
Abstract
Heart failure represents a systemic disease with profound effects on multiple peripheral tissues including skeletal muscle. Within the context of heart failure, perturbations in skeletal muscle physiology, structure, and function strongly contribute to exercise intolerance and the morbidity of this devastating disease. There is growing evidence that chronic heart failure imparts specific pathological changes within skeletal muscle beds resulting in muscle dysfunction and tissue atrophy. Mechanistically, systemic and local inflammatory responses drive critical aspects of this pathology. In this review, we will discuss pathological mechanisms that drive skeletal muscle inflammation and highlight emerging roles for distinct innate immune subsets that reside within damage muscle tissue focusing on the recently described embryonic and monocyte-derived macrophage lineages. Within this context, we will discuss how immune mechanisms can be differentially targeted to stimulate skeletal muscle inflammation, catabolism, fiber atrophy, and regeneration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
It is now recognized that heart failure is a systemic disease with profound effects on multiple peripheral tissues including skeletal muscle. Several clinical studies have convincingly demonstrated that impairments in skeletal muscle physiology and function are largely responsible for exercise intolerance in patients with chronic systolic heart failure. Structurally, cachexia and skeletal muscle atrophy represent important comorbidities associated with poor prognosis [2]. Within the chronic heart failure population, skeletal muscle atrophy and intrinsic reductions in oxidative metabolism are tightly associated with reduced exercise capacity [55]. Patients with chronic heart failure not only have smaller peripheral muscle mass but also display reduced oxidative metabolism during exercise. Moreover, measurement of skeletal muscle lactate production during exercise demonstrated metabolic abnormalities in patients with chronic heart failure independent of tissue perfusion. Consistent with this, skeletal mitochondria density is reduced in patients with chronic heart failure and the extent of mitochondrial loss correlates with peak exercise capacity [24, 110].
These observations demonstrate that while reductions in tissue perfusion may contribute to skeletal muscle dysfunction in patients with chronic systolic heart failure, there is substantial evidence indicating that pathologic and metabolic abnormalities intrinsic to skeletal muscle beds are the primary mediators of muscle atrophy and dysfunction [53, 54, 90]. Examination of muscle biopsies has demonstrated that compared to control subjects, patients with chronic systolic heart failure display multiple pathological findings including fiber atrophy, myocyte apoptosis, and increased interstitial inflammatory cells. In addition, immunostaining studies have revealed evidence of muscle fiber reprogramming favoring anaerobic muscle fiber types, namely, reduced type I slow twitch fibers and increased type II fast twitch fibers. Consistent with these observations, biochemical studies focused on mitochondrial function have revealed that subjects with chronic heart failure have significant deficits in oxidative metabolism and morphological studies have reported reduced capillary density [25, 40, 53, 55, 81, 90, 102, 110]. Together, these findings suggest that patients with heart failure display an intrinsic skeletal muscle myopathy, which likely contributes to the prominent muscle wasting, contractile dysfunction, and reduced exercise capacity observed in this patient population. While the molecular mechanisms that govern the development of heart failure-associated skeletal muscle myopathy have not yet been completely defined, recent findings have provided important and exciting insights.
Intriguingly, the pathology of heart failure-associated skeletal muscle myopathy closely resembles what has been observed in myopathies due to chronic inflammatory conditions, such as Crohn’s disease. Examination of muscle biopsies obtained from patients with Crohn’s disease demonstrated fiber atrophy, myocyte apoptosis, reduced type I fibers, increased type II fibers, and increased numbers of inflammatory cells. These findings were associated with elevated levels of the pro-inflammatory cytokines TNFα, sphingosine, and lipopolysaccharide (LPS) [17]. Importantly, numerous large studies have revealed that patients with systolic heart failure also have elevated serum levels of pro-inflammatory cytokines including TNFα, IL-1β, IL-6, and IL-2 [22, 29, 73, 96]. These findings highlight a potentially shared pathogenesis of skeletal muscle atrophy in Crohn’s disease and chronic systolic heart failure and implicate systemic and/or local inflammation as a potential causative mechanism in heart failure-associated skeletal muscle myopathy.
In this review, we will discuss pathologic mechanisms that drive skeletal muscle inflammation in the setting of chronic heart failure. In addition, we will highlight potential roles for distinct innate immune subsets that reside within damage muscle tissue focusing on the recently described embryonic-derived and monocyte-derived macrophage lineages. Specifically, we will discuss what these cell types represent and how they may differentially impact on skeletal muscle repair and inflammation. Finally, we will connect inflammatory mechanisms with pathways known to stimulate skeletal muscle atrophy, dysfunction, catabolism, and myokine production. Where appropriate, we will comment on potential contributions of aging to skeletal muscle inflammation and dysfunction.
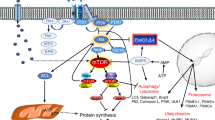
FormalPara Skeletal muscle injury and inflammation in heart failureWhether due to systolic or diastolic dysfunction, the systemic syndrome of heart failure imparts multiple insults on peripheral tissues including skeletal muscle. Collectively, these perturbations contribute to heart failure-associated skeletal myocyte injury and dysfunction. Within the context of heart failure, skeletal muscle injury can be divided into primary and secondary mechanisms. We will define primary injury mechanisms as those directly related to the syndrome of heart failure, namely, derangements in tissue perfusion, neurohormonal activation, disruption of skeletal muscle homeostasis, and systemic inflammation. Each of these primary mechanisms contributes to skeletal myocyte injury or death. Secondary injury mechanisms will be defined as the activation of local immune responses by injured or damaged skeletal muscle. In the following sections, we will outline the pathophysiological triggers of primary skeletal muscle injury in the setting of chronic heart failure (Fig. 1), describe the mechanisms by which these pathologies stimulate local inflammatory responses, and discuss the pathological and clinical implications of skeletal muscle inflammation as they relate to muscle atrophy and dysfunction.
Skeletal muscle insults in heart failure: primary injury mechanisms
Derangements in tissue perfusion
Reduced cardiac output (particularly during exercise) and increased venous pressure are central to all etiologies of heart failure and result in reduced skeletal muscle blood flow. Failure to augment blood flow during exercise due to reduced forward flow and impaired vasodilation leads to inadequate skeletal muscle perfusion and production of lactate, a hallmark of tissue ischemia [89]. Similarly, increased central venous pressures not only result in microvascular congestion and tissue edema but also reduced perfusion pressure across skeletal muscle capillary beds [84, 89]. As a consequence, skeletal myocytes receive inadequate nutrient supply including oxygen and are either stressed, reversibly injured, or in more extreme conditions undergo apoptotic and/or necrotic cell death.
Neurohormonal activation
In addition to episodic tissue ischemia, patients with heart failure display several systemic perturbations that contribute to skeletal muscle pathology including neurohormonal activation and systemic inflammation. Neurohormonal activation defined as exaggerated sympathetic tone and increased systemic epinephrine, norepinephrine, and angiotensin II levels are commonly observed in patients with acute and chronic heart failure [60]. In addition to promoting myocardial adverse remodeling, neurohormonal activation has direct effects on skeletal myocytes including induction of oxidative injury, sarcomere loss, and myocyte death. Chronic epinephrine and norepinephrine exposure is sufficient to promote oxidative stress, capillary rarefaction, and skeletal muscle atrophy. These effects appear to occur through a combination of chronic β1-adrenergic stimulation and downregulation of β2-adrenergic signaling [5, 107]. Moreover, norepinephrine may contribute to reduced tissue perfusion through exaggerated α-adrenergic-mediated vasoconstriction [67]. Lastly, increased local and systemic sympathetic nerve activity and reduced vagal tone have important effects on innate immune cell recruitment, pro-inflammatory signaling, and resolution of inflammatory responses. Sympathetic activation is an essential cue to mobilize monocytes from the bone marrow potentially through a β3-adrenergic mechanism, while parasympathetic signaling via nicotinic acetylcholine receptors is an important negative regulator of pro-inflammatory innate immune responses to sterile tissue injury [16, 26, 71].
Angiotensin II levels are also elevated in the setting of chronic heart failure and trigger the generation of damaging reactive oxygen species through direct stimulation of NADPH oxidase [38, 47]. In addition, angiotensin II signaling directly contributes to skeletal muscle inflammation through augmentation of inflammatory and inhibition of anti-inflammatory signaling mechanisms. Angiotensin II signaling activates canonical NFκB-dependent pro-inflammatory gene expression in skeletal muscle myocytes. Importantly, angiotensin II also suppresses insulin-like growth factor 1 (IGF1) signaling in skeletal myocytes, a central anti-inflammatory signaling mechanism that governs skeletal muscle homeostasis and growth [6, 9, 47, 76, 113].
Disruption of the IGF1 signaling axis
Patients with heart failure display reductions in circulating factors that are protective towards skeletal muscle, the most notable of which is IGF1 [7, 99]. Serum IGF1 levels are inversely associated with LV systolic dysfunction, neurohormonal activation, cytokine production, and muscle atrophy [3, 68, 74]. IGF1 is expressed by multiple tissues in the body including liver and skeletal and cardiac muscle. The exact mechanism by which systemic IGF1 is reduced in heart failure is not well defined. However, elevated angiotensin II activity may play an important role [76]. IGF1 signaling represents a well-established mechanism to stimulate muscle growth and regeneration [64]. IGF transgenic mice are resistant to angiotensin II and heart failure-induced muscle atrophy, and local IGF1 treatment can reverse disuse atrophy [83, 87, 88]. These protective properties of IGF1 signaling are mediated through an AKT1, mTOR, and FoxO axis supporting cell survival, myofibril growth, and protein synthesis, while inhibiting protein degradation [7]. Importantly, IGF1 also possesses significant immunomodulatory activity. Specifically, IGF1 is a robust inhibitor of pro-inflammatory responses through IL10-dependent suppression of monocyte activation [34].
Systemic inflammation
Numerous clinical studies have documented that patients with heart failure display signs of systemic inflammation [17, 22, 29, 73, 96]. Biomarker analysis of 1200 heart failure patients enrolled in the VEST trial demonstrated that serum levels of TNFα and IL6 are elevated in heart failure and prognostically important. Specifically, patients with elevated cytokine levels displayed higher mortality [22]. Moreover, serum TNFα and IL6 levels are predictive of the development of heart failure and are reduced in patients receiving optimal medical therapy for heart failure [97, 100]. The exact source of cytokines is not well established, but likely includes both the myocardium as well as peripheral tissues including but not limited to the gastrointestinal tract, liver, and circulating monocytes [49, 50, 100]. One proposed mechanism is that increased gut permeability leads to systemic inflammation through either systemic LPS exposure and/or dysregulated mucosal immune responses [69]. In either scenario, activation of innate immune cells results in elevated systemic levels of TNFα, IL-1β, IL-6, and IL-2 with deleterious effects on skeletal muscle. Cytokine signaling is sufficient to trigger skeletal myocyte injury, generation of reactive oxygen intermediates, and muscle atrophy [18, 59]. IL6 signaling via STAT3 has been reported to regulate myostatin, an important mediator of skeletal muscle catabolism [115]. In addition, muscle-specific TNFα signaling through NF-kB results in a severe muscle wasting phenotype that is mediated by the ubiquitin ligase MuRF1 and suppression of IGF1 signaling [10, 20]. The TNF superfamily member, TNF-like weak inducer of apoptosis (TWEAK), induces a similar phenotype by binding to the Fn14 receptor and activating NF-kB signaling [23].
Local skeletal muscle immune responses: secondary injury mechanisms
While primary mechanisms that contribute skeletal muscle dysfunction and injury are well established, less is known regarding the role of skeletal muscle cell death and local inflammatory responses. In the following section, we will highlight the importance of local inflammation and discuss the mechanistic basis by which myocyte cell death contributes to local activation of the innate immune system. In addition, we will introduce the concept of macrophage diversity and propose a conceptual framework explaining why local inflammation paradoxically contributes to both tissue repair and collateral tissue injury.
Immune recognition of injured and dying skeletal myocytes
In recent years, details have emerged clarifying the mechanisms by which tissue injury is recognized by the immune system and inflammatory responses are generated. It is now understood that cells that are injured or those that undergo cell death through necrosis, necroptosis, and aborted apoptosis release an array of intracellular signaling molecules with potent effects on tissue-resident immune cells [56]. Indeed, defective autophagocytic responses associated with skeletal muscle catabolism and atrophy also result in cell death and local inflammation [65]. Dying myocytes release an array of signals collectively referred to as danger-associated molecular patterns (DAMPs) that encompass a variety of mediators including alarmins (HMGB1, S100A8/9/12, S100B, IL1α, HSPs), nucleotides (ATP, CpG, dsRNA), bioactive lipids, extracellular matrix fragments (glycans, heparin sulfate, hyaluronan), and lectins [13].
The majority of DAMPs are recognized by a set of germ line-encoded pattern recognition receptors (PRRs) most prominently expressed on leukocytes, endothelial cells, and fibroblasts. PRRs are well known for their roles in recognizing pathogen-derived products referred to as pathogen-associated molecular patterns (PAMPs). Examples of PAMPs include gram-negative bacterial lipopolysaccharides (LPS), gram-positive bacterial teichoic acids, yeast zymosans, mycobacterial glycolipids, and viral dsRNAs [65]. Classically, engagement of PRRs such as toll-like receptor 4 (TLR4) by pathogen-derived products (i.e., LPS) results in activation of mononuclear phagocytes and generation of profound inflammation responses aimed at eliminating both pathogens and infected cells [57, 61, 95].
An array of recent studies has provided substantial evidence indicating that recognition of DAMPs by PRRs is responsible for sterile injury-associated inflammation and, in some scenarios, tissue repair. PRRs implicated in sterile injury responses include toll-like receptors (TLR2, 3, 4, and 9), NOD-like receptors (NOD, NLRs, NLRP3), RIG-I-like receptors, C-type lectin receptors (CLRs), RAGE receptors, and formyl peptide receptors (FPRs) [92, 101], all of which are present in healthy and diseased skeletal muscle [93, 109]. TLRs are found on either the plasma membrane (TLR2, 4) or endosomes (TLR3, 9) and recognize an array of DAMPs including HMGB1, S100A/B, HSPs, ECM fragments, and dsDNA including mitochondrial DNA and CpG fragments [92]. Engagement of TLR receptors by various DAMPS leads to the activation of a well-described cascade of signaling events culminating in the release of pro-inflammatory chemokines and cytokines [72]. Activation of the NOD-like receptors NOD1, NOD2, and NLRP3 by nucleotides and other metabolites including uric acid results in inflammasome activation and generation of robust inflammatory responses [35]. Formyl peptide receptors recognize products released from damaged mitochondria and have been shown to trigger robust activation of neutrophils in the setting of sterile liver injury [58]. At this point in time, considerably less is known regarding the roles of RIG-like receptors, C-type lectin receptors, and RAGE receptors in sterile tissue injury. However, it is likely that much will be learned in the near future.
With respect to skeletal muscle, multiple DAMPs have been implicated in skeletal myocyte injury, inflammation, and tissue repair. Traumatic or ischemic skeletal muscle injury elicits the release of mitochondria-associated DAMPs including formyl peptides and mitochondrial DNA. In this context, formyl peptides are recognized by FPR1 present on neutrophils, while mitochondrial DNA is recognized by neutrophils, monocytes, and macrophages expressing TLR9. Collectively, mitochondrial DAMPs appear to drive local and systemic inflammatory responses [114]. In contrast to mitochondrial-derived DAMPS, high-mobility group box 1 protein (HMGB1) release from injured skeletal muscle is required for tissue repair and regeneration. HMGB1 is a ubiquitous multifunctional protein, which under homeostatic conditions functions within the nucleus to augment interactions between transcription factors and chromatin. However, following tissue injury, HMGB1 is released into the extracellular matrix by injured or dying cells where it activates mononuclear phagocytes through TLR4 and RAGE receptor signaling [1, 32, 108]. HMGB1 may also signal directly to satellite cells and endothelial cells [21]. Importantly, HMGB1 is required for revascularization and myocyte regeneration following skeletal muscle injury [11, 21], suggesting that activation of local immune responses through a DAMP-mediated mechanism is a key determinant of not only inflammation but also tissue repair.
To date, no studies have investigated whether skeletal muscle DAMPs are activated in the setting of heart failure. Given data demonstrating increased skeletal myocyte death, inflammation, and the multiple insults imparted on skeletal muscle in the setting of chronic heart failure, it would not be surprising that heart failure-associated skeletal muscle inflammation is driven by DAMP signaling. Whether DAMP signaling contributes to heart failure-associated skeletal muscle atrophy, fiber type switching, metabolic derangements, fibrosis, and impaired microvasculature remains unclear. Moreover, whether DAMP signaling can be manipulated to favor satellite cell activation and regenerative responses is yet to be studied. Furthermore, the mechanistic determinants explaining why some DAMPs trigger inflammatory responses while others stimulate tissue repair is not fully understood; however, some clues are beginning to emerge.
Macrophage diversity
Immune cells represent the predominate cell type that utilizes PRRs to recognize and respond to tissue injury. Macrophages, neutrophils, dendritic cells, and B cells harbor an array of PRRs poised to recognize the release of an array of DAMPs. Within muscular tissues, macrophages represent the most abundant resident innate immune cell. Macrophages are evolutionary conserved phagocytes that were first discovered late in the nineteenth century by Ilya Metchnikoff [15, 94]. In the 1960s, Van Furth proposed that tissue macrophages originated from circulating blood monocytes, which has been the prevailing view for the last 40 years. However, evolving evidence has challenged this dogma and suggested that tissue macrophages may exist independent of circulating monocytes [80, 98, 106].
More recently, a series of studies performed in model systems have drastically revised our understanding of macrophage origin. Collectively, these publications demonstrated that many tissue-resident macrophages are established during embryonic development and persist into adulthood independent of blood monocyte input [27, 36, 39, 42, 48, 82, 112]. This advancement in knowledge was dependent on the development and utilization of genetic lineage tracing and parabiotic systems. Several detailed review articles have been published describing different aspects of macrophage ontogeny and related functions [19, 86, 111]. However, few publications have explored macrophage ontogeny within the skeletal muscle. In the following section, we will describe the currently available knowledge regarding tissue-resident macrophage subsets, origins, and potential functions in injured skeletal muscle.
In the mouse, embryonic and adult monocyte-derived macrophages can be readily distinguished through genetic lineage tracing. Adult monocyte-derived macrophages are derived from definitive hematopoietic stem cells, which express FLT3 as they differentiate. Based on this finding, a Flt3-Cre mouse was developed to track the progeny of adult definitive hematopoietic stem cells [8]. Using this genetic lineage tracing tool, several studies have identified embryonic-derived (Flt3-Cre negative) and adult monocyte-derived (Flt3-Cre positive) macrophages in multiple tissues including the brain, heart, lung, kidney, liver, and skin [28, 45]. Using a combination of immunostaining and genetic lineage tracing, we were able to similarly identify embryonic-derived and monocyte-derived macrophages in skeletal muscle (Fig. 2).

Schematic depicting mechanisms of skeletal muscle injury in the context of chronic heart failure. IGF-1 insulin-like growth factor 1, IL-6 interleukin 6, LPS lipopolysaccharide, NF-kb nuclear factor kappa-light chain-enhancer of activated B cells, TNFα tumor necrosis factor α, TWEAK TNF-related weak inducer of apoptosis

Skeletal muscle macrophages in heart failure. a Immunostaining for CD68 (green) and DAPI (blue) in skeletal muscle obtained from controls and a mouse model of dilated cardiomyopathy demonstrating increased macrophage abundance. b Diagram illustrating that macrophages are derived from distinct lineages including primitive and definitive hematopoietic progenitors. c Genetic lineage tracing with Flt3-Cre (red) revealing the presence of CD68+ macrophages derived from embryonic (Flt3-Cre-negative) and adult monocyte (Flt3-Cre-positive) origins. d Schematic describing the potential roles for embryonic-derived (blue) and monocyte-derived (red) macrophages in the context of skeletal muscle injury
Collectively, studies utilizing Flt3-Cre mice have revealed that adult monocyte-derived macrophages do fully replace embryonic-derived macrophages during steady state conditions [27, 42, 82]. In fact, macrophages of embryonic and adult monocyte origin coexist within tissues. Intriguingly, in the context of aging, adult monocyte-derived macrophages increase in number and progressively replace embryonic-derived macrophages in some tissues including the heart [62]. Furthermore, detailed studies have better refined the composition of Flt3-Cre-negative embryonic-derived macrophages suggesting that this population consists of a compellation of two lineages derived from progenitors located within the early yolk sac and those that migrate from the yolk sac to the fetal liver at later developmental stages [27].
Embryonic-derived macrophages are first observed during early gestation (embryonic day 6.5–8.5 in the mouse) in the extra-embryonic yolk sac where they arise from an early bipotent erythromyeloid progenitor. At this early stage of development, the yolk sac supports primitive hematopoiesis producing erythrocytes and yolk sac-derived macrophages [78]. In the mouse, primitive hematopoiesis can be distinguished from definitive hematopoiesis based on the requirement for the transcription factor MYB. Myb−/− embryos have a selective deficiency in definitive hematopoietic stems and are unable to support definitive hematopoiesis. While Myb−/− embryos lack definitive hematopoietic progeny (lymphocytes, granulocytes, monocytes, and megakaryocytes), their ability to produce early erythrocytes and yolk sac-derived macrophages is not perturbed [82]. Yolk sac-derived macrophages have a characteristic CX3CR1hi F4/80hi CD11blo expression pattern in the embryo and give rise to adult brain microglia, a subset of cardiac macrophages, and a transient population of skin Langerhans’s cells [27, 36, 42, 44, 82, 112].
At later stages during embryonic development (embryonic day 8.5–10.5 in the mouse), definitive hematopoietic stem cells emerge from the aorta-gonades mesonephros, hemogenic endothelium, and yolk sac giving rise to definitive fetal immune lineages. Beginning at E10.5, HSCs migrate to the fetal liver, which then serves as the major hematopoietic organ during the remainder of embryonic development [36, 79]. During this period, fetal monocytes emigrating from the liver take residence within multiple developing organs and likely constitute the primary origin of the majority of tissue-resident embryonic-derived macrophages [28, 45]. Within the skin, fetal monocytes out-compete yolk sac-derived macrophages and contribute to the overwhelming majority of adult Langerhans cells. Recent data suggests that a similar phenomenon occurs in the developing liver, kidney, and lung [39, 45, 48].
During the perinatal period, definitive hematopoietic stem cells take residence within the bone marrow where they establish the primary site of hematopoiesis for the remainder of life. Within this compartment, Flt3-Cre-positive definitive hematopoietic stem cells produce the full complement of immune lineages including adult monocyte-derived macrophages [70]. Adult monocytes arise from definitive hematopoietic stem cells through a well-characterized cascade involving the progressive differentiation of committed progenitor cells including the monocyte-macrophage dendritic cell progenitor (MDP) [30]. Recently, a Ly6C-positive monocyte-specific progenitor downstream of MDPs has been identified in the bone marrow and spleen [43]. In the mouse and human, there appear to be two principle monocyte subsets: classical Ly6Chigh monocytes and nonclassical Ly6Clow monocytes [46]. Ly6Chi monocytes are direct descendants of the Ly6C-positive monocyte progenitor and give rise to tissue-resident adult monocyte-derived macrophages [28, 48]. Nonclassical Ly6Clow monocytes differentiate from Ly6Chigh monocytes through an Nr4a1-dependent transcriptional program [41, 43, 112]. Within the vasculature, nonclassical Ly6Clow monocytes crawling over the endothelium in a LFA-1 integrin-dependent manner where they clear damaged endothelial cells and maintain vascular integrity (Auffray et al. 2007) [12]. While some have considered these monocytes to be “vascular macrophages,” they transcriptionally cluster with monocytes and do not express core macrophage transcripts, including Mer tyrosine kinase [33, 48].
Impact of local skeletal muscle immune responses
As discussed above, innate immune cells (predominately macrophages) recognize skeletal muscle injury and drive both damaging inflammatory as well as reparative responses. Until recently, it has remained relatively unclear how activation of the innate immune system can trigger such divergent responses. In fact, there are numerous examples of seemingly contradictory reports claiming that inflammation is both harmful following injury and an essential mediator of tissue repair [37]. This paradox is well established in models of ischemic cardiac injury. Macrophages within the infarcted heart not only drive robust inflammatory responses and pathological remodeling but also are required for the resolution of inflammation, tissue repair, and coronary angiogenesis [31]. One explanation for these findings is that distinct macrophage populations may mediate inflammatory (M1-like) and reparative (M2-like) macrophage behaviors [66]. Until recently, the exact identities of these proposed macrophage subsets have remained largely undefined.
Recently, we have demonstrated that distinct resident and recruited macrophage subsets derived from embryonic and definitive monocyte progenitors, respectively, govern tissue repair and inflammation in the context of acute cardiac injury [51]. Conceptually, whether these findings apply to other tissues such as skeletal muscle remains to be established. However, it is intriguing to postulate that in the context of chronic heart failure recognition of injured or stressed skeletal myocytes by resident and newly recruited inflammatory macrophages may similarly contribute to tissue repair and secondary pro-inflammatory responses.
In support of this concept, M2-like skeletal muscle macrophages are required for skeletal muscle regeneration following cardiotoxin injury through augmentation of satellite cell proliferation and angiogenesis [4, 91]. In distinction to the heart, monocyte recruitment was necessary for skeletal muscle regeneration [14, 85]. However, in this scenario, monocytes differentiated into anti-inflammatory M2-like macrophages through a mechanism involving phagocytosis [4]. How these M2-like macrophages compare to embryonic-derived macrophages resident in cardiac and skeletal muscle remains to be studied. These data do highlight the possibility that monocyte-derived macrophages can be instructed by tissue-derived factors to acquire a reparative phenotype and promote tissue regeneration. To date, it has not yet been explored whether macrophages resident within the skeletal muscle in the context of failure contribute to muscle regeneration.
Similar to following cardiac injury, monocyte-derived pro-inflammatory M1-like macrophages infiltrate injured skeletal muscle and secrete an array of inflammatory chemokines and cytokines including MCP1, IL1β, and TNFα [4]. Each of these factors is sufficient to drive inflammation through recruitment of additional pro-inflammatory monocytes and collateral tissue injury. In addition, IL1β and TNFα signaling to skeletal myocytes contribute to skeletal muscle atrophy and catabolism through reductions in oxidative metabolism and IGF1 signaling [20, 47, 52]. Furthermore, pro-inflammatory macrophages inhibit regeneration as persistence of these cells leads to ongoing damage and defective tissue healing [63, 75, 77]. Examination of a chronic model of skeletal muscle injury (i.e., muscular dystrophy) revealed the existence of pro-inflammatory M1-like and reparative alternatively activated “M2-like” macrophages [103–105], supporting the possibility that distinct macrophage subsets may also govern inflammation and regeneration in chronic skeletal muscle pathologies.
Could inflammation be a target for heart failure-associated skeletal muscle myopathy?
Based on the above findings, it is reasonable to postulate that manipulation of innate immune responses in the context of skeletal muscle injury or chronically disease may impact outcomes. With respect to heart failure, is it possible to bias the immune response to skeletal muscle injury in a manner that might slow the progression or reverse heart failure-associated skeletal muscle myopathy? Selective targeting of distinct macrophage subsets may represent one potential avenue for intervention. For example, manipulations favoring M2-like macrophages would likely bias the immune response towards muscle regeneration and ultimately restoration of skeletal muscle function. Such a manipulation may also minimize the collateral injury, metabolic perturbations, and muscle atrophy associated with excessive inflammation. At this point in time, the exact mechanisms that might be suitable for therapeutic intervention have not yet been identified. Future studies delineating specific mechanisms by which inflammatory M1-like and resident M2-like macrophages are recruited to sites of skeletal myocyte injury, become activated, and exert their functions will undoubtedly provide critical details that will likely be informative with respect to potential therapeutic intervention.
Conclusions
It has long been known that skeletal muscle wasting represents an important pathology contributing to the morbidity of heart failure. Recently, important insights have been made mechanistically linking inflammation to muscle atrophy and dysfunction. In addition, paradigm shifting studies have revealed that distinct components of the innate immune system may govern inflammation and tissue repair following sterile injury. Together, these advances have raised the question of whether it is possible to develop therapeutics that target systemic and local inflammatory responses in an effort to restore muscle mass and function. Future studies will be required to determine whether these exciting opportunities represent a reality and new hope for patients crippled by chronic heart failure.
References
Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M, Yang H, Tracey KJ (2000) High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med 192:565–570
Anker SD, Ponikowski P, Varney S, Chua TP, Clark AL, Webb-Peploe KM, Harrington D, Kox WJ, Poole-Wilson PA, Coats AJ (1997) Wasting as independent risk factor for mortality in chronic heart failure. Lancet 349:1050–1053
Anker SD, Volterrani M, Pflaum CD, Strasburger CJ, Osterziel KJ, Doehner W, Ranke MB, Poole-Wilson PA, Giustina A, Dietz R, Coats AJ (2001) Acquired growth hormone resistance in patients with chronic heart failure: implications for therapy with growth hormone. J Am Coll Cardiol 38:443–452
Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B (2007) Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med 204:1057–1069
Bacurau AV, Jardim MA, Ferreira JC, Bechara LR, Bueno CR Jr, Alba-Loureiro TC, Negrao CE, Casarini DE, Curi R, Ramires PR, Moriscot AS, Brum PC (2009) Sympathetic hyperactivity differentially affects skeletal muscle mass in developing heart failure: role of exercise training. J Appl Physiol (1985) 106:1631–1640
Bechara LR, Moreira JB, Jannig PR, Voltarelli VA, Dourado PM, Vasconcelos AR, Scavone C, Ramires PR, Brum PC (2014) NADPH oxidase hyperactivity induces plantaris atrophy in heart failure rats. Int J Cardiol 175:499–507
Bonaldo P, Sandri M (2013) Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 6:25–39
Boyer SW, Schroeder AV, Smith-Berdan S, Forsberg EC (2011) All hematopoietic cells develop from hematopoietic stem cells through Flk2/Flt3-positive progenitor cells. Cell Stem Cell 9:64–73
Brink M, Wellen J, Delafontaine P (1996) Angiotensin II causes weight loss and decreases circulating insulin-like growth factor I in rats through a pressor-independent mechanism. J Clin Invest 97:2509–2516
Cai D, Frantz JD, Tawa NE Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE (2004) IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 119:285–298
Campana L, Santarella F, Esposito A, Maugeri N, Rigamonti E, Monno A, Canu T, Del MA, Bianchi ME, Manfredi AA, Rovere-Querini P (2014) Leukocyte HMGB1 is required for vessel remodeling in regenerating muscles. J Immunol 192:5257–5264
Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, Hedrick CC, Cook HT, Diebold S, Geissmann F (2013) Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell 153:362–375
Chan JK, Roth J, Oppenheim JJ, Tracey KJ, Vogl T, Feldmann M, Horwood N, Nanchahal J (2012) Alarmins: awaiting a clinical response. J Clin Invest 122:2711–2719
Contreras-Shannon V, Ochoa O, Reyes-Reyna SM, Sun D, Michalek JE, Kuziel WA, McManus LM, Shireman PK (2007) Fat accumulation with altered inflammation and regeneration in skeletal muscle of CCR2−/− mice following ischemic injury. Am J Physiol Cell Physiol 292:C953–C967
Cooper MD, Alder MN (2006) The evolution of adaptive immune systems. Cell 124:815–822
Courties G, Herisson F, Sager HB, Heidt T, Ye Y, Wei Y, Sun Y, Severe N, Dutta P, Scharff J, Scadden DT, Weissleder R, Swirski FK, Moskowitz MA, Nahrendorf M (2015) Ischemic stroke activates hematopoietic bone marrow stem cells. Circ Res 116:407–417
Cuoco L, Vescovo G, Castaman R, Ravara B, Cammarota G, Angelini A, Salvagnini M, Dalla LL (2008) Skeletal muscle wastage in Crohn’s disease: a pathway shared with heart failure? Int J Cardiol 127:219–227
Dalla LL, Ravara B, Gobbo V, Danieli BD, Germinario E, Angelini A, Vescovo G (2005) Skeletal muscle myofibrillar protein oxidation in heart failure and the protective effect of Carvedilol. J Mol Cell Cardiol 38:803–807
Davies LC, Jenkins SJ, Allen JE, Taylor PR (2013) Tissue-resident macrophages. Nat Immunol 14:986–995
de Alvaro C, Teruel T, Hernandez R, Lorenzo M (2004) Tumor necrosis factor alpha produces insulin resistance in skeletal muscle by activation of inhibitor kappaB kinase in a p38 MAPK-dependent manner. J Biol Chem 279:17070–17078
De Mori R, Straino S, Di CA, Mangoni A, Pompilio G, Palumbo R, Bianchi ME, Capogrossi MC, Germani A (2007) Multiple effects of high mobility group box protein 1 in skeletal muscle regeneration. Arterioscler Thromb Vasc Biol 27:2377–2383
Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL (2001) Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 103:2055–2059
Dogra C, Changotra H, Wedhas N, Qin X, Wergedal JE, Kumar A (2007) TNF-related weak inducer of apoptosis (TWEAK) is a potent skeletal muscle-wasting cytokine. FASEB J 21:1857–1869
Drexler H, Riede U, Munzel T, Konig H, Funke E, Just H (1992) Alterations of skeletal muscle in chronic heart failure. Circulation 85:1751–1759
Duscha BD, Kraus WE, Keteyian SJ, Sullivan MJ, Green HJ, Schachat FH, Pippen AM, Brawner CA, Blank JM, Annex BH (1999) Capillary density of skeletal muscle: a contributing mechanism for exercise intolerance in class II-III chronic heart failure independent of other peripheral alterations. J Am Coll Cardiol 33:1956–1963
Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, Iwamoto Y, Thompson B, Carlson AL, Heidt T, Majmudar MD, Lasitschka F, Etzrodt M, Waterman P, Waring MT, Chicoine AT, van der Laan AM, Niessen HW, Piek JJ, Rubin BB, Butany J, Stone JR, Katus HA, Murphy SA, Morrow DA, Sabatine MS, Vinegoni C, Moskowitz MA, Pittet MJ, Libby P, Lin CP, Swirski FK, Weissleder R, Nahrendorf M (2012) Myocardial infarction accelerates atherosclerosis. Nature 487:325–329
Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, Schilling JD, Schwendener R, Sergin I, Razani B, Forsberg EC, Yokoyama WM, Unanue ER, Colonna M, Randolph GJ, Mann DL (2014a) Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40:91–104
Epelman S, Lavine KJ, Randolph GJ (2014b) Origin and functions of tissue macrophages. Immunity 41:21–35
Ferrari R, Bachetti T, Confortini R, Opasich C, Febo O, Corti A, Cassani G, Visioli O (1995) Tumor necrosis factor soluble receptors in patients with various degrees of congestive heart failure. Circulation 92:1479–1486
Fogg DK, Sibon C, Miled C, Jung S, Aucouturier P, Littman DR, Cumano A, Geissmann F (2006) A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 311:83–87
Frantz S, Nahrendorf M (2014) Cardiac macrophages and their role in ischaemic heart disease. Cardiovasc Res
Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, Rubartelli A (2002) The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep 3:995–1001
Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma’ayan A, Chua WJ, Hansen TH, Turley SJ, Merad M, Randolph GJ (2012) Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 13:1118–1128
Ge RT, Mo LH, Wu R, Liu JQ, Zhang HP, Liu Z, Liu Z, Yang PC (2015) Insulin-like growth factor-1 endues monocytes with immune suppressive ability to inhibit inflammation in the intestine. Sci Rep 5:7735
Geddes K, Magalhaes JG, Girardin SE (2009) Unleashing the therapeutic potential of NOD-like receptors. Nat Rev Drug Discov 8:465–479
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–845
Glaros T, Larsen M, Li L (2009) Macrophages and fibroblasts during inflammation, tissue damage and organ injury. Front Biosci (Landmark Ed) 14:3988–3993
Gomes-Santos IL, Fernandes T, Couto GK, Ferreira-Filho JC, Salemi VM, Fernandes FB, Casarini DE, Brum PC, Rossoni LV, de Oliveira EM, Negrao CE (2014) Effects of exercise training on circulating and skeletal muscle renin-angiotensin system in chronic heart failure rats. PLoS One 9:e98012
Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, Deswarte K, Malissen B, Hammad H, Lambrecht BN (2013) Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med 210:1977–1992
Hambrecht R, Fiehn E, Yu J, Niebauer J, Weigl C, Hilbrich L, Adams V, Riede U, Schuler G (1997) Effects of endurance training on mitochondrial ultrastructure and fiber type distribution in skeletal muscle of patients with stable chronic heart failure. J Am Coll Cardiol 29:1067–1073
Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA, Geissmann F, Hedrick CC (2011) The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nat Immunol 12:778–785
Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, Greter M, Mortha A, Boyer SW, Forsberg EC, Tanaka M, van Rooijen N, Garcia-Sastre A, Stanley ER, Ginhoux F, Frenette PS, Merad M (2013) Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38:792–804
Hettinger J, Richards DM, Hansson J, Barra MM, Joschko AC, Krijgsveld J, Feuerer M (2013) Origin of monocytes and macrophages in a committed progenitor. Nat Immunol 14:821–830
Hoeffel G, Wang Y, Greter M, See P, Teo P, Malleret B, Leboeuf M, Low D, Oller G, Almeida F, Choy SH, Grisotto M, Renia L, Conway SJ, Stanley ER, Chan JK, Ng LG, Samokhvalov IM, Merad M, Ginhoux F (2012) Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med 209:1167–1181
Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, Beaudin AE, Lum J, Low I, Forsberg EC, Poidinger M, Zolezzi F, Larbi A, Ng LG, Chan JK, Greter M, Becher B, Samokhvalov IM, Merad M, Ginhoux F (2015) C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 42:665–678
Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, Lang R, Haniffa M, Collin M, Tacke F, Habenicht AJ, Ziegler-Heitbrock L, Randolph GJ (2010) Comparison of gene expression profiles between human and mouse monocyte subsets. Blood 115:e10–e19
Inoue N, Kinugawa S, Suga T, Yokota T, Hirabayashi K, Kuroda S, Okita K, Tsutsui H (2012) Angiotensin II-induced reduction in exercise capacity is associated with increased oxidative stress in skeletal muscle. Am J Physiol Heart Circ Physiol 302:H1202–H1210
Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, Ivanov S, Duan Q, Bala S, Condon T, van Rooijen N, Grainger JR, Belkaid Y, Ma’ayan A, Riches DW, Yokoyama WM, Ginhoux F, Henson PM, Randolph GJ (2013) Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity 39:599–610
Kapadia SR, Oral H, Lee J, Nakano M, Taffet GE, Mann DL (1997) Hemodynamic regulation of tumor necrosis factor-alpha gene and protein expression in adult feline myocardium. Circ Res 81:187–195
Kapadia SR, Yakoob K, Nader S, Thomas JD, Mann DL, Griffin BP (2000) Elevated circulating levels of serum tumor necrosis factor-alpha in patients with hemodynamically significant pressure and volume overload. J Am Coll Cardiol 36:208–212
Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, Ornitz DM, Randolph GJ, Mann DL (2014) Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci U S A 111:16029–16034
Li W, Moylan JS, Chambers MA, Smith J, Reid MB (2009) Interleukin-1 stimulates catabolism in C2C12 myotubes. Am J Physiol Cell Physiol 297:C706–C714
Lipkin DP, Jones DA, Round JM, Poole-Wilson PA (1988) Abnormalities of skeletal muscle in patients with chronic heart failure. Int J Cardiol 18:187–195
Mancini DM, Coyle E, Coggan A, Beltz J, Ferraro N, Montain S, Wilson JR (1989) Contribution of intrinsic skeletal muscle changes to 31P NMR skeletal muscle metabolic abnormalities in patients with chronic heart failure. Circulation 80:1338–1346
Mancini DM, Walter G, Reichek N, Lenkinski R, McCully KK, Mullen JL, Wilson JR (1992) Contribution of skeletal muscle atrophy to exercise intolerance and altered muscle metabolism in heart failure. Circulation 85:1364–1373
Mann DL (2015) Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res 116:1254–1268
Mann DL, Topkara VK, Evans S, Barger PM (2010) Innate immunity in the adult mammalian heart: for whom the cell tolls. Trans Am Clin Climatol Assoc 121:34–50
McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P (2010) Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 330:362–366
Meadows KA, Holly JM, Stewart CE (2000) Tumor necrosis factor-alpha-induced apoptosis is associated with suppression of insulin-like growth factor binding protein-5 secretion in differentiating murine skeletal myoblasts. J Cell Physiol 183:330–337
Mentz RJ, O’Connor CM (2016) Pathophysiology and clinical evaluation of acute heart failure. Nat Rev Cardiol 13:28–35
Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, Kannan HR, Menna AC, Voelkel NF, Abbate A (2011) The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci U S A 108:19725–19730
Molawi K, Wolf Y, Kandalla PK, Favret J, Hagemeyer N, Frenzel K, Pinto AR, Klapproth K, Henri S, Malissen B, Rodewald HR, Rosenthal NA, Bajenoff M, Prinz M, Jung S, Sieweke MH (2014) Progressive replacement of embryo-derived cardiac macrophages with age. J Exp Med 211:2151–2158
Mounier R, Theret M, Arnold L, Cuvellier S, Bultot L, Goransson O, Sanz N, Ferry A, Sakamoto K, Foretz M, Viollet B, Chazaud B (2013) AMPKalpha1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab 18:251–264
Musaro A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, Sweeney HL, Rosenthal N (2001) Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet 27:195–200
Muth IE, Zschuntzsch J, Kleinschnitz K, Wrede A, Gerhardt E, Balcarek P, Schreiber-Katz O, Zierz S, Dalakas MC, Voll RE, Schmidt J (2015) HMGB1 and RAGE in skeletal muscle inflammation: implications for protein accumulation in inclusion body myositis. Exp Neurol 271:189–197
Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ (2007) The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med 204:3037–3047
Nazare Nunes Alves MJ, dos Santos MR, Nobre TS, Martinez DG, Pereira Barretto AC, Brum PC, Rondon MU, Middlekauff HR, Negrao CE (2012) Mechanisms of blunted muscle vasodilation during peripheral chemoreceptor stimulation in heart failure patients. Hypertension 60:669–676
Niebauer J, Pflaum CD, Clark AL, Strasburger CJ, Hooper J, Poole-Wilson PA, Coats AJ, Anker SD (1998) Deficient insulin-like growth factor I in chronic heart failure predicts altered body composition, anabolic deficiency, cytokine and neurohormonal activation. J Am Coll Cardiol 32:393–397
Niebauer J, Volk HD, Kemp M, Dominguez M, Schumann RR, Rauchhaus M, Poole-Wilson PA, Coats AJ, Anker SD (1999) Endotoxin and immune activation in chronic heart failure: a prospective cohort study. Lancet 353:1838–1842
Orkin SH, Zon LI (2008) Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132:631–644
Pereira MR, Leite PE (2016) The involvement of parasympathetic and sympathetic nerve in the inflammatory reflex. J Cell Physiol
Piccinini AM, Midwood KS (2010) DAMPening inflammation by modulating TLR signalling. Mediat Inflamm
Rauchhaus M, Doehner W, Francis DP, Davos C, Kemp M, Liebenthal C, Niebauer J, Hooper J, Volk HD, Coats AJ, Anker SD (2000) Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation 102:3060–3067
Ren J, Samson WK, Sowers JR (1999) Insulin-like growth factor I as a cardiac hormone: physiological and pathophysiological implications in heart disease. J Mol Cell Cardiol 31:2049–2061
Ruffell D, Mourkioti F, Gambardella A, Kirstetter P, Lopez RG, Rosenthal N, Nerlov C (2009) A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc Natl Acad Sci U S A 106:17475–17480
Russell ST, Wyke SM, Tisdale MJ (2006) Mechanism of induction of muscle protein degradation by angiotensin II. Cell Signal 18:1087–1096
Sag D, Carling D, Stout RD, Suttles J (2008) Adenosine 5’-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol 181:8633–8641
Samokhvalov IM (2014) Deconvoluting the ontogeny of hematopoietic stem cells. Cell Mol Life Sci 71:957–978
Samokhvalov IM, Samokhvalova NI, Nishikawa S (2007) Cell tracing shows the contribution of the yolk sac to adult haematopoiesis. Nature 446:1056–1061
Sawyer RT, Strausbauch PH, Volkman A (1982) Resident macrophage proliferation in mice depleted of blood monocytes by strontium-89. Lab Investig 46:165–170
Schaufelberger M, Eriksson BO, Grimby G, Held P, Swedberg K (1997) Skeletal muscle alterations in patients with chronic heart failure. Eur Heart J 18:971–980
Schulz C, Gomez PE, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, Frampton J, Liu KJ, Geissmann F (2012) A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336:86–90
Schulze PC, Fang J, Kassik KA, Gannon J, Cupesi M, MacGillivray C, Lee RT, Rosenthal N (2005) Transgenic overexpression of locally acting insulin-like growth factor-1 inhibits ubiquitin-mediated muscle atrophy in chronic left-ventricular dysfunction. Circ Res 97:418–426
Shiotani I, Sato H, Sato H, Yokoyama H, Ohnishi Y, Hishida E, Kinjo K, Nakatani D, Kuzuya T, Hori M (2002) Muscle pump-dependent self-perfusion mechanism in legs in normal subjects and patients with heart failure. J Appl Physiol (1985) 92:1647–1654
Shireman PK, Contreras-Shannon V, Ochoa O, Karia BP, Michalek JE, McManus LM (2007) MCP-1 deficiency causes altered inflammation with impaired skeletal muscle regeneration. J Leukoc Biol 81:775–785
Sieweke MH, Allen JE (2013) Beyond stem cells: self-renewal of differentiated macrophages. Science 342:1242974
Song YH, Li Y, Du J, Mitch WE, Rosenthal N, Delafontaine P (2005) Muscle-specific expression of IGF-1 blocks angiotensin II-induced skeletal muscle wasting. J Clin Invest 115:451–458
Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ (2004) The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 14:395–403
Sullivan MJ, Knight JD, Higginbotham MB, Cobb FR (1989) Relation between central and peripheral hemodynamics during exercise in patients with chronic heart failure. Muscle blood flow is reduced with maintenance of arterial perfusion pressure. Circulation 80:769–781
Sullivan MJ, Green HJ, Cobb FR (1990) Skeletal muscle biochemistry and histology in ambulatory patients with long-term heart failure. Circulation 81:518–527
Summan M, Warren GL, Mercer RR, Chapman R, Hulderman T, van Rooijen N, Simeonova PP (2006) Macrophages and skeletal muscle regeneration: a clodronate-containing liposome depletion study. Am J Physiol Regul Integr Comp Physiol 290:R1488–R1495
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140:805–820
Tamrakar AK, Schertzer JD, Chiu TT, Foley KP, Bilan PJ, Philpott DJ, Klip A (2010) NOD2 activation induces muscle cell-autonomous innate immune responses and insulin resistance. Endocrinology 151:5624–5637
Tauber AI (2003) Metchnikoff and the phagocytosis theory. Nat Rev Mol Cell Biol 4:897–901
Topkara VK, Evans S, Zhang W, Epelman S, Staloch L, Barger PM, Mann DL (2011) Therapeutic targeting of innate immunity in the failing heart. J Mol Cell Cardiol 51:594–599
Torre-Amione G, Kapadia S, Benedict C, Oral H, Young JB, Mann DL (1996) Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: a report from the Studies of Left Ventricular Dysfunction (SOLVD). J Am Coll Cardiol 27:1201–1206
Tsutamoto T, Hisanaga T, Wada A, Maeda K, Ohnishi M, Fukai D, Mabuchi N, Sawaki M, Kinoshita M (1998) Interleukin-6 spillover in the peripheral circulation increases with the severity of heart failure, and the high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. J Am Coll Cardiol 31:391–398
Van Furth R, Cohn ZA (1968) The origin and kinetics of mononuclear phagocytes. J Exp Med 128:415–435
Vasan RS, Sullivan LM, D’Agostino RB, Roubenoff R, Harris T, Sawyer DB, Levy D, Wilson PW (2003a) Serum insulin-like growth factor I and risk for heart failure in elderly individuals without a previous myocardial infarction: the Framingham Heart Study. Ann Intern Med 139:642–648
Vasan RS, Sullivan LM, Roubenoff R, Dinarello CA, Harris T, Benjamin EJ, Sawyer DB, Levy D, Wilson PW, D’Agostino RB (2003b) Inflammatory markers and risk of heart failure in elderly subjects without prior myocardial infarction: the Framingham Heart Study. Circulation 107:1486–1491
Venereau E, Ceriotti C, Bianchi ME (2015) DAMPs from cell death to new life. Front Immunol 6:422
Vescovo G, Serafini F, Facchin L, Tenderini P, Carraro U, Dalla LL, Catani C, Ambrosio GB (1996) Specific changes in skeletal muscle myosin heavy chain composition in cardiac failure: differences compared with disuse atrophy as assessed on microbiopsies by high resolution electrophoresis. Heart 76:337–343
Villalta SA, Nguyen HX, Deng B, Gotoh T, Tidball JG (2009) Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum Mol Genet 18:482–496
Villalta SA, Deng B, Rinaldi C, Wehling-Henricks M, Tidball JG (2011a) IFN-gamma promotes muscle damage in the mdx mouse model of Duchenne muscular dystrophy by suppressing M2 macrophage activation and inhibiting muscle cell proliferation. J Immunol 187:5419–5428
Villalta SA, Rinaldi C, Deng B, Liu G, Fedor B, Tidball JG (2011b) Interleukin-10 reduces the pathology of mdx muscular dystrophy by deactivating M1 macrophages and modulating macrophage phenotype. Hum Mol Genet 20:790–805
Volkman A, Chang NC, Strausbauch PH, Morahan PS (1983) Differential effects of chronic monocyte depletion on macrophage populations. Lab Investig 49:291–298
Voltarelli VA, Bechara LR, Bacurau AV, Mattos KC, Dourado PM, Bueno CR Jr, Casarini DE, Negrao CE, Brum PC (2014) Lack of beta2-adrenoceptors aggravates heart failure-induced skeletal muscle myopathy in mice. J Cell Mol Med 18:1087–1097
Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ (1999) HMG-1 as a late mediator of endotoxin lethality in mice. Science 285:248–251
Warren GL, Hulderman T, Liston A, Simeonova PP (2011) Toll-like and adenosine receptor expression in injured skeletal muscle. Muscle Nerve 44:85–92
Wilson JR, Mancini DM, Dunkman WB (1993) Exertional fatigue due to skeletal muscle dysfunction in patients with heart failure. Circulation 87:470–475
Wynn TA, Chawla A, Pollard JW (2013) Macrophage biology in development, homeostasis and disease. Nature 496:445–455
Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S (2013) Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38:79–91
Yoshida T, Semprun-Prieto L, Sukhanov S, Delafontaine P (2010) IGF-1 prevents ANG II-induced skeletal muscle atrophy via Akt- and Foxo-dependent inhibition of the ubiquitin ligase atrogin-1 expression. Am J Physiol Heart Circ Physiol 298:H1565–H1570
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464:104–107
Zhang L, Pan J, Dong Y, Tweardy DJ, Dong Y, Garibotto G, Mitch WE (2013) Stat3 activation links a C/EBPdelta to myostatin pathway to stimulate loss of muscle mass. Cell Metab 18:368–379
Acknowledgements
Lavine is supported by funding from Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CHII2015-462), Foundation of Barnes-Jewish Hospital (8038-88), Burroughs Foundation Welcome Fund, and NIH K08 HL123519.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Lavine, K.J., Sierra, O.L. Skeletal muscle inflammation and atrophy in heart failure. Heart Fail Rev 22, 179–189 (2017). https://doi.org/10.1007/s10741-016-9593-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-016-9593-0