
Conclusion and future perspectives
Considering all the evidence presented above, transient hypoxia and HIF signaling in acute events such as exercising or regeneration after muscle damage, play an important role in homeostasis promoting angiogenesis by vascular remodeling, the metabolic adaptation of muscle fibers, maintenance of stem cell stemness and immune response modulation. However, the role of sustained activation of hypoxic signaling in pro-fibrotic diseases has not been studied. Here, we propose that the chronic hypoxic state present in skeletal muscle pathologies is an important feature that contributes to the establishment of fibrosis. Moreover, based on the preliminary data presented here (Fig. 1), we hypothesize that HIF-mediated hypoxic signaling is determinant for the detrimental effects of chronic hypoxia resulting in CCN2 overexpression as we recently publish (Valle-Tenney et al. 2019)
Several reports have pointed to a relationship between capillary disfunction and skeletal muscle damage and fibrosis. This evidence leads us to hypothesize that hypoxic signaling through HIF-1α could be active in the muscle inducing the expression of several genes with HRE on their promoters that could be triggering a fibrotic response. Among these genes, CCN2 is a key pro-fibrotic factor that is regulated by hypoxia. We have recently shown that hypoxia through HIF-1α leads to overexpression of CTGF. Furthermore, HIF-1α synergizes with TGF-β on driving CCN2 overexpression (Valle-Tenney et al. 2019). In this way, HIF-1α can be acting together with other transcription factors that are also increased in pathological conditions, contributing to the establishment of fibrosis. The knowledge of these crosstalks can help to develop new combinatory therapies for skeletal muscle diseases where fibrosis is involved.
Abbreviations
- AP-1:
-
Activator Protein 1
- ALS:
-
Amyotrophic lateral sclerosis
- ACE:
-
Angiotensin Converting Enzyme
- Ang-II:
-
Angiotensin II
- Ang-1-7:
-
Angiotensin-(1–7)
- AT1:
-
Angiotensin Type I receptor
- ARBs:
-
Angiotensin Type I receptor antagonists
- CTGF/CCN2:
-
Connective Tissue Growth Factor
- CTX:
-
Cardiotoxin
- DMOG:
-
Dimethyloxalylglycine
- DMD:
-
Duchenne muscular dystrophy
- ECM:
-
Extracellular matrix
- FOG:
-
Fast-twitch oxidative glycolytic
- PHD:
-
HIF-prolyl hydroxylases domain-containing enzymes
- HIF-α:
-
Hypoxia-Inducible Factor α
- HRE:
-
Hypoxia Response Elements
- NO:
-
Nitric oxide
- nNOSμ:
-
Nitric oxide synthase
- NTX:
-
Notexin
- RAS:
-
Renin-angiotensin system
- SO:
-
Slow-twitch oxidative
- TGF-β:
-
Transforming growth factor β
- VEGF:
-
Vascular Endothelial Growth Factor
- VSMC:
-
Vascular smooth muscle cells
References
Acuña MJ, Pessina P, Olguin H, Cabrera D, Vio CP, Bader M, Munoz-Canoves P, Santos RA, Cabello-Verrugio C, Brandan E (2014) Restoration of muscle strength in dystrophic muscle by angiotensin-1-7 through inhibition of TGF-beta signalling. Hum Mol Genet 23:1237–1249
Acuña MJ, Brandan E, Motta-Santos D (2019) Skeletal muscle system. Angiotensin-(1-7): a comprehensive review. Springer International Publishing, Cham, pp 169–190
Akanji MA, Rotimi D, Adeyemi OS (2019) Hypoxia-inducible factors as an alternative source of treatment strategy for Cancer. Oxidative Med Cell Longev 2019:8547846
Alvarez K, Fadic R, Brandan E (2002) Augmented synthesis and differential localization of heparan sulfate proteoglycans in Duchenne muscular dystrophy. J Cell Biochem 85:703–713
Ameln H, Gustafsson T, Sundberg CJ, Okamoto K, Jansson E, Poellinger L, Makino Y (2005) Physiological activation of hypoxia inducible factor-1 in human skeletal muscle. FASEB J 19:1009–1011
Anderson JE (2000) A role for nitric oxide in muscle repair: nitric oxide-mediated activation of muscle satellite cells. Mol Biol Cell 11:1859–1874
Aragones J, Schneider M, Van Geyte K, Fraisl P, Dresselaers T, Mazzone M, Dirkx R, Zacchigna S, Lemieux H, Jeoung NH, Lambrechts D, Bishop T, Lafuste P, Diez-Juan A, Harten SK, Van Noten P, De Bock K, Willam C, Tjwa M, Grosfeld A, Navet R, Moons L, Vandendriessche T, Deroose C, Wijeyekoon B, Nuyts J, Jordan B, Silasi-Mansat R, Lupu F, Dewerchin M, Pugh C, Salmon P, Mortelmans L, Gallez B, Gorus F, Buyse J, Sluse F, Harris RA, Gnaiger E, Hespel P, Van Hecke P, Schuit F, Van Veldhoven P, Ratcliffe P, Baes M, Maxwell P, Carmeliet P (2008) Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet 40:170–180
Arendse LB, Danser AHJ, Poglitsch M, Touyz RM, Burnett JC Jr, Llorens-Cortes C, Ehlers MR, Sturrock ED (2019) Novel therapeutic approaches targeting the renin-angiotensin system and associated peptides in hypertension and heart failure. Pharmacol Rev 71:539–570
Arsic N, Zacchigna S, Zentilin L, Ramirez-Correa G, Pattarini L, Salvi A, Sinagra G, Giacca M (2004) Vascular endothelial growth factor stimulates skeletal muscle regeneration in vivo. Mol Ther 10:844–854
Au CG, Butler TL, Sherwood MC, Egan JR, North KN, Winlaw DS (2011) Increased connective tissue growth factor associated with cardiac fibrosis in the mdx mouse model of dystrophic cardiomyopathy. Int J Exp Pathol 92:57–65
Bauer R. Straub V. Blain A. Bushby K. MacGowan GA. (2009) Contrasting effects of steroids and angiotensin-converting-enzyme inhibitors in a mouse model of dystrophin-deficient cardiomyopathy. Eur J Heart Fail 11:463–71
Bentzinger CF, Wang YX, Dumont NA, Rudnicki MA (2013) Cellular dynamics in the muscle satellite cell niche. EMBO Rep 14:1062–1072
Biernacka A, Dobaczewski M, Frangogiannis NG (2011) TGF-beta signaling in fibrosis. Growth Factors 29:196–202
Billin AN, Honeycutt SE, McDougal AV, Kerr JP, Chen Z, Freudenberg JM, Rajpal DK, Luo G, Kramer HF, Geske RS, Fang F, Yao B, Clark RV, Lepore J, Cobitz A, Miller R, Nosaka K, Hinken AC, Russell AJ (2018) HIF prolyl hydroxylase inhibition protects skeletal muscle from eccentric contraction-induced injury. Skelet Muscle 8:35
Borisov AB, Huang SK, Carlson BM (2000) Remodeling of the vascular bed and progressive loss of capillaries in denervated skeletal muscle. Anat Rec 258:292–304
Borselli C, Storrie H, Benesch-Lee F, Shvartsman D, Cezar C, Lichtman JW, Vandenburgh HH, Mooney DJ (2010) Functional muscle regeneration with combined delivery of angiogenesis and myogenesis factors. Proc Natl Acad Sci U S A 107:3287–3292
Brink M, Price SR, Chrast J, Bailey JL, Anwar A, Mitch WE, Delafontaine P (2001) Angiotensin II induces skeletal muscle wasting through enhanced protein degradation and down-regulates autocrine insulin-like growth factor I. Endocrinology. 142:1489–1496
Buono R, Vantaggiato C, Pisa V, Azzoni E, Bassi MT, Brunelli S, Sciorati C, Clementi E (2012) Nitric oxide sustains long-term skeletal muscle regeneration by regulating fate of satellite cells via signaling pathways requiring Vangl2 and cyclic GMP. Stem Cells 30:197–209
Cabello-Verrugio C, Morales MG, Cabrera D, Vio CP, Brandan E (2012a) Angiotensin II receptor type 1 blockade decreases CTGF/CCN2-mediated damage and fibrosis in normal and dystrophic skeletal muscles. J Cell Mol Med 16:752–764
Cabello-Verrugio C, Santander C, Cofre C, Acuna MJ, Melo F, Brandan E (2012b) The internal region leucine-rich repeat 6 of decorin interacts with low density lipoprotein receptor-related protein-1, modulates transforming growth factor (TGF)-beta-dependent signaling, and inhibits TGF-beta-dependent fibrotic response in skeletal muscles. J Biol Chem 287:6773–6787
Caceres S, Cuellar C, Casar JC, Garrido J, Schaefer L, Kresse H, Brandan E (2000) Synthesis of proteoglycans is augmented in dystrophic mdx mouse skeletal muscle. Eur J Cell Biol 79:173–181
Chaillou T (2018) Skeletal muscle Fiber type in hypoxia: adaptation to high-altitude exposure and under conditions of pathological hypoxia. Front Physiol 9:1450
Chaillou T, Koulmann N, Meunier A, Pugniere P, McCarthy JJ, Beaudry M, Bigard X (2014) Ambient hypoxia enhances the loss of muscle mass after extensive injury. Pflugers Arch 466:587–598
Chang RL, Lin JW, Kuo WW, Hsieh DJ, Yeh YL, Shen CY, Day CH, Ho TJ, Viswanadha VP, Huang CY (2016) Angiotensin-(1-7) attenuated long-term hypoxia-stimulated cardiomyocyte apoptosis by inhibiting HIF-1alpha nuclear translocation via Mas receptor regulation. Growth Factors 34:11–18
Chaqour B, Yang R, Sha Q (2006) Mechanical stretch modulates the promoter activity of the profibrotic factor CCN2 through increased actin polymerization and NF-kappaB activation. J Biol Chem 281:20608–20622
Chavoshan B, Sander M, Sybert TE, Hansen J, Victor RG, Thomas GD (2002) Nitric oxide-dependent modulation of sympathetic neural control of oxygenation in exercising human skeletal muscle. J Physiol 540:377–386
Cheng Y, Lin CH, Chen JY, Li CH, Liu YT, Chen BC (2016) Induction of connective tissue growth factor expression by hypoxia in human lung fibroblasts via the MEKK1/MEK1/ERK1/GLI-1/GLI-2 and AP-1 pathways. PLoS One 11:e0160593
Cohn RD, van Erp C, Habashi JP, Soleimani AA, Klein EC, Lisi MT, Gamradt M, ap Rhys CM, Holm TM, Loeys BL, Ramirez F, Judge DP, Ward CW, Dietz HC (2007) Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med 13:204–210
Contreras O, Rebolledo DL, Oyarzun JE, Olguin HC, Brandan E (2016) Connective tissue cells expressing fibro/adipogenic progenitor markers increase under chronic damage: relevance in fibroblast-myofibroblast differentiation and skeletal muscle fibrosis. Cell Tissue Res 364:647–660
Cordova G, Rochard A, Riquelme-Guzman C, Cofre C, Scherman D, Bigey P, Brandan E (2015) SMAD3 and SP1/SP3 transcription factors collaborate to regulate connective tissue growth factor gene expression in myoblasts in response to transforming growth factor beta. J Cell Biochem 116:1880–1887
Costa MA, Lopez Verrilli MA, Gomez KA, Nakagawa P, Pena C, Arranz C, Gironacci MM (2010) Angiotensin-(1-7) upregulates cardiac nitric oxide synthase in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 299:H1205–H1211
Cozzoli A, Nico B, Sblendorio VT, Capogrosso RF, Dinardo MM, Longo V, Gagliardi S, Montagnani M, De Luca A (2011) Enalapril treatment discloses an early role of angiotensin II in inflammation- and oxidative stress-related muscle damage in dystrophic mdx mice. Pharmacol Res 64:482–492
Dadgar S, Wang Z, Johnston H, Kesari A, Nagaraju K, Chen YW, Hill DA, Partridge TA, Giri M, Freishtat RJ, Nazarian J, Xuan J, Wang Y, Hoffman EP (2014) Asynchronous remodeling is a driver of failed regeneration in Duchenne muscular dystrophy. J Cell Biol 207:139–158
Darby IA, Hewitson TD (2016) Hypoxia in tissue repair and fibrosis. Cell Tissue Res 365:553–562
Delafontaine P, Yoshida T (2016) The renin-angiotensin system and the biology of skeletal muscle: mechanisms of muscle wasting in chronic disease states. Trans Am Clin Climatol Assoc 127:245–258
Dias-Peixoto MF, Santos RA, Gomes ER, Alves MN, Almeida PW, Greco L, Rosa M, Fauler B, Bader M, Alenina N, Guatimosim S (2008) Molecular mechanisms involved in the angiotensin-(1-7)/mas signaling pathway in cardiomyocytes. Hypertension. 52:542–548
Distler JH, Jungel A, Pileckyte M, Zwerina J, Michel BA, Gay RE, Kowal-Bielecka O, Matucci-Cerinic M, Schett G, Marti HH, Gay S, Distler O (2007) Hypoxia-induced increase in the production of extracellular matrix proteins in systemic sclerosis. Arthritis Rheum 56:4203–4215
Doria C, Toniolo L, Verratti V, Cancellara P, Pietrangelo T, Marconi V, Paoli A, Pogliaghi S, Fano G, Reggiani C, Capelli C (2011) Improved VO2 uptake kinetics and shift in muscle fiber type in high-altitude trekkers. J Appl Physiol (1985) 111:1597–1605
Drouin G, Couture V, Lauzon MA, Balg F, Faucheux N, Grenier G (2019) Muscle injury-induced hypoxia alters the proliferation and differentiation potentials of muscle resident stromal cells. Skelet Muscle 9:18
Duncan MR, Frazier KS, Abramson S, Williams S, Klapper H, Huang X, Grotendorst GR (1999) Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: down-regulation by cAMP. FASEB J 13:1774–1786
Ennen JP, Verma M, Asakura A (2013) Vascular-targeted therapies for Duchenne muscular dystrophy. Skelet Muscle 3:9
Euler-Taimor G, Heger J (2006) The complex pattern of SMAD signaling in the cardiovascular system. Cardiovasc Res 69:15–25
Ferreira AJ, Castro CH, Santos RAS (2019) Heart - coronary vessels and Cardiomyocytes. Angiotensin-(1-7): a comprehensive review. Springer International Publishing, Cham, pp 73–81
Forristal CE, Winkler IG, Nowlan B, Barbier V, Walkinshaw G, Levesque JP (2013) Pharmacologic stabilization of HIF-1alpha increases hematopoietic stem cell quiescence in vivo and accelerates blood recovery after severe irradiation. Blood. 121:759–769
Fujii M, Toyoda T, Nakanishi H, Yatabe Y, Sato A, Matsudaira Y, Ito H, Murakami H, Kondo Y, Kondo E, Hida T, Tsujimura T, Osada H, Sekido Y (2012) TGF-beta synergizes with defects in the hippo pathway to stimulate human malignant mesothelioma growth. J Exp Med 209:479–494
Gargioli C, Coletta M, De Grandis F, Cannata SM, Cossu G (2008) PlGF-MMP-9-expressing cells restore microcirculation and efficacy of cell therapy in aged dystrophic muscle. Nat Med 14:973–978
Giani JF, Burghi V, Veiras LC, Tomat A, Munoz MC, Cao G, Turyn D, Toblli JE, Dominici FP (2012) Angiotensin-(1-7) attenuates diabetic nephropathy in Zucker diabetic fatty rats. Am J Physiol Ren Physiol 302:F1606–F1615
Gomes ER, Lara AA, Almeida PW, Guimaraes D, Resende RR, Campagnole-Santos MJ, Bader M, Santos RA, Guatimosim S (2010) Angiotensin-(1-7) prevents cardiomyocyte pathological remodeling through a nitric oxide/guanosine 3′,5′-cyclic monophosphate-dependent pathway. Hypertension. 55:153–160
Gonzalez D, Contreras O, Rebolledo DL, Espinoza JP, van Zundert B, Brandan E (2017) ALS skeletal muscle shows enhanced TGF-beta signaling, fibrosis and induction of fibro/adipogenic progenitor markers. PLoS One 12:e0177649
Gonzalez D, Rebolledo DL, Correa LM, Court FA, Cerpa W, Lipson KE, van Zundert B, Brandan E (2018) The inhibition of CTGF/CCN2 activity improves muscle and locomotor function in a murine ALS model. Hum Mol Genet 27:2913–2926
Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, Ruas JL, Poellinger L, Lendahl U, Bondesson M (2005) Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell 9:617–628
Gutpell KM, Hoffman LM (2015) VEGF induces stress fiber formation in fibroblasts isolated from dystrophic muscle. J Cell Commun Signal 9:353–360
Hardy D, Besnard A, Latil M, Jouvion G, Briand D, Thepenier C, Pascal Q, Guguin A, Gayraud-Morel B, Cavaillon JM, Tajbakhsh S, Rocheteau P, Chretien F (2016) Comparative study of injury models for studying muscle regeneration in mice. PLoS One 11:e0147198
Higgins DF, Biju MP, Akai Y, Wutz A, Johnson RS, Haase VH (2004) Hypoxic induction of Ctgf is directly mediated by Hif-1. Am J Physiol Ren Physiol 287:F1223–F1232
Hong KS, Kim K (2017) Skeletal muscle contraction-induced vasodilation in the microcirculation. J Exerc Rehabil 13:502–507
Hong KH, Yoo SA, Kang SS, Choi JJ, Kim WU, Cho CS (2006) Hypoxia induces expression of connective tissue growth factor in scleroderma skin fibroblasts. Clin Exp Immunol 146:362–370
Hotta K, Behnke BJ, Masamoto K, Shimotsu R, Onodera N, Yamaguchi A, Poole DC, Kano Y (2018) Microvascular permeability of skeletal muscle after eccentric contraction-induced muscle injury: in vivo imaging using two-photon laser scanning microscopy. J Appl Physiol (1985) 125:369–380
Huang H, Fan Y, Gao Z, Wang W, Shao N, Zhang L, Yang Y, Zhu W, Chen Z, Hu J, Ding G (2019) HIF-1alpha contributes to Ang II-induced inflammatory cytokine production in podocytes. BMC Pharmacol Toxicol 20:59
Hudlicka O (1967) Blood flow and oxygen consumption in muscles after section of ventral roots. Circ Res 20:570–577
Ismaeel A, Kim JS, Kirk JS, Smith RS, Bohannon WT, Koutakis P (2019) Role of transforming growth factor-beta in skeletal muscle fibrosis: a review. Int J Mol Sci 20
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr (2001) HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 292:464–468
Jendzjowsky NG, DeLorey DS (2013) Role of neuronal nitric oxide in the inhibition of sympathetic vasoconstriction in resting and contracting skeletal muscle of healthy rats. J Appl Physiol (1985) 115:97–106
Joharapurkar AA, Pandya VB, Patel VJ, Desai RC, Jain MR (2018) Prolyl hydroxylase inhibitors: a breakthrough in the therapy of Anemia associated with chronic diseases. J Med Chem 61:6964–6982
Ke Q, Costa M (2006) Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol 70:1469–1480
Kimura K, Iwano M, Higgins DF, Yamaguchi Y, Nakatani K, Harada K, Kubo A, Akai Y, Rankin EB, Neilson EG, Haase VH, Saito Y (2008) Stable expression of HIF-1alpha in tubular epithelial cells promotes interstitial fibrosis. Am J Physiol Ren Physiol 295:F1023–F1029
Koh MY, Spivak-Kroizman TR, Powis G (2009) HIF-1 regulation: not so easy come, easy go. Trends Biochem Sci 33:526–534
Kunz M, Ibrahim SM (2003) Molecular responses to hypoxia in tumor cells. Mol Cancer 2:23
Leask A, Abraham DJ (2004) TGF-beta signaling and the fibrotic response. FASEB J 18:816–827
Leask A, Sa S, Holmes A, Shiwen X, Black CM, Abraham DJ (2001) The control of ccn2 (ctgf) gene expression in normal and scleroderma fibroblasts. Mol Pathol 54:180–183
Leu T, Schutzhold V, Fandrey J, Ferenz KB (2019) When the brain yearns for oxygen. Neurosignals. 27:50–61
Lunde IG, Anton SL, Bruusgaard JC, Rana ZA, Ellefsen S, Gundersen K (2011) Hypoxia inducible factor 1 links fast-patterned muscle activity and fast muscle phenotype in rats. J Physiol 589:1443–1454
Mahdy MAA (2018) Skeletal muscle fibrosis: an overview. Cell Tissue Res
Majmundar AJ, Skuli N, Mesquita RC, Kim MN, Yodh AG, Nguyen-McCarty M, Simon MC (2012) O(2) regulates skeletal muscle progenitor differentiation through phosphatidylinositol 3-kinase/AKT signaling. Mol Cell Biol 32:36–49
Majmundar AJ, Lee DS, Skuli N, Mesquita RC, Kim MN, Yodh AG, Nguyen-McCarty M, Li B, Simon MC (2015) HIF modulation of Wnt signaling regulates skeletal myogenesis in vivo. Development. 142:2405–2412
Mann CJ, Perdiguero E, Kharraz Y, Aguilar S, Pessina P, Serrano AL, Munoz-Canoves P (2011) Aberrant repair and fibrosis development in skeletal muscle. Skelet Muscle 1:21
Mason S, Johnson RS (2007) The role of HIF-1 in hypoxic response in the skeletal muscle. Adv Exp Med Biol 618:229–244
Mason SD, Howlett RA, Kim MJ, Olfert IM, Hogan MC, McNulty W, Hickey RP, Wagner PD, Kahn CR, Giordano FJ, Johnson RS (2004) Loss of skeletal muscle HIF-1alpha results in altered exercise endurance. PLoS Biol 2:e288
Matsakas A, Yadav V, Lorca S, Narkar V (2013) Muscle ERRgamma mitigates Duchenne muscular dystrophy via metabolic and angiogenic reprogramming. FASEB J 27:4004–4016
Messina S, Mazzeo A, Bitto A, Aguennouz M, Migliorato A, De Pasquale MG, Minutoli L, Altavilla D, Zentilin L, Giacca M, Squadrito F, Vita G (2007) VEGF overexpression via adeno-associated virus gene transfer promotes skeletal muscle regeneration and enhances muscle function in mdx mice. FASEB J 21:3737–3746
Mingyuan X, Qianqian P, Shengquan X, Chenyi Y, Rui L, Yichen S, Jinghong X (2018) Hypoxia-inducible factor-1alpha activates transforming growth factor-beta1/Smad signaling and increases collagen deposition in dermal fibroblasts. Oncotarget. 9:3188–3197
Morales MG, Cabrera D, Cespedes C, Vio CP, Vazquez Y, Brandan E, Cabello-Verrugio C (2013a) Inhibition of the angiotensin-converting enzyme decreases skeletal muscle fibrosis in dystrophic mice by a diminution in the expression and activity of connective tissue growth factor (CTGF/CCN-2). Cell Tissue Res 353:173–187
Morales MG, Gutierrez J, Cabello-Verrugio C, Cabrera D, Lipson KE, Goldschmeding R, Brandan E (2013b) Reducing CTGF/CCN2 slows down mdx muscle dystrophy and improves cell therapy. Hum Mol Genet 22:4938–4951
Mounier R, Pedersen BK, Plomgaard P (2010) Muscle-specific expression of hypoxia-inducible factor in human skeletal muscle. Exp Physiol 95:899–907
Niemi H, Honkonen K, Korpisalo P, Huusko J, Kansanen E, Merentie M, Rissanen TT, Andre H, Pereira T, Poellinger L, Alitalo K, Yla-Herttuala S (2014) HIF-1alpha and HIF-2alpha induce angiogenesis and improve muscle energy recovery. Eur J Clin Investig 44:989–999
Norman JT, Clark IM, Garcia PL (2000) Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int 58:2351–2366
Oh CD, Yasuda H, Zhao W, Henry SP, Zhang Z, Xue M, de Crombrugghe B, Chen D (2016) SOX9 directly regulates CTGF/CCN2 transcription in growth plate chondrocytes and in nucleus Pulposus cells of intervertebral disc. Sci Rep 6:29916
Olenchock BA, Moslehi J, Baik AH, Davidson SM, Williams J, Gibson WJ, Chakraborty AA, Pierce KA, Miller CM, Hanse EA, Kelekar A, Sullivan LB, Wagers AJ, Clish CB, Vander Heiden MG, Kaelin WG Jr (2016) EGLN1 inhibition and rerouting of alpha-Ketoglutarate suffice for remote ischemic protection. Cell. 165:497
Olfert IM, Howlett RA, Wagner PD, Breen EC (2010) Myocyte vascular endothelial growth factor is required for exercise-induced skeletal muscle angiogenesis. Am J Phys Regul Integr Comp Phys 299:R1059–R1067
Ono Y, Sensui H, Sakamoto Y, Nagatomi R (2006) Knockdown of hypoxia-inducible factor-1alpha by siRNA inhibits C2C12 myoblast differentiation. J Cell Biochem 98:642–649
Orphanides C, Fine LG, Norman JT (1997) Hypoxia stimulates proximal tubular cell matrix production via a TGF-beta1-independent mechanism. Kidney Int 52:637–647
Pawlik MW, Kwiecien S, Pajdo R, Ptak-Belowska A, Brzozowski B, Krzysiek-Maczka G, Strzalka M, Konturek SJ, Brzozowski T (2014) Esophagoprotective activity of angiotensin-(1-7) in experimental model of acute reflux esophagitis. Evidence for the role of nitric oxide, sensory nerves, hypoxia-inducible factor-1alpha and proinflammatory cytokines. J Physiol Pharmacol 65:809–822
Pessina P, Cabrera D, Morales MG, Riquelme CA, Gutierrez J, Serrano AL, Brandan E, Munoz-Canoves P (2014) Novel and optimized strategies for inducing fibrosis in vivo: focus on Duchenne muscular dystrophy. Skelet Muscle 4:7
Petty WJ, Aklilu M, Varela VA, Lovato J, Savage PD, Miller AA (2012) Reverse translation of phase I biomarker findings links the activity of angiotensin-(1-7) to repression of hypoxia inducible factor-1alpha in vascular sarcomas. BMC Cancer 12:404
Pisani DF, Dechesne CA (2005) Skeletal muscle HIF-1alpha expression is dependent on muscle fiber type. J Gen Physiol 126:173–178
Podkalicka P, Mucha O, Dulak J, Loboda A (2019) Targeting angiogenesis in Duchenne muscular dystrophy. Cell Mol Life Sci 76:1507–1528
Preisser F, Giehl K, Rehm M, Goppelt-Struebe M (2016) Inhibitors of oxygen sensing prolyl hydroxylases regulate nuclear localization of the transcription factors Smad2 and YAP/TAZ involved in CTGF synthesis. Biochim Biophys Acta 1863:2027–2036
Qi D, Wei M, Jiao S, Song Y, Wang X, Xie G, Taranto J, Liu Y, Duan Y, Yu B, Li H, Shah YM, Xu Q, Du J, Gonzalez FJ, Qu A (2019) Hypoxia inducible factor 1alpha in vascular smooth muscle cells promotes angiotensin II-induced vascular remodeling via activation of CCL7-mediated macrophage recruitment. Cell Death Dis 10:544
Rana MK, Srivastava J, Yang M, Chen CS, Barber DL (2015) Hypoxia increases the abundance but not the assembly of extracellular fibronectin during epithelial cell transdifferentiation. J Cell Sci 128:1083–1089
Rasbach KA, Gupta RK, Ruas JL, Wu J, Naseri E, Estall JL, Spiegelman BM (2010) PGC-1alpha regulates a HIF2alpha-dependent switch in skeletal muscle fiber types. Proc Natl Acad Sci U S A 107:21866–21871
Rebolledo DL, Gonzalez D, Faundez-Contreras J, Contreras O, Vio CP, Murphy-Ullrich JE, Lipson KE, Brandan E (2019) Denervation-induced skeletal muscle fibrosis is mediated by CTGF/CCN2 independently of TGF-beta. Matrix Biol
Reischl S, Li L, Walkinshaw G, Flippin LA, Marti HH, Kunze R (2014) Inhibition of HIF prolyl-4-hydroxylases by FG-4497 reduces brain tissue injury and edema formation during ischemic stroke. PLoS One 9:e84767
Ren Y, Garvin JL, Carretero OA (2002) Vasodilator action of angiotensin-(1-7) on isolated rabbit afferent arterioles. Hypertension. 39:799–802
Richard DE, Berra E, Pouyssegur J (2000) Nonhypoxic pathway mediates the induction of hypoxia-inducible factor 1alpha in vascular smooth muscle cells. J Biol Chem 275:26765–26771
Richardson RS, Noyszewski EA, Kendrick KF, Leigh JS, Wagner PD (1995) Myoglobin O2 desaturation during exercise. Evidence of limited O2 transport. J Clin Invest 96:1916–1926
Rigamonti E, Touvier T, Clementi E, Manfredi AA, Brunelli S, Rovere-Querini P (2013) Requirement of inducible nitric oxide synthase for skeletal muscle regeneration after acute damage. J Immunol 190:1767–1777
Riquelme C, Acuna MJ, Torrejon J, Rebolledo D, Cabrera D, Santos RA, Brandan E (2014) ACE2 is augmented in dystrophic skeletal muscle and plays a role in decreasing associated fibrosis. PLoS One 9:e93449
Russo V. Papa AA. Williams EA. Rago A. Palladino A. Politano L. Nigro G. (2018) ACE inhibition to slow progression of myocardial fibrosis in muscular dystrophies. Trends Cardiovasc Med 28:330–337
Sabharwal R, Chapleau MW (2014) Autonomic, locomotor and cardiac abnormalities in a mouse model of muscular dystrophy: targeting the renin angiotensin system. Exp Physiol
Sampaio WO, Souza dos Santos RA, Faria-Silva R, da Mata Machado LT, Schiffrin EL, Touyz RM (2007) Angiotensin-(1-7) through receptor mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension. 49:185–192
Sanchez-Elsner T, Botella LM, Velasco B, Langa C, Bernabeu C (2002) Endoglin expression is regulated by transcriptional cooperation between the hypoxia and transforming growth factor-beta pathways. J Biol Chem 277:43799–43808
Sanghani NH, Haase, VH (2019) Hypoxia-Inducible Factor Activators in Renal Anemia: Current Clinical Experience. Adv Chronic Kidney Dis 26:253–266
Santos RAS, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M, Campagnole-Santos MJ (2018) The ACE2/angiotensin-(1-7)/MAS Axis of the renin-angiotensin system: focus on angiotensin-(1-7). Physiol Rev 98:505–553
Santos RAS, Oudit GY, Verano-Braga T, Canta G, Steckelings UM, Bader M (2019) The renin-angiotensin system: going beyond the classical paradigms. Am J Physiol Heart Circ Physiol 316:H958–H970
Scheerer N, Dehne N, Stockmann C, Swoboda S, Baba HA, Neugebauer A, Johnson RS, Fandrey J (2013) Myeloid hypoxia-inducible factor-1alpha is essential for skeletal muscle regeneration in mice. J Immunol 191:407–414
Semprun-Prieto LC, Sukhanov S, Yoshida T, Rezk BM, Gonzalez-Villalobos RA, Vaughn C, Michael Tabony A, Delafontaine P (2011) Angiotensin II induced catabolic effect and muscle atrophy are redox dependent. Biochem Biophys Res Commun 409:217–221
Serrano AL, Munoz-Canoves P (2017) Fibrosis development in early-onset muscular dystrophies: mechanisms and translational implications. Semin Cell Dev Biol 64:181–190
Serrano AL, Mann CJ, Vidal B, Ardite E, Perdiguero E, Munoz-Canoves P (2011) Cellular and molecular mechanisms regulating fibrosis in skeletal muscle repair and disease. Curr Top Dev Biol 96:167–201
Shimizu-Motohashi Y, Asakura A (2014) Angiogenesis as a novel therapeutic strategy for Duchenne muscular dystrophy through decreased ischemia and increased satellite cells. Front Physiol 5:50
Silva SJD. Rassi S. Pereira ADC. (2017) Angiotensin-Converting Enzyme ID Polymorphism in Patients with Heart Failure Secondary to Chagas Disease. Arq Bras Cardiol 109:307–312
Smith LR, Barton ER (2018) Regulation of fibrosis in muscular dystrophy. Matrix Biol 68-69:602–615
Sukhanov S, Semprun-Prieto L, Yoshida T, Michael Tabony A, Higashi Y, Galvez S, Delafontaine P (2011) Angiotensin II, oxidative stress and skeletal muscle wasting. Am J Med Sci 342:143–147
Sun G, Haginoya K, Wu Y, Chiba Y, Nakanishi T, Onuma A, Sato Y, Takigawa M, Iinuma K, Tsuchiya S (2008) Connective tissue growth factor is overexpressed in muscles of human muscular dystrophy. J Neurol Sci 267:48–56
Tang K, Breen EC, Gerber HP, Ferrara NM, Wagner PD (2004) Capillary regression in vascular endothelial growth factor-deficient skeletal muscle. Physiol Genomics 18:63–69
Thomas GD (2013) Functional muscle ischemia in Duchenne and Becker muscular dystrophy. Front Physiol 4:381
Thomas GD, Shaul PW, Yuhanna IS, Froehner SC, Adams ME (2003) Vasomodulation by skeletal muscle-derived nitric oxide requires alpha-syntrophin-mediated sarcolemmal localization of neuronal nitric oxide synthase. Circ Res 92:554–560
Tran CM, Markova D, Smith HE, Susarla B, Ponnappan RK, Anderson DG, Symes A, Shapiro IM, Risbud MV (2010) Regulation of CCN2/connective tissue growth factor expression in the nucleus pulposus of the intervertebral disc: role of Smad and activator protein 1 signaling. Arthritis Rheum 62:1983–1992
Valle-Tenney R, Rebolledo DL, Lipson KE, Brandan E (2019) Role of hypoxia in skeletal muscle fibrosis: synergism between hypoxia and TGF-beta signaling upregulates CCN2/CTGF expression specifically in muscle fibers. Matrix Biol
Vial C, Zuniga LM, Cabello-Verrugio C, Canon P, Fadic R, Brandan E (2008) Skeletal muscle cells express the profibrotic cytokine connective tissue growth factor (CTGF/CCN2), which induces their dedifferentiation. J Cell Physiol 215:410–421
Wynn TA, Ramalingam TR (2012) Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18:1028–1040
Xie L, Yin A, Nichenko AS, Beedle AM, Call JA, Yin H (2018) Transient HIF2A inhibition promotes satellite cell proliferation and muscle regeneration. J Clin Invest 128:2339–2355
Yan X, Zhao R, Feng X, Mu J, Li Y, Chen Y, Li C, Yao Q, Cai L, Jin L, Han C, Zhang D (2020) Sialyltransferase7A promotes angiotensin II-induced cardiomyocyte hypertrophy via HIF-1alpha-TAK1 signalling pathway. Cardiovasc Res 116:114–126
Yang X, Yang S, Wang C, Kuang S (2017) The hypoxia-inducible factors HIF1alpha and HIF2alpha are dispensable for embryonic muscle development but essential for postnatal muscle regeneration. J Biol Chem 292:5981–5991
Yang Y, Yu X, Zhang Y, Ding G, Zhu C, Huang S, Jia Z, Zhang A (2018) Hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat (FG-4592) protects against cisplatin-induced acute kidney injury. Clin Sci (Lond) 132:825–838
Zhang Q, Yan Q, Yang H, Wei W (2019) Oxygen sensing and adaptability won the 2019 Nobel prize in physiology or medicine. Genes Dis 6:328–332
Zhu Q, Wang Z, Xia M, Li PL, Van Tassell BW, Abbate A, Dhaduk R, Li N (2011) Silencing of hypoxia-inducible factor-1alpha gene attenuated angiotensin II-induced renal injury in Sprague-Dawley rats. Hypertension. 58:657–664
Acknowledgments
The authors acknowledge the following funding sources: FONDECYT 1150106 and 1190144 grants and CONICYT AFB170005 grant to E.B. Beca de Doctorado Nacional Folio 21150188 from CONICYT to R.V-T. FONDECYT 3140357 and 11181090 to D.R.L. FONDECYT 11170623 to M.J.A.. The authors acknowledge the services provided by the UC CINBIOT Animal Facility funded by PIA CONICYT* ECM-07 Program for Associative Research, of the Chilean National Council for Science and Technology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Valle-Tenney, R., Rebolledo, D., Acuña, M.J. et al. HIF-hypoxia signaling in skeletal muscle physiology and fibrosis. J. Cell Commun. Signal. 14, 147–158 (2020). https://doi.org/10.1007/s12079-020-00553-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12079-020-00553-8