
Abstract
The renin-angiotensin system (RAS) plays a critical role in skeletal muscle biology. Angiotensin-(1-7) (Ang-(1-7)) has beneficial effects in skeletal muscle biology and disease. During exercise, Ang-(1-7) can modulate different organs helping to prevent and treat diseases such as chronic heart failure and diabetes. In skeletal muscle diseases, it has been found that Ang-(1-7) plays a protective role, counteracting the deleterious effects of angiotensin II (Ang-II) and transforming growth factor type beta (TGF-β), improving muscle function, and reducing fibrosis and inflammation.
Ang-(1-7) and Mas receptor antagonists have a tremendous potential for the treatment of different muscle pathologies such as muscular dystrophies and skeletal muscle wasting.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
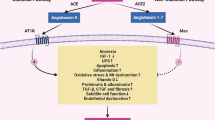

Keywords
Skeletal Muscle Overview
Skeletal muscle (SM) is the most abundant tissue spread through the entire body, plays critical role in keeping the postural control, has a vital function for making movements, both voluntary and nonvoluntary; and is also involved in the balance of energy metabolism.
SM is composed of muscle fibers that are long multinucleated cells surrounded by a specialized form of connective tissue the endomysium, and the fibers are organized as bundles of fibers called fascicles that are surrounded by the perimysium, and groups of fascicles are forming the muscles that are surrounded by the epimysium. Within the muscle fibers, capillaries that are oxygenating and nurturing the tissue are found, and each fiber is innervated by a motor neuron at the neuromuscular junction (NMJ), where the fibers are connected with the peripheral nervous system for the control of the muscle contraction. The quality of the skeletal muscle fibers is strongly influenced by the nerve. Another component of SM are the satellite cells that lie under the fibers basal lamina, these cells are stem cells committed to the muscle lineage. Other cell types present in SM are the fibroadipogenic progenitors (FAPs) and some monocytes residents in the muscle [1,2,3,4,5]. These cell types play important roles in the regeneration of SM. Figure 1 shows a schematic representation of SM architecture.

Left: Schematic representation of skeletal muscle tissue, showing the basic components of SM. Right: Upper panel shows a transversal cross-section from gastrocnemius of wild-type mice stained with hematoxylin/eosin. Lower panel shows an immunofluorescence for CD31 that is present in capillaries and laminin that marks the basal lamina delineating the muscle fiber; note that there are several capillaries surrounding one fiber (photograph taken and donated by Dr. Daniela Rebolledo)
RAS Components in Skeletal Muscle (SM)
There is increasing evidence that the renin-angiotensin system (RAS) plays a direct role in different tissues and the SM does not seem to be an exception. Several works have shown that components of both arms of the RAS are present in this tissue. Among the components of the classic RAS axis, it has been described that there is angiotensin- converting enzyme (ACE) expression and activity [6,7,8] and immunohistochemical analyses show that (ACE) is present in the endothelium and in the neuromuscular junction [8, 9] and its levels are increased in SM from Duchenne muscular dystrophy (DMD) patients [9], a myopathy that occurs by the lack of the dystrophin protein and is characterized by muscle weakness and fibrosis [10].
ACE exerts a key role in SM metabolism; in humans, there is a polymorphism on ACE gene, ACE I and D (insertion or deletion of a sequence in intron 16 of the ACE gene), and the ACE I polymorphism is characterized by reduction of ACE levels [11]. This brings more resistance to exercise performance because it can influence the blood flow and increase the oxidative metabolism in the SM [12].
The angiotensin type 1 (AT1) receptors are expressed in the vasculature of skeletal muscle of normal adult tissue [13] and its levels are increased in muscle byopsies obtained from DMD patients and AT1 can be found in the degenerating muscle fibers and fibroblasts as well [9]. AT1 receptors are also present in the SM satellite cells and its activation by angiotensin-II (Ang-II) reduces satellite cells proliferation and reduced regeneration after cardiotoxin (CTX) injury [14].
Angiotensin type 2 receptor (AT2) transcripts are detected in human fetal skeletal muscle tissues, but not in adult human skeletal muscle [13]; on the other hand, in mice, AT2 is detected in SM and shown to be increased by transforming growth factor beta (TGF-β1) injection in wild-type and in the mdx mice (the DMD murine model) [15]. AT2 receptor is increased in the regenerating muscle after CTX injury, and is necessary for proper satellite cell differentiation [16]. With regard to the opposite RAS axis, ACE2/angiotensin-(1-7)/Mas receptor (ACE2/Ang-(1-7)/Mas), there is evidence for ACE2 and Mas receptor presence in the SM [17,18,19,20,21,22].
ACE2 activity was present in mice SM, and it was localized in the sarcolemma and connective tissue of SM and was increased in the dystrophic muscle [18]. ACE2 is also playing an important role in the muscle metabolism, since its deletion leads to a reduced exercise performance [17]. Mas receptor is also found in mice SM and reported to increase in different muscle wasting conditions [20]. Interestingly, in mdx mice its expression was increased compared to wild-type mice (Fig. 1), which indicates that its levels are induced in this muscle pathology. At the cellular level, Mas receptor expression was found in myoblast and myotubes and also in SM-derived fibroblasts [23]. Mas receptor is downregulated by TGF-β1 in fibroblasts but not in myoblast or myotubes, suggesting differential regulation in the different SM cell types; interestingly, Mas expression was not modulated by Ang-II or connective tissue growth factor (CTGF/CCN2) [23]. Taken together, all these evidences support that the RAS is present and is an active player with critical and complex functions in the pathophysiology of SM.
RAS and SM Disease and Regeneration: Angiotensin-(1-7) Protective Role
Muscular Dystrophies
The classic arm of the RAS is related to skeletal muscle pathologies. In muscular dystrophies such as DMD, there are continuing cycles of progressive damage and regeneration of the fibers leading to a chronic inflammatory response that ultimately leads to the replacement of the fibers by fibrotic tissue. The main pro-fibrotic molecules in SM are (transforming growth factor type-beta) TGF-β [24,25,26], connective tissue growth factor (CTGF/CCN2) [26,27,28,29], and classic RAS components ACE/Ang-II/AT1 [25, 26].
Several studies have indicated that Ang-II plays an important role in the muscle fibrosis, since treatment with angiotensin type 1 receptor blockers (ARBs) and angiotensin-converting enzyme inhibitors (ACEi) general outcome is a reduction in the damage and fibrosis [30,31,32,33]. Losartan reduces TGF-β expression levels and smad-dependent signaling, an intracellular canonical branch activated upon TGF-β binding to its receptor [30, 31]. Both losartan and enalapril treatment reduce CTGF levels in mdx mice, and reduce the fibrotic response induced by CTGF overexpression [31, 32]. Ang-II through AT1 receptor leads to fibrosis by increasing TGF-β levels via activation of NAD(P)H oxidase-induced ROS and p38 phosphorylation that results in increased CTGF levels, increased extracellular matrix (ECM) proteins such as fibronectin, and collagen III [34, 35]; interestingly, the activation of NAD(P)H oxidase is PKC-dependent [35].
In muscular dystrophy, Ang-(1-7) systemic infusion by osmotic pumps or delivered orally by the formulation, cyclodextrin-conjugated Ang-(1-7) (CD-Ang-(1-7)), plays a protective role, reducing the damage, fibrosis, and restoring muscle strength in the mdx mice [21]; also in the model of sarcoglican-δ null muscular dystrophy, CD-Ang-(1-7) improves function and reduces the oxidative stress [36]. The mechanisms mediating the beneficial effects on dystrophy involve the reduction in TGF-β levels and smad-dependent signaling, observed by the reduction in phosphorylated smad3, decrease in the fibrotic miR-21, resulting in a reduction of TCF-4 positive fibroblasts. The Ang-(1-7) effects are mediated by the Mas receptor signaling since treatment with the Mas antagonist A-779 or the genetic deletion of Mas receptor in the mdx mice model results in a worse dystrophic phenotype, dramatically increasing damage, fibrosis, and smad3 signaling and reducing skeletal muscle strength [21], resembling the SM phenotype observed in DMD.
Another mechanism for the reduction of damage and fibrosis mediated by Ang-(1-7) in dystrophic muscle is by the decrease in CTGF levels (Fig. 3). Interestingly, Ang-(1-7) can also reduce muscle fibrosis in a model of radiation-induced fibrosis by reducing TGF-β and CTGF [37].

Mas receptor expression is increased in skeletal muscle of mdx mice. The relative expression of Mas receptor was evaluated by qPCR using taqman probes for Mas1 and GAPDH as control, cDNA was obtained from total RNA extracts from wild-type (WT) and mdx mice Gastrocnemius, tibial anterior, and diaphragm muscles. T-test ∗p < 0.05 vs WT

CTGF/CCN2 expression is reduced by Ang-(1-7) treatment. The relative expression of CTGF was evaluated by qPCR using taqman probes for CTGF and GAPDH as control; cDNA was obtained from total RNA extracts of gastrocnemius from wild-type, mdx, and mdx treated with Ang-(1-7) mice. One-way ANOVA ∗p < 0.001 vs WT, ∗∗p < 0.001 vs mdx
Overall, Ang-(1-7) protects muscle from damage and fibrosis, and since the mdx KO Mas mice had a worse phenotype [21], it seems that the endogenous Ang-(1-7) protects from further damage, maybe by compensatory mechanisms, because ACE2 and MasR are increased in the mdx mice. It would be interesting to measure ACE2/Ang-(1-7)/MasR in DMD patient’s samples. Interestingly, in the sarcoglican-δ null mice, there is a reduction of MasR levels compared with wild type that are restored by CD-Ang-(1-7) infusion [36], suggesting that the reduction in Ang-(1-7) receptor could lead to damage in this model.
Atrophy and Muscle Wasting Conditions
Muscle wasting, a condition observed in disuse, immobilization, aging, and some neuromuscular diseases, occurs by the increase of the catabolic pathways and reduction of the anabolic pathways. The muscle catabolic pathways upregulation is the result of increased activity of growth factors such as myostatin, which leads to protein degradation, and also of the induction of transcription factors such as NF-κB and FoxO3, which results in the expression of E3 ligases MuRF-1 and atrogin1 (also known as MAFbx) with consequent protein degradation by the ubiquitin proteasome pathway (UPS ubiquitin proteasome system) [38,39,40,41]. On the other hand, there is a reduction of anabolic pathways such as IGF-1 and its downstream signaling cascade PI3K/AKT/mTOR, leading to decreased protein synthesis and cell growth [39, 41, 43], the reduction of IGF-1 also activates the FoxO 1/3 transcription factors that induce atrogin1 [38, 42] tilting the balance toward the catabolic pathways, finally resulting in a decrease of SM mass.
Ang-II drives the muscle wasting by inducing the catabolism and inhibiting IGF-1 signaling [44]; it induces NF-κB, which in turns induces Atrogin1. Ang-II drives muscle wasting by increasing oxidative pathways [45, 46], leading to activation of catabolic pathways [47, 48]. Also by inducing protein phosphatase 2C alpha (PP2Cα), Ang-II reduces AMPK signaling also leading to muscle wasting [49]. Interestingly, the knockdown of PP2Cα reduces Ang-II-mediated atrophy by a mechanism involving mitochondrial recycling [50].
Cabello–Verrugio’s group has studied Ang-(1-7) effects in several models of muscle atrophy. In a model of atrophy induced by Ang-II, the treatment with Ang-(1-7) prevented the reduction in fiber size, the reduction in strength, and maintained the levels of myosin heavy chain; these effects were dependent on Mas receptor since, in the presence of A-779 (the Mas receptor antagonist), the protective Ang-(1-7) effects were lost [51]. Ang-(1-7) prevented the increase of the expression of atrogin1 and MuRF1. Also, it was found that Ang-(1-7) induces the phosphorylation of AKT in a Mas-dependent fashion. In vitro, the inhibition of AKT inhibited the anti-atrophic effects of Ang-(1-7) [51]. Further experiments showed that Ang-(1-7) reduces the myonuclear apoptosis induced by Ang-II, as observed by the reduction in the apoptotic nuclei, reduction in the caspases 8 and 9, and reduction in the activity of caspase 3 and the Bax/Bcl-2 ratio. These effects were Mas-dependent because A-779 prevented Ang-(1-7) protection [52]. In a model of atrophy induced by limb immobilization, Ang-(1-7) also prevented the wasting effects, such as the reduction of fiber size decreased MHC levels, and reduced the E3 Ligases Atrogin-1 and MuRF-1 protein levels, by a mechanism that involved the phosphorylation of the IGF-1 receptor, AKT, and p70S6K resulting in the inhibition of FoxO3. The activation of IGF receptor (IGF-R) was necessary for the anti-atrophic effects of Ang-(1-7). Importantly, all these anti-atrophic effects of Ang-(1-7) were lost in the KO Mas mice [53]. Interestingly, a dendrimer carrying Ang-(1-7) (PAMAM-OH-Ang-(1-7)) that can encapsulate two molecules of Ang-(1-7) was administered intraperitoneally to mice, and also prevented the inmobilization-induced atrophy [54].
In a model of atrophy induced by the endotoxin lipopolysaccharide (LPS), Ang-(1-7) treatment prevented the atrophy induced by LPS, as seen by the maintenance of fiber size and MHC levels and the prevention in the reduction muscle force. Ang-(1-7) reduced the levels of atrogin-1 and MuRF-1 induced by LPS, and these effects were Mas receptor-dependent since A-779 inhibited the Ang-(1-7) effects. Interestingly, Ang-(1-7) reduced the p38 phosphorylation mediated by LPS and this was necessary for the anti-atrophic effects of Ang-(1-7) [55].
Ang-(1-7) through Mas receptor, also prevented the atrophy induced by TGF-β in vivo and in vitro, as observed by maintenance of fiber size, MHC levels, and a reduction in the MuRF-1 levels. Interestingly, Ang-(1-7) reduced ROS production induced by TGF-β preventing its atrophic effects [56].
Skeletal Muscle Regeneration
Muscle regeneration occurs after muscle injury or damage, and is a regulated process: different cell types participate in the regeneration – first, there is an acute inflammatory response that in turns activates the resident fibroblasts and FAPS that start producing ECM molecules to seal the injury site, then this ECM is degraded and fibroblasts die by apoptosis. During this process, the satellite cells begin to proliferate and ultimately differentiate to form new muscle fibers [2, 57]. Ang-II impairs muscle regeneration by reducing satellite cell proliferation and differentiation capacity [14]. Losartan treatment showed an improved regeneration capacity in CTX injury in a fibrilin-1 deficient mice, mdx mice, and in old mice with sarcopenia by a mechanism that involve TGF-β signaling reduction [30, 58].
The potential role of Ang-(1-7) participating in muscle regeneration has not been fully elucidated, since there are no reports to our knowledge. Nevertheless, this issue is being studied by us and so far we have found that systemic infusion with Ang-(1-7) accelerates the regeneration by modulating macrophages population and increasing satellite cells number, by a mechanism dependent of iNOS activation (Ramirez et al., ms in preparation)). Interestingly, in a model of chronic injury, by repeated cycles of BaCl2 injection there are increased levels and activity of ACE2 [18], suggesting that Ang-(1-7) is present in the chronic damage mice model. What is the function played by Ang-(1-7) in skeletal muscle regeneration is still an open question.
Future Directions
There are still open questions about Ang-(1-7) role in skeletal muscle biology and disease. For example, we still don’t know the mechanisms by which Ang-(1-7) has a protective role in SM disease downstream Mas receptor activation, we know that there are some signaling pathways involved like IGF-1 and AKT in the prevention of atrophy [51, 53], but it is still unclear if the same pathways are activated in the protection of damage and fibrosis in DMD. In regeneration, Ang-(1-7) induces iNOS but we do not know if it occurs directly or indirectly by activating another route. Recently, the Kallikrein Kinin system (KKS), a hypotensor and anti-fibrotic system [59], has been involved in DMD [60], so it would be interesting to elucidate if Ang-(1-7) could also be synergizing with Bradykinin, the main product of the KKS, to exert a beneficial effect.
Ang-(1-7) in SM has the opposite actions to Ang-II, and can reduce TGF-β and its signaling molecules, so it would be of interest to search what is the effect of Ang-(1-7) in other neuro- muscular diseases that have increased levels of TGF-β, such as ALS [61] or Marfan Syndrome [30]. Since Ang-(1-7) has anti-atrophic effects, there is also an interesting question if this peptide has any effect on sarcopenia or aged muscles.
There are efforts being made to test Ang-(1-7) analogs or Mas receptor agonists or different ways to deliver Ang-(1-7), that are less prone to degradation, since it could have a therapeutic potential; in this regard, cyclodextrin-delivered Ang-(1-7) has been proved and could be a good candidate [21, 36]; other way of delivering Ang-(1-7) was tested in a model of atrophy by the use of PAMAM-OH with promising results [54]. There are analogs, such as A-1317, being tested together with Robson Santo’s group in DMD mice model to see if the same beneficial effects as for Ang-(1-7) can be achieved using this molecule. Other candidate to prove could be the cyclic Ang-(1-7) that has been tested in mice model of cardiac infarction [62]. AVE0991 a Mas agonist that has been tested in different pathologies such as heart failure, arthritis, gastric ulcers, and asthma [63,64,65,66,67] could also be tested in SM diseases.
Physical Exercise and ACE2/Angiotensin-(1-7)/Mas Axis
Physical exercise represents an important nonpharmacological tool for prevention and treatment of cardiovascular and metabolic disease. Skeletal muscle is the main organ affected by acute and chronic exercise and can act as an endocrine organ producing and secreting myokines [68]. The skeletal muscle contraction is a potent stimulus to induce exercise adaptations in many other organs such as brain, heart, lung, kidney, and liver. Here, we will describe:
-
Angiotensin-(1-7) effects in trained animals.
-
Modulation of ACE2/Ang-(1-7)/MAS axis in response to physical exercise.
-
Angiotensin-(1-7) activation (treatment) or inactivation (Mas-deficiency) and exercise training.
Angiotensin-(1-7) Effects in Trained Animals
The angiotensin-(1-7) effects in trained and sedentary brains and vessels have shown the role of this peptide in response to aerobic training exercise. These studies demonstrated that chronic exercise affects the acute actions of Ang-(1-7) (hemodynamic and vascular responses). In some pathological conditions as in hypertensive rats (SHR), these effects are more evident.
Brain
The first study showing the effects of angiotensin-(1-7) associated with exercise was published in 2005 by Becker et al. The authors demonstrated an involvement of Ang-(1-7) in physical exercise adaptations [69]. In this study, the rats performed 20 swimming exercise sessions, 1 hour per day, 5 days a week. After 4 weeks of training, cardiovascular effects produced by microinjections in rostroventrolateral medulla (RVLM) of the angiotensin peptides, Ang-II and Ang-(1-7) were evaluated in trained and sedentary animals. The exercise training enhanced Ang-II and attenuated Ang-(1-7) pressor effect (Fig. 4).

Changes in mean arterial pressure (mm Hg) produced by microinjection of saline, Ang-II (50 ng) or Ang-(1-7) (50 ng) into the RVLM of sedentary and trained rats. ∗p < 0.05 as compared with saline
The spontaneous hypertensive rats (SHR) presented altered renin-angiotensin system in the RLVM and the effects of 12 weeks of running training (5 days per week; 60 min per day at 15–20 m/min) were evaluated by Ren et al. [70]. Aerobic training significantly reduced sarthan (antagonist of Ang-II) or increased A-779 (antagonist of Ang-(1-7)) cardiovascular responses to central application of each antagonist, respectively. The protein expression of MasR in the RVLM was significantly elevated in SHR following aerobic training. These results suggest that the central effect in the pressor response for Ang-(1-7) is modulated by physical exercise and future research needs to be made to understand the mechanisms involved in this modulation.
Vessels
The vascular effects of Ang-(1-7) in trained animals (8-weeks of overload 5% of the body weight swimming training) were investigated in the aorta of SHR-trained animals [71]. Untrained SHR had an impaired vasodilator response to Ang-(1-7) and exercise training could reverse this response (Fig. 5). The Ang-(1-7) vasodilator effect was abrogated by A-779 and d-Pro(7)-Ang-(1-7) (selective Ang-(1-7) receptor antagonists) and by removal of the endothelium.

Vasodilator effect of Ang-(1-7) in endothelium-intact aortic rings from (a) trained wistar and untrained wistar rats, (b) untrained SHR and untrained and (c) untrained and trained SHR rats, ∗p < 0.05; ∗∗∗p < 0.001
This study also showed that only in SHR-trained animals, the aorta MasR protein expression was substantially increased and correlated with the Ang-(1-7) effect (Fig. 6).

Representative immunoblots of MasR in untrained and trained SHR aortas. Top shows representative Western blot signals. ∗p < 0.05 untrained vs. trained SHR
The cardioprotective effects promoted by exercise training could implicate the activation of ACE2/Ang-(1-7)/Mas axis, especially in pathological conditions such arterial hypertension.
Modulation of ACE2/ANG-(1-7)/MAS Axis in Response to Physical Exercise
The effects of physical exercise on ACE2/Ang-(1-7) /Mas activation in different pathologies/organs will be discussed in the following sections.
Heart, Hypertension, and Heart Failure
Aerobic exercise training induces several cardiac effects that culminate in an aerobic capacity improvement due in part to the increased ventricular stroke volume, cardiac output, and left ventricle hypertrophy [72]. The left-ventricle hypertrophy is dependent on volume training; therefore, Fernandes et al. investigated the modulation of ACE2 and Ang-(1-7) in rats following distinct swimming training protocols [73]:
-
Low-intensity, moderate-volume exercise: 60-minute per session, 5 days a week, for 10 weeks
-
Low-intensity, high-volume exercise: the same swimming training protocol described until the end of the eighth week and on the ninth week, rats were trained twice a day (60 min per session and an interval of 4 hours between sessions).
All exercise session was carried out with caudal dumbbells weighing 5% of animal body weight. ACE2, Ang-(1-7), and the ratio Ang-(1-7) in left ventricle increase significantly in both exercise protocols and involve regulatory MicroRNAs (miR-27a, miR-27b, and miR-143).
Physical training effect on MasR expression in hearts under different physiological and pathological conditions has been evaluated by Dias-Peixoto et al. [74]. The physiological stimulus was the swimming training performed 40–60 min per day, 5 days per week over 10 weeks. No changes in MasR expression in the trained-left ventricle Sprague–Dawley (SD) rats were observed. However, in some pathological conditions such as isoproterenol treatment and infarction, MasR downregulated responses have been evidenced. Since in hypertensive rats (SHR), the role of Ang-(1-7) mediating cardioprotective effects has been consistently demonstrated (please see Actions of Angiotensin-(1-7): Heart), the reduction of MasR expression showed by Dias-Peixoto et al. could explain in part the cardiac damage in these model.
The cardiac effects of 8 weeks period of 5% overload swimming training (1 hour per day, 5 days a week) and the role of Ang-(1-7) in normotensive (Wistar) and hypertensive (SHR) rats were evaluated by Filho et al. [75]. Interestingly, the plasma levels of Ang II reduced in both trained groups but only SHR-trained had a significant increase in left ventricle levels of Ang-(1-7). Cardiac (left ventricle) gene expression of MasR was significantly increased in trained SHR, but not in trained Wistar rats (Fig. 7).

Left ventricle Ang(1-7) levels in sedentary and trained Wistar rats and SHR (upper panel) and relative levels of MasR mRNA in left ventricle assessed by semi-quantitative RT-PCR (lower panel), ∗∗∗p < 0.001 compared with sedentary rats, ∗p < 0.01 compared with sedentary SHR. SWR sedentary Wistar, SSHR sedentary SHR, TSHR trained-SHR, TWR trained-Wistar
Additionally, it has been described that heart failure reduction of ACE and ACE2 levels in the brain can be normalized through chronic running exercise (30 min per day, 6 days a week for 3 weeks) [76].
Recently, Tyrankiewicz et al. analyzed systemic (plasma) and local (heart/aorta) changes in ACE/ACE-2 balance in Tgαq∗44 mice in course of heart failure (HF) [77]. Tgαq∗44 mice present cardiomyocyte 38 specific Gαq overexpression and develop late onset of HF. The HF development in this animal model is associated with systemic and local activation of ACE/Ang-II axis and this effect is counterbalanced by an important ACE2/Ang-(1-7) activation. In this study, they also evaluated voluntary wheels running performance in young and aged animals, but only in 12-month-old Tgαq∗44 mice, the mean distance and time were significantly decreased. However, running wheels is not the best “model” to assess cardiovascular performance considering that neurotransmitter systems are involved in wheel-running behavior. In this line, running-wheel training in a ACE2-deficiency mice affects physical performance and impairs cardiac and skeletal muscle adaptations to exercise [17].
Another effect of ACE2 could involve tryptophan that was recently shown to stimulate the expression of myogenic genes [78]. The muscle strength in ACE2-knockout mice could be impaired by the reduction of tryptophan action, considering that ACE2 has an important effect on tryptophan uptake [79].
Moderate- intensity running training for 12 weeks modulates RAS axis by reducing AngII and increasing Ang-(1-7) in aorta of trained SHR [80]. Confirming the role of Ang-(1-7) promoting vasoprotective effect induced by physical exercise, Ang-(1-7) and ACE2 protein levels were normalized (Fig. 8).

Effects of exercise training on aortic Ang-(1-7) content and ACE2 expression in SHR. Values are represented as mean ± S.D. ∗p < 0.05 versus WKY; †p < 0.05 versus SHR; n = 10–12 in each group
The effects of running aerobic exercise training (60 min at 60% of peak VO2, 5 days a week for 8 week) in a ischemic model of cardiac heart failure (CHF) confirm that exercise training causes a shift in the Ang-(1-7)/Mas axis in skeletal muscle of CHF rats [81]. CHF (left coronary artery ligation) reduced ACE2 serum activity; however, exercise training restored and increased the Ang-(1-7)/Ang-II ratio. Skeletal muscle ACE and ACE2 activity and protein did not change, but Ang-(1-7) in plantaris and MasR in the soleus of CHF mice significantly increased. It is important to note that the local RAS (skeletal muscle) is not directly affected by circulation levels of angiotensin peptides.
Another elegant study using SHR provides strong evidence that low-intensity aerobic training downregulates RAS not only in vessels but also in the kidney and plasma of normotensive and hypertensive rats [82].
Preeclampsia and Estrogen Deficiency
Exercise training can attenuate/prevent preeclampsia and these protective effects could be associated with RAS modulation [83]. In a previous study on preeclampsia mouse model (overexpressing human angiotensinogen and human renin), it was demonstrated that voluntary running is effective in attenuating blood pressure increase [84]. Preeclamptic mice presented lower MasR protein expression in aorta and placenta compared to normotensive, and the voluntary running is able to “normalize” the MasR expression.
In estrogen deficiency and hypertensive conditions, some RAS components are altered. Endlich et al. demonstrated that ovariectomized spontaneously hypertensive trained rats (60 min per day, 5 days a week, lasted 8 weeks) increased constrictor responses to Ang-II and decreased dilatory responses to Ang-(1-7) independently of estrogen therapy [85]. A significant increase in Ang-(1-7) aorta was found in exercise trained-groups and in the estrogen therapy group. This study has demonstrated that in a model of menopause in rodents, the estrogen therapy and swimming training are able to decrease systolic blood pressure and increase Ang-(1-7) (Fig. 9).

ANG-(1-7) plasma levels in ovariectomized SHRs subjected to regular swimming or estrogen therapy. SH (n = 7), OVX (n = 4), OE2 (n = 4), OSW (n = 6), and OE2 + SW (n = 5) groups. Data are expressed as the means ± SEM. One-way ANOVA with Tukey’s post-hoc test was used. ∗p < 0.05 vs. SH group, #p < 0.05 vs. OE2 group, †p < 0.05 vs. OE2 + SW group
Diabetes and Obesity
It is quite well established the protective metabolic effects of Ang-(1-7) in diabetes and obesity. However, little is known about the involvement of this heptapeptide in metabolic protective responses induced by chronic exercise. Somineni et al. investigated whether exercise training and/or metformin improve glucose homeostasis and downregulate renal ADAM17 and ACE2 shedding in db/db mice [86]. The training consisted 10 weeks of aerobic training and daily exercise (1 hour per day of forced exercise on walking wheel). Exercise training alone and in combination with metformin prevented shedding of renal ACE2 by decreasing ADAM17 protein, and this may partially contribute to renal protection.
The exercise training (8 weeks of training, 50–75% of maximal running speed, 60 min per day and 4 days a week) normalizes (prevents the increase) angiotensin-converting enzyme, ACE (activity and protein) and Ang-II in hepatic tissue in fructose overloaded rats [87]. In addition, ACE2, Ang(1-7), and Mas receptor increase in the liver leading to ACE/ACE2, Ang-II/Ang(1-7), and AT1R/Mas receptor ratios towards normal values. Interestingly, no changes in the systemic RAS components were detected.
The effects of High Intensity Interval Training (HIIT) mice fed high-fat or high-fructose in RAS components were recently evaluated [88]. The exercise training consisted of HIIT protocol: 2 min of high-intensity 45 m/min (90% VO2) running and 1 min of low intensity at 15 m/min (30% VO2) running 3 days a week on alternate days over 12 weeks. The authors showed that the exercise training enhanced the insulin sensitivity and these results could be related to reduced levels of the classic RAS components and increase in ACE2 and MasR in the HIIT mice compared with the nontrained group.
Asthma
Unpublished data suggested that the protective effects of chronic aerobic exercise in asthma involves the activation of MasR in a model of chronic asthma. In their study, Gregório et al. (2016), induced asthma through OVA albumin challenge and submitted the animals to running exercise (1 hour per day, 5 days a week, during 6 weeks). The IgG circulating levels in “asthmatic” animals were significantly elevated but running training was able to abolish this increase. If physical exercise is associated with MasR blockade (A-779 antagonist), the levels of IgG are significantly higher compared to asthmatic animals even when associated with exercise training. These results suggested that the beneficial effects of exercise could be missing when MasR actions were blocked.
Angiotensin-(1-7) Activation (Treatment) or Inactivation (Mas-Deficiency) and Exercise Training
Some studies investigated the additive effects of Ang-(1-7) treatment associated with physical training and the effects of exercise training in Mas-deficiency animals (Mas−/−) [89, 90]. Additionally, the protective effects induced by exercise training appear to be MasR-dependent. The Mas−/− mice did not decrease blood pressure and improve body composition (observed in Mas+/+ mice) when submitted to voluntary running through 6 weeks (Motta-Santos, unpublished data).
The Ang-(1-7) infusion effects associated or not to swimming training was evaluated in hypertensive rats (2K1C) [91]. Exercise training consisted of swimming training performed 1 hour per day, 5 days a week through 4 weeks. Intriguing, Ang-(1-7) treatment attenuated hypertension and cardiac hypertrophy only 2K1C-trained rats and MasR was upregulated only in the left-ventricles of trained 2K1C rats. These results suggest that the beneficial effect of Ang-(1-7) is potentiated by physical performance.
The treatment with orally active Ang-(1-7) included in hydroxy-propyl-beta-cyclodextrin produces several cardioprotective effects (see Heart Chapter) and its effects combined or not to physical training in spontaneously hypertensive rats (SHR) were investigated by Bertagnolli et al. [90]. The SHR were divided in control (tap water) or treated with HPβ-CD/Ang-(1-7) with or without running exercise training (1 hour per day, 5 days per week, 10 weeks). Similar beneficial effects to the ones produced by exercise training were observed in HPβ-CD/Ang-(1-7) nontrained SHR. These effects include decreased arterial blood pressure (BP) and heart rate, improved left ventricular (LV) end-diastolic pressure, restored the maximum and minimum derivatives (dP/dT), and decreased cardiac hypertrophy index. Additionally, an improvement in autonomic control by attenuating sympathetic modulation on heart and vessels and the SAP variability, as well as increasing parasympathetic modulation and HR variability were observed in trained and Ang-(1-7)-treated SHR animals.
While activation of Ang-(1-7)/MAS promotes effects similar to those seen in trained animals, on the other hand, the MasR deficiency (Mas−/−) abolishes some benefits of the physical exercise.
Mas−/− mice presented heart and skeletal muscle remodeling alterations (please see Heart and Skeletal muscle chapter). Since Ang-(1-7) could be involved in the physiological cardiac remodeling induced by exercise training, Guimaraes et al. aimed to investigate the cardiac physical exercise effects in Mas−/− [89]. Six weeks of swimming training (5 days per week, once a day for 1 hour with an 80% of maximal load workload attached to the tail) induced similar increase (∼10%) in cardiomyocyte diameter in Mas−/− and Wild-Type (WT) animals. However, in sedentary groups, circulating levels of Ang-(1-7) were significantly lower in Mas−/− as compared to WT. Also, Ang-II levels in blood and LV increase only in the Mas-KO-trained (Fig. 10). Additionally, to a null increase in the cardiac Ang-(1-7) levels of Mas−/−trained group, they presented a higher collagen I and II gene expression compared to WT (Fig. 11). The authors concluded that exercise training was able to induce an increase in the Ang II/Ang-(1-7) blood ratio only in Mas-deficiency-trained, suggesting strong imbalance in circulating RAS with a predominance of Ang-II in Mas−/−.

Ang-II and Ang-(1-7) levels in the total blood and left ventricle (LV) of sedentary and trained Mas-KO and WT mice as determined by RIA. Statistically significant differences between the groups are indicated as ∗p < 0.05 in comparison to sedentary control and #p < 0.05 in comparison to WT

Left ventricle (LV) mRNA expression of extracellular matrix proteins in sedentary and trained Mas-KO and WT mice. Data are presented as means ± SE. Statistically significant differences between the groups are indicated as ∗p < 0.05 in comparison to sedentary control and #p < 0.05 in comparison to WT
The physical performance associated with asthma model in Mas−/− mice also suggests that the asthmatic Mas−/− animals presented a worse performance in a test of maximum physical exercise compared with WT asthmatic group [92].
Human Studies
A series of human studies have been conducted to verify the effects of acute and chronic exercise in Ang-(1-7) peptides levels. The preliminary and unpublished results suggest that Ang-(1-7) plasmatic levels after exercise (acute and chronic) is dependent of duration, intensity, and mode (resistance, aerobic, and combined exercise). The status of training is another point that can interfere in the systemic RAS modulation.
Acute eccentric physical exercise can promote some microlesions, inflammation, and pain (DOMS) in skeletal muscle but after a chronic period important adaptations can be reached. This include strength gain, power increase, and remodeling (hypertrophy). The effects of Ang-(1-7) oral compound associated with eccentric exercise induced muscle damage (squat exercise) was recently tested in young healthy subjects. Surprisingly, the Ang-(1-7)-treated group presented less pain and higher strength compared to placebo group [93]. The inflammatory marker responses suggest that the Ang-(1-7) can attenuate inflammation or maybe accelerate the recovery following eccentric exercise protocol. Further studies can elucidate the mechanism involved in this effect including local analysis directly to skeletal muscle.
Abbreviations
- ACE:
-
Angiotensin converting enzyme
- ACE2:
-
Angiotensin converting enzyme 2
- Ang-(1-7):
-
Angiotensin-(1-7)
- Ang-II:
-
Angiotensin II
- AT1:
-
Angiotensin receptor 1
- AT2:
-
Angiotensin receptor 2
- CTGF/CCN-2:
-
Connective tissue growth factor
- DMD:
-
Duchenne muscular dystrophy
- ECM:
-
Extracellular matrix
- MasR:
-
Mas receptor
- NMJ:
-
Neuromuscular junctions
- RAS:
-
Renin-angiotensin system
- SM:
-
Skeletal muscle
- TGF-β:
-
Transforming growth factor type-β
Bibliography
Judson RN, Zhang RH, Rossi FM. Tissue-resident mesenchymal stem/progenitor cells in skeletal muscle: collaborators or saboteurs? FEBS J. 2013;280(17):4100–8.
Zammit PS. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin Cell Dev Biol. 2017;72:19–32.
Pannerec A, Marazzi G, Sassoon D. Stem cells in the hood: the skeletal muscle niche. Trends Mol Med. 2012;18(10):599–606.
Latroche C, et al. Skeletal muscle microvasculature: a highly dynamic lifeline. Physiology (Bethesda). 2015;30(6):417–27.
Mounier R, Chretien F, Chazaud B. Blood vessels and the satellite cell niche. Curr Top Dev Biol. 2011;96:121–38.
Reneland R, Lithell H. Angiotensin-converting enzyme in human skeletal muscle. A simple in vitro assay of activity in needle biopsy specimens. Scand J Clin Lab Invest. 1994;54(2):105–11.
Ward PE, Russell JS, Vaghy PL. Angiotensin and bradykinin metabolism by peptidases identified in skeletal muscle. Peptides. 1995;16(6):1073–8.
Schaufelberger M, et al. Angiotensin-converting enzyme gene expression in skeletal muscle in patients with chronic heart failure. J Card Fail. 1998;4(3):185–91.
Sun G, et al. Intramuscular renin-angiotensin system is activated in human muscular dystrophy. J Neurol Sci. 2009;280(1–2):40–8.
Fairclough RJ, Wood MJ, Davies KE. Therapy for Duchenne muscular dystrophy: renewed optimism from genetic approaches. Nat Rev Genet. 2013;14(6):373–8.
Tiret L, et al. Evidence, from combined segregation and linkage analysis, that a variant of the angiotensin I-converting enzyme (ACE) gene controls plasma ACE levels. Am J Hum Genet. 1992;51(1):197–205.
Vaughan D, et al. The angiotensin converting enzyme insertion/deletion polymorphism modifies exercise-induced muscle metabolism. PLoS One. 2016;11(3):e0149046.
Malendowicz SL, et al. Angiotensin II receptor subtypes in the skeletal muscle vasculature of patients with severe congestive heart failure. Circulation. 2000;102(18):2210–3.
Yoshida T, et al. Angiotensin II inhibits satellite cell proliferation and prevents skeletal muscle regeneration. J Biol Chem. 2013;288(33):23823–32.
Painemal P, et al. Transforming growth factor type beta 1 increases the expression of angiotensin II receptor type 2 by a SMAD- and p38 MAPK-dependent mechanism in skeletal muscle. Biofactors. 2013;39(4):467–75.
Yoshida T, Huq TS, Delafontaine P. Angiotensin type 2 receptor signaling in satellite cells potentiates skeletal muscle regeneration. J Biol Chem. 2014;289(38):26239–48.
Motta-Santos D, et al. Effects of ACE2 deficiency on physical performance and physiological adaptations of cardiac and skeletal muscle to exercise. Hypertens Res. 2016;39(7):506–12.
Riquelme C, et al. ACE2 is augmented in dystrophic skeletal muscle and plays a role in decreasing associated fibrosis. PLoS One. 2014;9(4):e93449.
Echeverria-Rodriguez O, Del Valle-Mondragon L, Hong E. Angiotensin 1-7 improves insulin sensitivity by increasing skeletal muscle glucose uptake in vivo. Peptides. 2014;51:26–30.
Morales MG, et al. Expression of the Mas receptor is upregulated in skeletal muscle wasting. Histochem Cell Biol. 2015;143(2):131–41.
Acuna MJ, et al. Restoration of muscle strength in dystrophic muscle by angiotensin-1-7 through inhibition of TGF-beta signalling. Hum Mol Genet. 2014;23(5):1237–49.
Fernandes T, Hashimoto NY, Oliveira EM. Characterization of angiotensin-converting enzymes 1 and 2 in the soleus and plantaris muscles of rats. Braz J Med Biol Res. 2010;43(9):837–42.
Cofre C, et al. Transforming growth factor type-beta inhibits Mas receptor expression in fibroblasts but not in myoblasts or differentiated myotubes; relevance to fibrosis associated to muscular dystrophies. Biofactors. 2015;41(2):111–20.
Pessina P, et al. Novel and optimized strategies for inducing fibrosis in vivo: focus on Duchenne muscular dystrophy. Skelet Muscle. 2014;4:7.
Serrano AL, Munoz-Canoves P. Fibrosis development in early-onset muscular dystrophies: mechanisms and translational implications. Semin Cell Dev Biol. 2017;64:181–90.
Smith LR, Barton ER. Regulation of fibrosis in muscular dystrophy. Matrix Biol. 2018;602:68–9.
Morales MG, et al. The pro-fibrotic connective tissue growth factor (CTGF/CCN2) correlates with the number of necrotic-regenerative foci in dystrophic muscle. J Cell Commun Signal. 2018;12(1):413–21.
Morales MG, et al. Reducing CTGF/CCN2 slows down mdx muscle dystrophy and improves cell therapy. Hum Mol Genet. 2013;22(24):4938–51.
Morales MG, et al. CTGF/CCN-2 over-expression can directly induce features of skeletal muscle dystrophy. J Pathol. 2011;225(4):490–501.
Cohn RD, et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med. 2007;13(2):204–10.
Morales MG, et al. Inhibition of the angiotensin-converting enzyme decreases skeletal muscle fibrosis in dystrophic mice by a diminution in the expression and activity of connective tissue growth factor (CTGF/CCN-2). Cell Tissue Res. 2013;353(1):173–87.
Cabello-Verrugio C, et al. Angiotensin II receptor type 1 blockade decreases CTGF/CCN2-mediated damage and fibrosis in normal and dystrophic skeletal muscles. J Cell Mol Med. 2012;16(4):752–64.
Cozzoli A, et al. Enalapril treatment discloses an early role of angiotensin II in inflammation- and oxidative stress-related muscle damage in dystrophic mdx mice. Pharmacol Res. 2011;64(5):482–92.
Morales MG, et al. Angiotensin II-induced pro-fibrotic effects require p38MAPK activity and transforming growth factor beta 1 expression in skeletal muscle cells. Int J Biochem Cell Biol. 2012;44(11):1993–2002.
Cabello-Verrugio C, et al. Fibrotic response induced by angiotensin-II requires NAD(P)H oxidase-induced reactive oxygen species (ROS) in skeletal muscle cells. Biochem Biophys Res Commun. 2011;410(3):665–70.
Sabharwal R, Chapleau MW. Autonomic, locomotor and cardiac abnormalities in a mouse model of muscular dystrophy: targeting the renin-angiotensin system. Exp Physiol. 2014;99(4):627–31.
Willey JS, et al. Angiotensin-(1-7) attenuates skeletal muscle fibrosis and stiffening in a mouse model of extremity sarcoma radiation therapy. J Bone Joint Surg Am. 2016;98(1):48–55.
Sandri M, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117(3):399–412.
von Haehling S, et al. Muscle wasting and cachexia in heart failure: mechanisms and therapies. Nat Rev Cardiol. 2017;14(6):323–41.
Ibebunjo C, et al. Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol Cell Biol. 2013;33(2):194–212.
Meng SJ, Yu LJ. Oxidative stress, molecular inflammation and sarcopenia. Int J Mol Sci. 2010;11(4):1509–26.
Brioche T, et al. Muscle wasting and aging: experimental models, fatty infiltrations, and prevention. Mol Asp Med. 2016;50:56–87.
Dennison EM, Sayer AA, Cooper C. Epidemiology of sarcopenia and insight into possible therapeutic targets. Nat Rev Rheumatol. 2017;13:340.
Brink M, et al. Angiotensin II induces skeletal muscle wasting through enhanced protein degradation and down-regulates autocrine insulin-like growth factor I. Endocrinology. 2001;142(4):1489–96.
Semprun-Prieto LC, et al. Angiotensin II induced catabolic effect and muscle atrophy are redox dependent. Biochem Biophys Res Commun. 2011;409(2):217–21.
Sukhanov S, et al. Angiotensin II, oxidative stress and skeletal muscle wasting. Am J Med Sci. 2011;342(2):143–7.
Delafontaine P, Yoshida T. The renin-angiotensin system and the biology of skeletal muscle: mechanisms of muscle wasting in chronic disease states. Trans Am Clin Climatol Assoc. 2016;127:245–58.
Yoshida T, et al. Molecular mechanisms and signaling pathways of angiotensin II-induced muscle wasting: potential therapeutic targets for cardiac cachexia. Int J Biochem Cell Biol. 2013;45(10):2322–32.
Tabony AM, et al. Angiotensin II upregulates protein phosphatase 2Calpha and inhibits AMP-activated protein kinase signaling and energy balance leading to skeletal muscle wasting. Hypertension. 2011;58(4):643–9.
Tabony AM, et al. Protein phosphatase 2C-alpha knockdown reduces angiotensin II-mediated skeletal muscle wasting via restoration of mitochondrial recycling and function. Skelet Muscle. 2014;4:20.
Cisternas F, et al. Angiotensin-(1-7) decreases skeletal muscle atrophy induced by angiotensin II through a Mas receptor-dependent mechanism. Clin Sci (Lond). 2015;128(5):307–19.
Meneses C, et al. The angiotensin-(1-7)/ Mas axis reduces myonuclear apoptosis during recovery from angiotensin II-induced skeletal muscle atrophy in mice. Pflugers Arch. 2015;467(9):1975–84.
Morales MG, et al. Angiotensin-(1-7) attenuates disuse skeletal muscle atrophy in mice via its receptor, Mas. Dis Model Mech. 2016;9(4):441–9.
Marquez-Miranda V, et al. The complex of PAMAM-OH dendrimer with angiotensin (1-7) prevented the disuse-induced skeletal muscle atrophy in mice. Int J Nanomedicine. 2017;12:1985–99.
Morales MG, et al. Endotoxin-induced skeletal muscle wasting is prevented by angiotensin-(1-7) through a p38 MAPK-dependent mechanism. Clin Sci (Lond). 2015;129(6):461–76.
Abrigo J, et al. Angiotensin-(1-7) prevents skeletal muscle atrophy induced by transforming growth factor type Beta (TGF-beta) via Mas receptor activation. Cell Physiol Biochem. 2016;40(1–2):27–38.
Bentzinger CF, et al. Cellular dynamics in the muscle satellite cell niche. EMBO Rep. 2013;14(12):1062–72.
Burks TN, et al. Losartan restores skeletal muscle remodeling and protects against disuse atrophy in sarcopenia. Sci Transl Med. 2011;3(82):82ra37.
Regoli D, Gobeil F. Kinins and peptide receptors. Biol Chem. 2016;397(4):297–304.
Acuna MJ, et al. Blockade of Bradykinin receptors worsens the dystrophic phenotype of mdx mice: differential effects for B1 and B2 receptors. J Cell Commun Signal. 2018;12(3):589–601.
Gonzalez D, et al. ALS skeletal muscle shows enhanced TGF-beta signaling, fibrosis and induction of fibro/adipogenic progenitor markers. PLoS One. 2017;12(5):e0177649.
Seva Pessoa B, et al. Effect of a stable angiotensin-(1-7) analogue on progenitor cell recruitment and cardiovascular function post myocardial infarction. J Am Heart Assoc, 2015;4(2).
Cunha TM, et al. The nonpeptide ANG-(1-7) mimic AVE 0991 attenuates cardiac remodeling and improves baroreflex sensitivity in renovascular hypertensive rats. Life Sci. 2013;92(4–5):266–75.
Ferreira AJ, et al. The nonpeptide angiotensin-(1-7) receptor Mas agonist AVE-0991 attenuates heart failure induced by myocardial infarction. Am J Physiol Heart Circ Physiol. 2007;292(2):H1113–9.
da Silveira KD, et al. Anti-inflammatory effects of the activation of the angiotensin-(1-7) receptor, MAS, in experimental models of arthritis. J Immunol. 2010;185(9):5569–76.
Pawlik MW, et al. The renin-angiotensin system and its vasoactive metabolite angiotensin-(1-7) in the mechanism of the healing of preexisting gastric ulcers. The involvement of Mas receptors, nitric oxide, prostaglandins and proinflammatory cytokines. J Physiol Pharmacol. 2016;67(1):75–91.
Rodrigues-Machado MG, et al. AVE 0991, a non-peptide mimic of angiotensin-(1-7) effects, attenuates pulmonary remodelling in a model of chronic asthma. Br J Pharmacol. 2013;170(4):835–46.
Giudice J, Taylor JM. Muscle as a paracrine and endocrine organ. Curr Opin Pharmacol. 2017;34:49–55.
Becker LK, Santos RA, Campagnole-Santos MJ. Cardiovascular effects of angiotensin II and angiotensin-(1-7) at the RVLM of trained normotensive rats. Brain Res. 2005;1040(1–2):121–8.
Ren CZ, et al. Exercise training improves the altered renin-angiotensin system in the rostral ventrolateral medulla of hypertensive rats. Oxidative Med Cell Longev. 2016;2016:7413963.
Silva DM, et al. Swimming training improves the vasodilator effect of angiotensin-(1-7) in the aorta of spontaneously hypertensive rat. J Appl Physiol (1985). 2011;111(5):1272–7.
Bernardo BC, et al. Understanding key mechanisms of exercise-induced cardiac protection to mitigate disease: current knowledge and emerging concepts. Physiol Rev. 2018;98(1):419–75.
Fernandes T, et al. Aerobic exercise training-induced left ventricular hypertrophy involves regulatory MicroRNAs, decreased angiotensin-converting enzyme-angiotensin ii, and synergistic regulation of angiotensin-converting enzyme 2-angiotensin (1-7). Hypertension. 2011;58(2):182–9.
Dias-Peixoto MF, et al. The cardiac expression of Mas receptor is responsive to different physiological and pathological stimuli. Peptides. 2012;35(2):196–201.
Filho AG, et al. Selective increase of angiotensin(1-7) and its receptor in hearts of spontaneously hypertensive rats subjected to physical training. Exp Physiol. 2008;93(5):589–98.
Kar S, Gao L, Zucker IH. Exercise training normalizes ACE and ACE2 in the brain of rabbits with pacing-induced heart failure. J Appl Physiol (1985). 2010;108(4):923–32.
Tyrankiewicz U, et al. Activation pattern of ACE2/Ang-(1-7) and ACE/Ang II pathway in course of heart failure assessed by multiparametric MRI in vivo in Tgαq∗44 mice. J Appl Physiol (1985). 2018;124(1):52–65.
Dukes A, et al. The aromatic amino acid tryptophan stimulates skeletal muscle IGF1/p70s6k/mTor signaling in vivo and the expression of myogenic genes in vitro. Nutrition. 2015;31(7–8):1018–24.
Hashimoto T, et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature. 2012;487(7408):477–81.
Gu Q, et al. Contribution of renin-angiotensin system to exercise-induced attenuation of aortic remodeling and improvement of endothelial function in spontaneously hypertensive rats. Cardiovasc Pathol. 2014;23(5):298–305.
Gomes-Santos IL, et al. Effects of exercise training on circulating and skeletal muscle renin-angiotensin system in chronic heart failure rats. PLoS One. 2014;9(5):e98012.
Silva SD, et al. Downregulation of the vascular renin-angiotensin system by aerobic training - focus on the balance between vasoconstrictor and vasodilator axes. Circ J. 2015;79(6):1372–80.
Genest DS, et al. Impact of exercise training on preeclampsia: potential preventive mechanisms. Hypertension. 2012;60(5):1104–9.
Genest DS, et al. Novel role of the renin-angiotensin system in preeclampsia superimposed on chronic hypertension and the effects of exercise in a mouse model. Hypertension. 2013;62(6):1055–61.
Endlich PW, et al. Exercise modulates the aortic renin-angiotensin system independently of estrogen therapy in ovariectomized hypertensive rats. Peptides. 2017;87:41–9.
Somineni HK, Boivin GP, Elased KM. Daily exercise training protects against albuminuria and angiotensin converting enzyme 2 shedding in db/db diabetic mice. J Endocrinol. 2014;221(2):243–59.
Frantz EDC, et al. Exercise training modulates the hepatic renin-angiotensin system in fructose-fed rats. Exp Physiol. 2017;102(9):1208–20.
de Oliveira Sá G, et al. High-intensity interval training has beneficial effects on cardiac remodeling through local renin-angiotensin system modulation in mice fed high-fat or high-fructose diets. Life Sci. 2017;189:8–17.
Guimaraes GG, et al. Exercise induces renin-angiotensin system unbalance and high collagen expression in the heart of Mas-deficient mice. Peptides. 2012;38(1):54–61.
Bertagnolli M, et al. An orally active angiotensin-(1-7) inclusion compound and exercise training produce similar cardiovascular effects in spontaneously hypertensive rats. Peptides. 2013;51C:65–73.
Shah A, et al. Angiotensin-(1-7) attenuates hypertension in exercise-trained renal hypertensive rats. Am J Physiol Heart Circ Physiol. 2012;302(11):H2372–80.
Magalhaes GS, et al. Angiotensin-(1-7) attenuates airway remodelling and hyperresponsiveness in a model of chronic allergic lung inflammation. Br J Pharmacol. 2015;172(9):2330–42.
Becker LK, et al. Eccentric overload muscle damage is attenuated by a novel angiotensin- (1-7) treatment. Int J Sports Med. 2018;39(10):743–8.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Acuña, M.J., Brandan, E., Motta-Santos, D. (2019). Skeletal Muscle System. In: Santos, R. (eds) Angiotensin-(1-7). Springer, Cham. https://doi.org/10.1007/978-3-030-22696-1_11
Download citation
DOI: https://doi.org/10.1007/978-3-030-22696-1_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-22695-4
Online ISBN: 978-3-030-22696-1
eBook Packages: MedicineMedicine (R0)