
Abstract
Oxytosis/ferroptosis is an iron-dependent oxidative form of cell death triggered by lethal accumulation of phospholipid hydroperoxides (PLOOHs) in membranes. Failure of the intricate PLOOH repair system is a principle cause of ferroptotic cell death. Glutathione peroxidase 4 (GPX4) is distinctly vital for converting PLOOHs in membranes to non-toxic alcohols. As such, GPX4 is known as the master regulator of oxytosis/ferroptosis. Ferroptosis has been implicated in a number of disorders such as neurodegenerative diseases (amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD), etc.), ischemia/reperfusion injury, and kidney degeneration. Reduced function of GPX4 is frequently observed in degenerative disorders. In this study, we examine how diminished GPX4 function may be a critical event in triggering oxytosis/ferroptosis to perpetuate or initiate the neurodegenerative diseases and assess the possible therapeutic importance of oxytosis/ferroptosis in neurodegenerative disorders. These discoveries are important for advancing our understanding of neurodegenerative diseases because oxytosis/ferroptosis may provide a new target to slow the course of the disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cell death is an essential process for mammalian growth, homeostasis, and mitigation of hyperproliferative disorders like cancer, and it is intricately linked to other biological processes [1, 2]. Over several decades numerous regulated or accidental cell death mechanisms have been discovered. Among these, apoptosis is regarded as the most regulated cell death pathway in different disease models, and it was assumed that mammalian cells mostly die via activation of caspase-dependent apoptosis [2, 3]. However, new discoveries of non-apoptotic forms of cell death including pyroptosis [4], necroptosis [5], and more recently oxytosis/ferroptosis [6] have perhaps challenged this notion. Oxytosis/ferroptosis is an iron-dependent oxidative cell death hallmarked by reduced cystine intake, glutathione deficiency, and fatal amounts of phospholipid hydroperoxides (PLOOHs) [1, 6]. Formation of PLOOH is a complex process that involves multiple factors and pathways. One of the main sources of PLOOHs is the action of lipoxygenases (LOXs), enzymes that catalyze the oxidation of polyunsaturated fatty acids (PUFA) [1, 6]. These enzymes play a crucial role in the production of PLOOHs and are tightly regulated to prevent excessive accumulation of these harmful molecules [1, 6]. Additionally, reactive oxygen species (ROS), such as hydroxyl radicals and singlet oxygen, can also contribute to the formation of PLOOHs [1, 6]. The presence of cellular iron ions further amplifies the production of PLOOHs, as these ions catalyze the generation of highly reactive oxygen species through the Fenton reaction [1, 6]. This reaction involves the conversion of hydrogen peroxide into hydroxyl radicals, which are potent oxidizing agents and react with PUFA to form PLOOHs [1, 6]. PLOOHs directly react with PUFAs in cell membranes, leading to the production of lipid hydroperoxides [1, 6]. These lipid hydroperoxides can further propagate the oxidative damage and trigger a cascade of events that ultimately result in cell death through oxytosis/ferroptosis [1, 6]. Additionally, PLOOH can also inhibit the activity of key antioxidant enzymes such as glutathione peroxidase 4 (GPX4), further promoting lipid peroxidation and exacerbating ferroptotic cell death [1, 6].
Based on cell death patterning and biochemical hallmarks, this form of cell death is distinctive from apoptosis, necrosis, and autophagy [6]. There is no nuclear condensation; the nuclei of the cell remain intact [7], and no DNA fragmentation; the chromatin is not aggregated [8]. In comparison to physiological conditions, it is morphologically characterized by a reduction in mitochondrial cisternae, disruption of the outer mitochondrial membrane, condensed mitochondrial density, and overall reduced mitochondria size [8]. Similarly, the biochemical features differ in the hemochromatosis, aggregation of lipid peroxidation products, and fatal rise of ROS derived from sources such as iron overload, activation of oxygenases, and inhibition of GPX4 [1]. Oxytosis/ferroptosis has been implicated in many neurodegenerative diseases and neoplastic conditions like Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), ischemic stroke/reperfusion injury, cardiovascular disorders, acute renal failure due to rhabdomyolysis, B cell lymphoma, and hepatic diseases have all been linked to oxytosis/ferroptosis [1, 9,10,11,12]. Oxytosis/ferroptosis could be induced in a number of different ways and in fact, it was discovered while screening novel chemical candidates for targeting oncogenic Ras-expressing human foreskin fibroblast cells [13]. These compounds include erastin and Ras-selective lethal small molecule 3 (RSL 3) [6, 13]. Iron chelators (e.g., deferoxamine mesylate) were shown to suppress this erastin-induced cell death mechanism, which was accompanied by an increase in lipid hydroperoxide concentration [14, 15]. The antioxidant defense enzyme, GPX4, is capable of reducing PLOOHs in membranes directly, and as such, GPX4 is now regarded as the master regulator of oxytosis/ferroptosis [16]. Indeed, the Nomenclature Committee on Cell Death (NCCD) in 2018 described oxytosis/ferroptosis as “a type of regulated cell death triggered by oxidative disturbances of the intracellular milieu that is regulated by GPX4” [16]. Knockout of GPX4 in mice has been documented to cause severe pathological conditions such as neurodegeneration, fetal death [17, 18], and acute renal failure [7]. Because of GPX4’s vital role in oxytosis/ferroptosis, the findings suggest that oxytosis/ferroptosis is a driving force in pathogenesis of disorders such as neurodegenerative diseases.
Herein, we review the importance of oxytosis/ferroptosis and the underlying molecular pathways involved in oxytosis/ferroptosis in neurodegenerative diseases, highlighting the significance of GPX4 as a key player. Of note, GPX4 acts by converting lipid peroxides into harmless lipid alcohols, effectively neutralizing their toxic effects [1, 6]. This pivotal role of GPX4 in maintaining cellular homeostasis and preventing neurodegeneration has sparked immense interest in exploring its therapeutic potential [1, 6]. Accordingly, enhancing the GPX4 activity or developing drugs that target its regulation, it may be possible to halt the progression of these devastating diseases. Importantly, recent research has linked GPX4 deficiency to increased sensitivity to oxidative stress, dysregulated cellular processes, and neurodegeneration [1, 6]. Therefore, investigating how GPX4 interacts with other cellular processes such as autophagy and apoptosis may provide new treatment options for these debilitating diseases. Thus, understanding the precise mechanisms by which GPX4 functions could lead to the development of novel therapeutic strategies for treating these devastating diseases.
Oxytosis/Ferroptosis Regulatory Mechanisms
Oxytosis/ferroptosis is implicated in diseases, like cancer and neurodegeneration [1, 6]. Oxytosis/ferroptosis is primarily caused by iron-catalyzed lipid peroxidation, initiated by both non-enzyme (Fenton reaction) and enzymatic mechanisms [1, 6]. The buildup of lipid hydroperoxides, primarily phosphatidylethanolamine-OOH (PE-OOH), ultimately leads to ferroptosis, with iron acting as a catalyst or vital mediator of ferroptosis [1, 6]. PUFAs are the primary targets of membrane lipid breakdown during ferroptosis [1, 6]. ROS generation from different sources, particularly iron metabolism, the NADPH oxidase (NOX) isoforms, and mitochondrial electron transport chain, is thought to be the initial and critical signal of oxytosis/ferroptosis induction [1, 6]. The main pathways that regulate oxytosis/ferroptosis are briefly outlined below.
Fenton Reaction
The mechanism of the Fenton reaction involves the generation of highly reactive hydroxyl radicals (•OH) through the interaction between hydrogen peroxide (H2O2) and ferrous ions (Fe2+) [19]. This reaction is highly dependent on the pH of the solution, with acidic conditions being more favorable for the production of hydroxyl radicals [19]. In the presence of Fe2+, H2O2 is first oxidized to hydroperoxyl radicals (HO2•) through a homolytic cleavage of the O-O bond [19]. This reaction proceeds through a series of steps, starting with the formation of an iron-hydroperoxo-complex, followed by its decomposition to produce hydroxyl radicals [19]. These hydroxyl radicals are known for their strong oxidizing properties, resulting in the oxidation of lipid molecules and other cellular components [1, 6]. These lipid hydroperoxides can further propagate the oxidative damage and trigger a cascade of events that ultimately result in cell death through ferroptosis [1, 6]. Furthermore, this oxidative damage can also lead to cell membrane disruption, protein denaturation, DNA strand breaks, and cell death [1, 6]. Therefore, understanding the mechanisms of iron mediated hydroxyl radical production and its effects on cellular components is crucial in developing strategies to mitigate oxidative damage and protect cellular integrity in oxytosis/ferroptosis.
Lipid Peroxidation and Enzymatic Mechanisms
Lipid peroxidation refers to the oxidative degradation of lipids, resulting in the formation of ROS and lipid peroxidation products (LPPs) [1]. This process plays a crucial role in various physiological and pathological conditions, including aging, inflammation, and neurodegeneration [1, 6]. The enzyme acyl-CoA synthetase long-chain family member 4 (ACSL4) is required for the incorporation of PUFA into phospholipids, an important event of ferroptosis [1, 6, 20]. ACSL4 has been identified as a significant predictor of ferroptosis sensitivity in a number of studies [1, 6, 20,21,22,23]. In order to promote the esterification of PUFA to phospholipids, ACSL4 catalyzes the addition of CoA to the long-chain polyunsaturated bonds of arachidonic acid [1, 6, 20]. Following the activation of ACSL4, lysophosphatidylcholine acyltransferase 3 (LPCAT3) contributes to ferroptotic lipid signaling by adding acyl groups to lysophospholipids, particularly to the phospholipids phosphatidylcholine and phosphatidylethanolamine (PE) [1, 6, 20, 21]. Additionally, ferroptosis can occur independently of ACSL4 [1, 6, 20]. However, if other ACSL family members participate in ACSL4-independent ferroptosis, is still unexplored.
Furthermore, the enzyme lipoxygenase (LOX), which catalyzes the oxidation of PUFA, is implicated in lipid peroxidation and contributes to the generation of lethal lipid peroxidation products [1, 6]. LOXs are non-heme, iron-containing dioxygenases that have six subtypes in humans [1, 6]. These subtypes include ALOX5 (arachidonate 5-lipoxygenase), ALOX12 (arachidonate 12-lipoxygenase, 12S type), ALOX12B (arachidonate 12-lipoxygenase, 12 R type), ALOX15 (arachidonate 15-lipoxygenase), ALOX15B (arachidonate 15-lipoxygenase type B), and ALOXE3 (arachidonate lipoxygenase 3) [24]. LOXs oxidizes arachidonic acid (AA) and adrenoyl-(AdA-)-phosphatidylethanolamines (PE) [25]. This process is further controlled by PEBP1/RKIP (phosphatidylethanolamine binding protein 1) as it interacts with ALOX15 and potentiates its catalytic efficacy for PUFA-PE, thereby promoting ferroptosis [26]. Furthermore, studies have also suggested the involvement of ALOX15 and ALOX5 in the regulation of the proinflammatory, anti-inflammatory, and ferroptosis pathways [24, 27]. Moreover, ALOX12 inhibition prevented glutamate-induced cell death in the primary cortical neurons and hippocampal HT22 cells [28]. Thus, LOXs play a crucial role in the regulation of cellular redox balance, as they convert PUFA into reactive lipid species [1, 6]. This conversion process not only leads to the generation of lipid peroxides but also triggers a cascade of events that ultimately culminate in cell death through oxytosis/ferroptosis.
Another important enzyme system involved in lipid peroxidation is cytochrome P450 oxidoreductase (POR) [1, 6]. POR is an endoplasmic membrane bound enzyme that associates with two cofactors (flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN)) to directly deliver electrons to P450 enzymes from NADPH thereby facilitate the peroxidation of PUFA [29, 30]. Several studies have suggested that POR initiate lipid peroxidation by converting ferric iron (Fe3+) to ferrous iron (Fe2+), a step essential for the Fenton reaction in ferroptosis mediated cell death [25, 31, 32]. For instance, inhibition of POR activity by galangin and isosylbin mitigated microglia ferroptosis-mediated neurodegeneration in PD [33]. In addition to reducing PUFA-induced ferroptosis susceptibility, constitutive or inducible POR deletion reduced intrinsic ferroptosis sensitivity in ccRCC cells 786-O and 769-P [34]. In summary, the involvement of lipid peroxidation and its enzymatic regulation offers novel insights into the intricate biology of oxidative damage and ferroptotic cell death, opening the door for future developments in the prevention and treatment of diseases associated with oxytosis/ferroptosis.
The System Xc –-GSH-GPX4 Axis
System Xc– is made up of a regulatory subunit (SLC3A2) and a catalytic component (SLC7A11) that facilitates the interchange of extracellular cystine and intracellular glutamate across the plasma membrane [1, 6, 30]. SLC7A11 is a key functional component that transports cystine into cells for GSH production to maintain the anti-oxidant pool of the cell [1, 6, 30]. Furthermore, the disruption of the cystine/glutamate transporter results in the depletion of GSH, rendering the GPX4 inactive, which leads to the build-up of lipid peroxide and ferroptosis [1, 6, 30]. Moreover, SLC7A11 deletion resulted in tumor-selective ferroptosis and prevented the pathogenesis of pancreatic ductal adenocarcinoma (PDAC) [35]. Of note, the fibroblasts isolated from SLC7A11 knockout animals undergo ferroptotic cell death, which could be reversed by the addition of anti-oxidants/ROS scavengers like N-acetylcysteine (NAC), β-mercaptoethanol (β-ME), or vitamin E [25, 30]. These observations corroborate the indispensable role of system Xc–-GSH-GPX4 axis in ferroptosis (Fig. 1). The detailed involvement of this pathway, with the emphasis on the GPX4 as a master regulator of oxytosis/ferroptosis, will be discussed in the later sections.

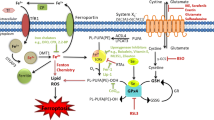
Schematic illustration of the oxytosis/ferroptosis regulatory mechanisms. Oxytosis/ferroptosis starts with the binding of Fe3+-loaded transferrin with transferrin receptor leading to endosomal uptake and release of Fe3+ from Tf–TfR1 complex into the cytosol. Endosomal metalloprotease, STEAP3 reduces Fe3+ to Fe2+ which can be transported and stored in ferritin by DMT1 or remain in the cytoplasm as a labile iron (Fe2+) pool. Increased Fe2+ induces the production of ROS using both enzymatic (lipoxygenase and P450 oxidoreductase) and non-enzymatic (fenton reaction) pathways, ultimately leading to ferroptosis through ROS-mediated lipid peroxidation of membranal PUFAs. Ferroptosis is alleviated by antioxidant systems such as glutathione peroxidase 4 (GPX4), CoQ10, and NRF2 signaling. Cystine is required for the synthesis of GSH, which is transported into the cell by Xc− system for an exchange of glutamate. Levels of glutamate in cell are maintained by SLC1A5 which imports glutamine, a precursor of various amino acids. GSH activates GPX4, and activated GPX4 prevents lipid peroxidation and reduces PUFA-OOH to PUFA-OH. AIFM2/FSP1-mediated CoQ10 synthesis prevents ferroptosis by inhibiting lipid peroxidation. NRF2 signaling is involved in the synthesis of GSH, SCL7A11 directly inhibits ferroptosis, and HO-1 inhibits ferroptosis by limiting ROS production. Tf, transferrin; STEAP3, six-transmembrane epithelial antigen of the prostate 3; DMT1, divalent metal transporter 1; GSH, glutathione; GLS, glutaminases; GPX4, glutathione peroxidase 4; CoQ10, ubiquinone; AIFM2/FSP1, apoptosis-inducing factor mitochondria-associated 2/Ferroptosis suppressor protein 1; HO-1, heme oxygenase-1; NRF2, nuclear factor erythroid 2–related factor 2. Figure created with Biorender
GPX4 Independent Pathways
Apoptosis-inducing factor mitochondria-associated 2 (AIFM2, also known as ferroptosis suppressor protein 1 (FSP1))-mediated coenzyme Q10 (CoQ10) synthesis has recently been identified as an endogenous antioxidant mechanism, independent of GPX4 regulation, to modulate oxytosis/ferroptosis [1, 6, 36, 37] (Fig. 1). CoQ10, also known as panquinone, is a naturally occurring endogenously produced isoprenoid benzoquinone molecule [1, 6, 36, 37]. CoQ10 is an essential component of the mitochondrial electron transport chain, which can prevent lipid peroxidation by trapping free radical intermediates [36, 37]. By using genetic screening assays, it was determined that independent of GPX4 or GSH, AIFM2 by replenishing reduced CoQ10 levels, prevented ferroptosis [36, 37]. Similarly, independent of GPX4 or GSH regulation, tetrahydrobiopterin/dihydrobiopterin (BH4/BH2), a biochemical byproduct of enzyme GTP hydrolase 1, by regulating CoQ10 synthesis inhibits ferroptosis [25, 30].
The NRF2-HO-1 Signaling Pathway
Nuclear factor erythroid 2–related factor 2 (NRF2) is a transcription factor that, when phosphorylated, enters the nucleus and transactivates both NRF2 and its target gene, such as heme oxygenase-1 (HO-1) [30, 38]. HO-1 has both stress-reducing and anti-inflammatory properties [30, 38, 39]. The potent ROS scavenger biliverdin and its reduced counterpart bilirubin are produced as a result of heme breakdown by HO-1, which also inhibits ferroptosis and reduces the production of peroxides, peroxynitrites, hydroxyl radicals, and superoxide radicals [30, 38, 39]. Furthermore, NRF2 also regulates iron homeostasis by modulating ferroportin (an iron transporter), and the light and heavy chains of ferritin (crucial for iron accumulation) [30, 40]. In addition to iron homeostasis, NRF2 also modulates the GSH antioxidant system by regulating the production of GSH synthesis-related enzymes such as glutathione synthase (GSS), glutamate cysteine ligase (GCLC/GCLM), and SLC7A11 [30, 41]. Thus, NRF2/HO-1 axis is the primary signal mechanism controlling oxytosis/ferroptosis, through the regulation of iron metabolism, GSH synthesis, and mitigation of oxidative stress [30, 38,39,40] (Fig. 1).
Glutathione Peroxidase 4 (GPX4)
Glutathione peroxidases (GPXs) are evolutionarily conserved enzymes that convert peroxides (e.g., R–OOH) to their respective alcohols using glutathione (GSH) as reducing agent, thereby preventing the generation of harmful radicals (e.g., R–O•) [42]. GPX4 is a selenocysteine (Sec)-containing glutathione peroxidase that works as a phospholipid hydroperoxidase to reduce lipid peroxides to lipid alcohols [43]. In humans, GPX4 is a single-copy gene on chromosome 19 [44] that is transcriptionally controlled by stimulating proteins 1 and 3 (SP1/3) and nuclear factor Y (NF-Y) [45]. Mitochondrial (mGPX4), Cytosolic (cGPX4), and sperm nuclear GPX4 are its three distinct isoforms (snGPX4). Among these, cGPX4 is ubiquitous in cells, and snGPX4 and mGPX4 are primarily expressed in the testis [46,47,48]. The reduction of complex organic hydroperoxides is catalyzed by GPX4 by oxidizing the active site selenol (Se–H) to selenenic acid (Se–OH), which is subsequently reduced back to the active selenol utilizing GSH [49].
GPX4 has high affinity for ROOH substrates and takes either simple ROOHs like H2O2 or even more complicated ones like phosphatidylcholine hydroperoxide (PCOOH) or cholesterol and cholesterol ester hydroperoxides [50]. Furthermore, when GSH levels are low, GPX4 has been shown to accept structurally unrelated low-molecular-weight thiols and specific protein thiols [51]. GPX4-mediated protein thiol oxidation has been identified as a crucial redox event in sperm maturation [51], and it could also be linked to other redox events in which ROOH flux surpasses the reducing capability capped by GSH concentration [52]. The appearance of a seleninic acid (Se–OO—) in the active site of enzyme was demonstrated by crystal lattice of seleno–GPX4 [8] and depending on the degree of oxidative stress experienced by the cell, this may induce GPX4 to undergo mild oxidation cycles of selenol to selenenic acid and strong oxidation cycles of selenenic acid to seleninic acid [53]. GPX4 plays necessary functions during embryonic development, inflammatory processes, male fertility, and cell survival [54, 55]. In fact, mice lacking GPX4 died shortly after gastrulation (E7.5) [56, 57]. Interestingly, mice lacking the enzyme’s catalytic component phenotypically resembled GPX4 knockout animals [58], indicative of the central role of enzyme activity for its overall physiological function. We have found that adult mice with conditional GPX4 deletion exclusively in forebrain neurons displayed significantly impaired spatial learning and memory functioning when compared to age matched control mice [18]. Furthermore, these mice revealed hippocampal neurodegeneration and ferroptosis associated signs like increased lipid peroxidation levels, ERK activation, and enhanced neuroinflammation [18]. More recently, we found that overexpression of GPX4 in ALS model (SOD1G93A) of neurodegeneration was able to ameliorate spinal motor neuron degeneration and reduce lipid peroxidation levels [12]. Moreover, in another study, we found that GPX4 overexpression in 5xFAD mice was able to attenuate ferroptosis markers like increased lipid peroxidation, elevated lyso-phospholipids, and ameliorate cognitive impairment and neurodegeneration of 5xFAD mice [59]. GPX4 plays an indispensable role in maintaining lipid homeostasis within the cell by attenuating the toxic lipid ROS accumulation which eventually prevents the onset of ferroptosis [6] (Figs. 1 and 2).

The oxytosis/ferroptosis pathway and the arsenal of therapeutic candidates targeting proteins/signaling molecules to mitigate ferroptotic cell death. Of these proteins/signaling molecules, GPX4, lipid peroxidation, and system xc are the most important therapeutic targets in the context of drug discovery research for treating oxytosis/ferroptosis (see “Oxytosis/Ferroptosis Inhibiting Strategies” section for additional details)
Oxytosis/Ferroptosis and GPX4
Oxytosis/ferroptosis is an iron-dependent oxidative form of cell death characterized by impaired cystine uptake, GSH depletion, iron overload, and accumulation of PLOOH to lethal levels [6]. Although ferroptosis was coined in (2012) by Stockwell’s group [6], but its basis was well-known long before when Murphy’s group reported that glutamate is able to induce neuronal cell death by inhibiting system xc –/GSH/GPX4 [60], which was later named “oxytosis” by Maher’s group in 2001 [61]. Recently, it has been found that oxytosis and ferroptosis had similar pathological hallmarks like ROS production and activation of LOXs [62]. The accumulation of particular PLOOH, increased ferrous ion (Fe2+) concentration, and excessive lipid ROS are the most remarkable features of ferroptosis [7, 63]. This cascade is endogenously neutralized by the system Xc−/GSH/GPX4 axis [7, 63]. The most upstream component of this axis is system Xc− [64], where an extremely precise uptake mechanism for cystine and cystathionine takes place with exchange of glutamate at 1:1 M ratio [65, 66]. As cysteine is the rate-limiting substrate in GSH biosynthesis and GSH itself being predominantly an antioxidant in mammalian cells, conditions that hamper on intracellular cysteine and subsequently GSH levels have shown to impact directly GPX4 activity and thus influence ferroptotic cascade [67] (Fig. 1). Pharmacological inhibition of system xc- with erastin or GCL inhibitor butylthioninesulfoximine have reported depleted GSH levels which in-turn triggers inactivation of GPX4 and increase in lipid ROS [63]. GPX4 has an active site consisting of selenocysteine, which forms a catalytic quadruplex with tryptophan, glutamine, and asparagine [49]. Even though this quadruplex is conserved across GPXs family, GPX4 is the only member to act as a gatekeeper of ferroptosis [49]. The major structural difference is that GPX4 is a 20–22 kDa monomer protein, whereas the other GPXs are tetramers, and having a flexible site allowing it to use PLOOH as substrates [49]. GPX4 catalyzes the reduction of lipid hydroperoxides involving a ping-pong mechanism, in which selenocysteine shuttles between reduced selenol (SeH) and oxidized selenic acid (SeOH) states [49]. Genetic mutation of selenocysteine in the active site of GPX4 to serine (U46S) or alanine (U46A) has revealed embryonic lethality, implying the importance of its catalytic activity for normal embryonic development [43, 68, 69]. The application of oxidizing substrates demonstrates GPX4’s versatility in relation to a variety of reducing substrates. GPX4 may easily utilize additional low molecular thiols and even protein thiols in complement to its major cofactor GSH [70]. In comparison to the glutathione peroxidase family enzymes, GPX3, GPX7, and Peroxiredoxin 6 have shown to reduce esterified lipid hydroperoxide in vitro but none of them could signify the antioxidant potential of GPX4 in reducing PLOOH in membranes and inhibition of microsomal lipid peroxidation [70]. The inhibition or genetic deletion of GPX4 has indicated that GPX4 requires particular oxidation of PE comprising PUFAs like arachidonic and adrenic acid to prevent cell death and the inhibition of GPX4 during the ferroptotic cascade is triggered by the particular oxidation of arachidonoyl-PE, a mechanism which has been provisionally linked to non-heme iron containing LOXs that insert stereo-specific molecular oxygen in free or esterified PUFAs [21, 22] (Figs. 1 and 2). PE oxidation has been specifically associated with mitochondrial damage during GPX4 inactivation or related ferroptotic signaling [71] and the role of LOX in the ferroptosis process is still inadequate and has been recently questioned. We have recently found that 5×FAD neural stem cells (NSCs) had reduced level of GPX4, and this reduction appeared to be caused by enhanced autophagy-lysosomal degradation of GPX4 protein [72]. Reduced GPX4 levels led to functional deficits like decreased neuronal differentiation in these NSC’s, whereas increased GPX4 levels were able restore differentiation potential and increase the number of neuronal populations upon differentiation [72].
Oxytosis/Ferroptosis Modulators
With the discovery and recognition of ferroptosis as a vital process in the pathogenesis of neurodegenerative diseases, therapeutic targeting of this pathway presented an opportunity and an alternative way to cure these incurable diseases. From the point of drug discovery and involvement of oxytosis/ferroptosis in several other diseases, ferroptosis is being modulated in two ways: use of ferroptosis inhibitors to prevent neurodegeneration and tissue atrophy in degenerative diseases, or ferroptosis inducers to kill cancerous cells [3, 5]. The comprehensive overview of these ferroptosis modulators has been extensively described previously [1, 9, 15, 62, 73, 74]. Since the central theme of this article is degenerative diseases which are associated with ferroptotic cell death, therefore, we will focus on oxytosis/ferroptosis inhibiting strategies as a therapeutic avenue for treating these debilitating diseases.
Oxytosis/Ferroptosis Inhibiting Strategies
Oxytosis/ferroptosis inhibition by preventing cellular loss and tissue damage presents an innovative and alternate approach in combating neurodegenerative diseases. The drug discovery horizon for oxytosis/ferroptosis inhibition encompasses the small molecule inhibitors (Ferrostatin-1), natural products or molecules (vitamin E), and genetic approaches (GPX4 overexpression) (Fig. 2) [1, 73]. The first documented small molecule ferroptosis inhibitor, ferrostatin-1, prevented erastin-induced cell death [73, 75]. Liproxstatin-1 was found to be the first in vivo effective ferroptosis inhibitor through a phenotypic screening effort in cells experiencing Gpx4 knockout-induced ferroptosis [73, 75]. Liproxstatin-1 demonstrated low nanomolar IC50 values in vitro, markedly increased longevity in a genetic model of abrupt renal failure, and reduced the severity of hepatic ischemia/reperfusion damage in rodents [7, 76]. Furthermore, the first reported receptor interacting protein kinase 1 inhibitor, Necrostatin-1, showed beneficial outcome due to inhibition of ferroptosis as opposed to necroptosis inhibition as had been previously reported [1, 73]. Recently, the mechanism of action of lipoxstatin has been described, indicating that these molecules act as superior antioxidant traps by breaking the autoxidation chain reaction [1, 73]. Liproxstatins can capture several peroxyl radicals per molecule and are produced during the reduction of fatty hydroperoxides [1, 73]. LOX inhibitors like zileuton, baicalein, LOXBlock-1-3, MK886, PD146176, and BWA4C have demonstrated anti-ferroptotic effects; however, caution must be exercised when using these inhibitors as the majority of them are non-specific or even exhibit broad antioxidant effects [1, 73].
The identification of ACSL4 as a significant oxytosis/ferroptosis downstream component and its draggability with thiazolidinediones (TZD) provides an important avenue for targeting the ferroptosis pathway [1, 73]. The lipidomic profile produced by cells treated with the TZD, rosiglitazone, was identical to that of ACSL4 knockout cells, and TZDs prevented the buildup of lipid hydroperoxides and ferroptosis triggered by GPX4 inhibition/genetic disruption [1, 73]. Besides these, compelling antiferroptotic candidates, such as phenoxazines, and an array of nitroxide-based compounds (mito-targeted or untargeted), such as XJB-5-131, JP4-039, 2,2,6,6-tetramethylpiperidin-N-oxyl (TEMPO), N-arylnitroxide, N, N-diarylnitroxide, and phenoxazine-N-oxyl, have been reported [1, 73, 77]. A distinct strategy using computation- and structure-based techniques located an allosteric site in GPX4 that made it possible to find the first GPX4 allosteric activators [1, 73, 77]. These molecules in addition to GPX4 activation suppress NFkB activation and prevent ferroptosis induced by erastin and cholesterol hydroperoxides [1, 73, 77]. The molecular mechanisms and therapeutic prospects of ferroptosis as a druggable pathway have been highlighted by the discovery of an ever-expanding number of agonists and antagonists of ferroptosis as an alternate therapeutic target for degenerative diseases (Fig. 2).
Antioxidants and Oxytosis/Ferroptosis
Since oxytosis/ferroptosis is characterized by the accumulation of lipid peroxides and iron-dependent ROS [1, 6]. By scavenging these ROS and limiting lipid peroxidation, antioxidants play a vital role in reducing oxytosis/ferroptosis. Antioxidants can also help restore cellular redox balance and protect against oxidative stress, which further contributes to the prevention of ferroptosis [1, 6]. Furthermore, the oxidative stress is intricately associated with ferroptotic cell death in neurodegenerative disorders, and the use of antioxidant molecules has gained tremendous attention as an effective therapeutic strategy to mitigate these debilitating diseases [1, 6]. For instance, in Friedreich’s ataxia, a metal-induced neurodegenerative disease, the nuclear translocation of the NRF2 signaling pathway is disrupted by ferroptosis, followed by the reduced expression of genes involved in antioxidant defense [78, 79]. Dimethyl fumarate, an FDA-approved drug for treating multiple sclerosis, due to its strong antioxidant properties, is also being tested and is under clinical trials for the treatment of Friedrich’s ataxia [80]. Similarly, dimethyl fumarate administration after TBI boosted NRF2 activation and exhibited neuroprotection against TBI-induced ferroptosis [81]. Furthermore, antioxidant, Insamgobonhwan (GBH), mitigated GPX4 suppression and enhanced ROS scavenging activity and suppressed lipid-peroxidation in ferroptosis-mediated neurodegeneration in both in vitro and in vivo conditions [82]. Similarly, NAC, a thiol-containing redox modulatory drug, has been shown to reduce hemin-induced ferroptosis by removing toxic lipids generated by arachidonate-dependent ALOX5 activity in hemorrhagic stroke [83]. Additionally, NAC serves as a cysteine donor for reduced GSH biosynthesis, boosting cellular antioxidant defenses, thereby lowering lipid peroxidation and subsequently suppressing ferroptosis [84]. Similarly, ROS inhibitors such as apocynin and diphenyleneiodonium mitigated NOX stimulated ferroptosis by decreasing MDA concentration and enhancing the expression of GPX4 levels (Fig. 2) [85].
Another strategy to mitigate ferroptosis linked to lipid peroxide built up involves the use of antioxidant molecules that have the ability to trap radical oxidants [86]. A recently developed chemical molecule, DT-PTZ-C, has demonstrated promising outcomes in protecting brain tissue against oxidative stress [86]. The N10-carbonyl-substituted phenothiazine family member DT-PTZ-C reduces ferroptosis in the hippocampus slice culture toxicity model caused by thermolabile azo initiator ABAP, by scavenging hydroxyl radicals and restricting lipid peroxidation via generating a stable phenothiazine radical cation [86]. Plethora of small-molecule radical-trapping agents (RTAs) such as Fer-1, liproxstatin-1, and α-tocopherol inhibit ferroptosis by halting the lipid peroxidation process [73]. Other compounds such as Zileuton, CoQ10, deferiprone, DFO, BH4, and FSP1 are effective by working through pathways, such as reversion of ROS production, GPX4 induction, and production of GSH, and have been identified as potent inhibitor of ferroptosis in several neurodegenerative studies [84]. Furthermore, radical-sequestering antioxidant, vitamin A, modulates the expression of ferroptosis regulators, such as GPX4 and ACSL3, via heterodimeric nuclear receptor complex retinoic acid receptor (RAR)/retinoid X receptor (RXR), thereby suppressing ferroptosis [87]. Additionally, cycloheximide and β-mercaptoethanol have been observed to ameliorate ferroptosis by primarily targeting system xc− to increase GSH levels for the efficient removal of lipid peroxides [1, 88]. The iron chelator and ferroptosis inhibitor DFO, inhibited ferroptosis in primary cortical neurons treated with erastin, involved the upregulation of both GPX4 and system Xc-, the antioxidant defense pathways and primary ferroptotic regulators [6, 89]. FSP1, a newly discovered ferroptotic antagonist, exerts its anti-ferroptosis and antioxidant effects by reducing ubiquitin (CoQ) to antioxidative dihydroubiquitin (CoQH2) which quinches ROS [37, 90]. Melatonin (MT), an agonist of MT2 receptor, provides anti-inflammatory, antioxidant, and anti-ferroptosis effects post-TBI by activating MT2/IL-33/FTH pathway, thereby alleviate edema and neurological deficits [91]. Exogenous melatonin was reported to suppress hemin or erastin-induced ferroptosis in various cells [92].
Tetrahydrobiopterin (BH4), a naturally occurring nutrient, acts as free radical scavenger and displays antioxidant effects through GTP cyclohydrolase-1 (GCH1) and dihydrofolate reductase (DHFR) enzymes [93]. GCH1-induced synthesis of BH4 selectively inhibits PUFA-PL depletion-mediated membrane lipid remodeling [94]. DHFR is important for the regeneration of BH4, and its inhibition may synergize with GPX4 inhibitors to induce ferroptosis [95]. BH4 has also been reported to quench ROS by inducing the synthesis of CoQ10 [93]. Baicalin, a flavonoid, inhibits ferroptosis by increasing the expression of SLC7A11 and GPX4 in hemin-treated PC12 cells, thereby alleviated hematoma, motor dysfunctions, and neurodegeneration in the peri-hematoma brain tissues [96]. Baicalin also reduced the expression of SLC11A2 to inhibit iron transport in the peri-hematoma brain tissue [96]. Additionally, Baicalein also acts as a selective ALOX12/15 inhibitor and prevented lipid peroxidation and ROS production, and improved traumatic brain injury [97]. Likewise, Sterubin, a flavanone, functions as an iron chelator and protected against ferroptosis in neuronal cells by modulating GSH levels and activating NRF2/ATF4 signaling [98, 99]. Moreover, tetrahydroxy stilbene glycoside (TSG), a traditional Chinese medicine, provided protection against ferroptosis and oxidative stress against Aβ-induced hippocampal neuron injury in APP/PS1 mice by activating Keap1/NRF2/ARE and GSH/GPX4/ROS signaling pathways [100]. Morachalcone D and morachalcone E are the two prenylated flavonoids and provide protection against erastin-induced ferroptosis in mouse hippocampal neuronal cell line (HT-22) [101]. Mechanistically, morachalcone D decreased ROS production, iron accumulation, and GSH depletion, as well as increased the expression of many antioxidant genes, such as GPX4, SOD, CAT, NRF2, SLC7A11, and HMOX1, leading to the inhibition of ferroptosis and neuroprotection [102, 103]. Artepillin C, a cinnamic acid, has been reported to alleviate neurotoxicity by decreasing ROS generation and elevating HO-1 expression, which in turn prevented ferroptosis and protected against erastin-mediated cell damage [104]. Additionally, the hybrid compound 7-O-cinnamoyltaxifolin, a derivative of cinnamic acid, shown synergistic neuroprotective benefits against ferroptosis by activating NRF2 in both in vitro and in vivo studies (Fig. 2) [105, 106].
GPX4 Modulators
Following the identification of erastin, a small-molecule inducer of oxytosis/ferroptosis, RSL3 was discovered, which operated separately from system XC, through GPX4 inhibition, a master regulator of ferroptosis [15, 62]. Because of its reactive chloroacetamide moiety, RSL3 is a strong and permanent inhibitor of GPX4 [9]. However, because of its poor solubility and unfavorable ADME (absorption, distribution, metabolism, and elimination) characteristics, it is not appropriate for application in vivo [9]. Contrarily, to investigate the function of GPX4 and lipid peroxidation in a variety of biological processes, in particular ferroptosis, RSL3 has been indispensable tool in the in vitro setup [1]. Thus, the development of better RSL3 compounds for use in vivo has been the main emphasis of drug discovery pipeline targeting GPX4. Subsequently, more GPX4 inhibitors containing chloroacetamide have been discovered, such as DPI12, DPI13, DPI15, DPI6, DPI7/ML162, DPI8, and DPI19 [62]. These compounds have all been demonstrated to induce ferroptosis by C11-BODIPY oxidation, butylated hydroxytoluene (BHT)-sensitive cell killing, and buthionine sulfoximine-enhanced cell killing in BJ-derived cell lines [62]. In BJeLR cell lysates, DPI12, DPI13, DPI7/ML162, and DPI19 have been shown to suppress GPX4 activity [62]. Although these candidates are not as thoroughly characterized as RSL3, however, the best-characterized of these chloroacetamide GPX4 inhibitors, DPI7/ML162, can be used to confirm RSL3’s GPX4 dependency [73]. Apart from the chloroacetamide moiety, RSL3 and ML162 have very distinct structures and are therefore likely to have different off-target effects [73].
There have been reports of three new structural groups of GPX4 antagonists. First, the chloromethyltriazines DPI17, DPI18 [62], and altretamine [107] function as covalent GPX4 inhibitors. Similar in structure to the chloroacetamide compounds, DPI17 and DPI18 have been demonstrated to display ferroptotic-like cell-killing actions [73]. In BJeLR cell lysates, DPI17 has been demonstrated to suppress GPX4 activity [62, 73]. Therefore, it is probable that DPI17 and DPI18 are covalent GPX4 inhibitors [62]. Second, the related candidates JKE-1674 and ML210 [108] contain a nitroisoxazole moiety that has been shown to produce a nitrile oxide electrophile that interacts with GPX4 in a cellular setting [109]. Additionally, GPX4 is inhibited by nitrile oxide electrophiles produced by similar substances, such as JKE-1674 and JKE-1716, the furoxan-containing NSC144988, and related diacylfuroxans [110]. Lastly, it has been observed that the natural substance withaferin A, a steroidal lactone and epoxide, inhibits GPX4 by means of its electrophilic groups (Fig. 2) [111].
Oxytosis/Ferroptosis, GPX4, and Amyotrophic Lateral Sclerosis (ALS)
ALS is a neurodegenerative disorder affecting motor neurons and results in weakness, atrophy of voluntary skeletal muscles, paralysis, and eventually patients die due to respiratory failure [112,113,114]. Majority of ALS cases are sporadic with indefinite etiology and around 15% of cases present familial cause with dominant inheritance [115, 116]. Both of these patients share several pathogenetic pathways and are clinically indistinguishable [117]. Around 50% of ALS patients have cognitive impairments, with a subgroup of patients (~15%) displaying hallmarks of frontotemporal dementia (FTD), frontotemporal lobar degeneration (FTLD), and progressive social, behavioral, or language dysfunction [118,119,120]. Genetic factors consisting of mutations in the C9orf72, TARDBP (Transactive Response DNA-Binding Protein 43), SOD1 (Superoxide Dismutase-1), and FUS (Fused in Sarcoma) genes may account for up to 50% of familial cases [121]. Several pathophysiological mechanisms have been reported that alter the microenvironment of motor neurons which include perturbed RNA processing, protein aggregation, mitochondrial dysfunctions, glutamate excitotoxicity, Golgi dysfunctions, impaired neuronal transport and glial functioning, oxidative stress, and metal imbalance [122,123,124]. At present, there are no effective treatments for this debilitating disease and the development of effective therapies is impeded by the lack of suitable targets, particularly for sporadic ALS, which do not have identifiable genetic causes. Oxidative stress characterized by increased ROS levels has been identified as one of the contributing factors in ALS and iron accumulation via Fenton reaction has been established as a key factor related with the production and presence of this ROS, producing highly reactive hydroxyl radicals [125] (Fig. 3). Ferroptosis being an iron dependent oxidative form of cell death has been found to be a contributing factor in various neurodegenerative diseases as evidenced from rodent models where affected neurons revealed an accumulation of iron and reduced GSH levels [126]. Studies have found iron accumulation in spinal cord motor neurons in SOD1 mutant-mediated mice model of ALS [127]. Recent studies on the human-induced pluripotent stem cell (hiPSC)-derived motor neuron (hiMNs), a well-known model of numerous motor neuron disorders, reported that motor neurons are vulnerable to ferroptosis with almost all hiMNs dying after treating with GPX4 inhibitors and interestingly, lipid peroxidase inhibitors, iron chelators, and vitamin E acetate all reduced cell death, thereby implying a role for ferroptosis [127,128,129]. A longitudinal study from 512 patients aimed at deciphering the prognostic biomarkers for ALS has identified four biomarkers and intriguingly all of them are closely associated with ferroptosis [130]. A study using edaravone (clinically approved drug for ALS) has shown to attenuate ferroptosis in cystine-deprived mouse hepatoma Hepa 1-6 cells and further suppressed ferroptosis in xCT-knockout mouse–derived embryonic fibroblasts [131]. Interestingly, this attenuation was not because of change in intracellular levels of Cys and GSH levels rather edaravone suppressed ferrous iron and lipid peroxide levels that were significantly high due to initial cystine and GSH deprivation [131]. Another clinical trial candidate, Diacetyl-bis(4-methyl-3-thiosemicarbazonato) copperII (CuII(atsm)), has shown to protect primary and immortalized neuronal cells against lipid peroxidation and ferroptotic lethality by attenuating elevated Fe and low GSH levels [132]. Studies from our laboratory have found that GPX4 is crucial for motor neuron health and survival more specifically during oxidative stress [18]. Conditional ablation of GPX4 in neurons of adult mice caused rapid onset and progression of paralysis followed by death and interestingly, these mice showed remarkable spinal cord motor neuron degeneration but no explicit degeneration in cerebral cortex [18]. This degeneration followed ferroptotic mode of cell death as evidenced by enhanced spinal cord inflammation, ERK activation, and no signs of caspase-3 activation and TUNEL staining [18]. Moreover, we found GPX4 ablation resulted in mitochondrial dysfunctions and elevated lipid peroxidation in motor neurons which are hallmarks of ferroptosis and these functionalities were attenuated by supplementation with vitamin E, a lipophilic antioxidant and inhibitor of ferroptosis [18]. The onset of paralysis and mortality caused by GPX4 ablation was further delayed with this supplementation, indicating importance of GPX4 in suppressing ferroptosis and motor neuron death [18]. Recently, we found that GPX4 impedes motor neuron disease of SOD1G93A mice [133]. SOD1G93A was the first mutation in the SOD1 gene to be reported in cases of familial ALS, and even this model demonstrates paralysis and motor neuron death in the same way that ALS patients do [133]. We aimed to uncover how enhanced GPX4 function in SOD1G93A mice affected their lifespan and motor neuron atrophy. We generated SOD1G93AGPX4 double transgenic mice and found that overexpression of GPX4 delayed loss of motor function, disease onset, and extended life span [12]. We found compromised anti-ferroptosis defense in SOD1G93A mice and ALS patient samples due to decreased GPX4 expression in symptomatic SOD1G93A mice and ALS patient samples compared to control [12]. Moreover, GPX4 upregulation reduced SOD1G93A toxicity and motor neuron degeneration in SOD1G93A, similar to pharmacologic ferroptosis inhibitors, demonstrating that GPX4 can prevent motor neuron degeneration in SOD1G93A mice, most likely by a mechanism that prevents motor neuron ferroptosis [12] (Fig. 3). Our findings therefore emphasize on the importance of oxytosis/ferroptosis as a possible target for intervention in the development of ALS therapies [12].

Central role of ferroptosis in the pathogenesis of neurodegenerative diseases like amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD). The characteristic features of ferroptosis, including iron accumulation, reduced cystine intake, glutathione deficiency, inhibition of GPX4, and fatal amounts of phospholipid hydroperoxides, have all been observed in the distinct brain regions in these degenerative diseases. In PD, neurodegeneration is associated with neuromelanin accumulation in dopaminergic neurons, GPX4 inactivation, lipid peroxidation, and mitochondrial dysfunction. This degeneration is associated with the abnormal aggregation of α-synuclein and the formation of Lewy bodies. β-amyloid deposition, hyperphosphorylation of tau proteins, GPX4 inactivation, mitochondrial dysfunction, and iron-mediated oxidative stress promote neurodegeneration in AD. In MS, iron dysregulation and iron accumulation in the grey matter contribute to the destruction of oligodendrocytes and myelin subsequently to demyelination and axonal damage. Increased iron levels in basal ganglia appear to be a potential contributor to the pathogenesis in HD. In ALS, there is a potential relation between iron accumulation in the basal ganglia, GPX4 and cysteine depletion, oxidative stress, and neurodegeneration
Oxytosis/Ferroptosis, GPX4, and Alzheimer’s Disease (AD)
AD is the leading form of dementia and accounts for 60 to 80% of total cases [134,135,136,137,138,139,140]. AD affects more than 6 million people in the USA [134]. This figure is expected to reach around 13 million by 2050 [134]. The economic costs of AD are $260 billion and are projected to reach $1167 billion by 2050 [135, 141]. AD pathology is characterized by progressive degeneration in cerebral cortex and histopathologically hallmarked by intracellular neurofibrillary tangles and extracellular amyloid beta (Aβ) plaques [136, 137, 139]. At present, there are no effective therapeutics that significantly slow the progression of the disease. A myriad of evidence implies that ferroptosis could have a role in neuropathological disorders including AD. Recent reports are demonstrating that high oxidative stress that correlates with abnormal levels or overload of brain metals also results in progressive neuronal loss and death [135]. The activation of ferroptotic cascade is being defined as ferrosenescence and associated with iron dyshomeostasis as indicated in some aging and neurodegenerative disorders [142]. However, the characterized features based on morphological and biochemical changes in AD brains are increased lipid peroxidation, mitochondrial dysfunctions, GSH depletion, and inactivation of GPX4 [9, 18, 143,144,145]. Iron dysregulation in the brain along with diminished levels of endogenous antioxidant regulator of ferroptosis GPX4 has been found to be linked with AD pathology [146,147,148,149] (Fig. 3). Cortical iron concentrations were observed to be elevated in patients with mild cognitive impairment and a high Aβ plaque burden, which might raise the prevalence of AD [146,147,148,149]. Studies using in vivo and in vitro models have shown that ferroptosis inducers like erastin and RSL-3 are able to accumulate massive lipid peroxidation levels and deplete GSH within the cells and these levels could be attenuated using lipophilic antioxidants like vitamin E or ferroptosis inhibitors like Ferrostatin-1 or Liproxtatin-1 [75]. Among the protective mechanisms against ferroptosis and attenuation of AD pathology, GPX4 is a novel anti-peroxidant enzyme that prevents lipid peroxidation by converting hazardous membrane lipid hydroperoxides to non-toxic lipid alcohols [150]. Since GSH/GPX4 is a crucial anti-ferroptotic axis, studies have found reduced levels of GSH in animal models [151] as well as in postmortem AD brains [152]. Moreover, the rodent moles of AD revealed GPX4 downregulation compared to control mice [153]. On contrary, we have shown that its overexpression protects cortical neurons against Aβ-induced cytotoxicity by attenuating lipid peroxidation levels [150]. Since, neurons are exceptionally vulnerable to oxidative damage due to presence of their high content of PUFA, we exposed neurons cultured from GPX4 overexpressing transgenic mice and WT control mice with t-butyl hydroperoxide, hydrogen peroxide, and Aβ25–35 and Aβ1–40 peptides and found that neurons that from transgenic mice that had upregulated levels of GPX4 had significantly lower oxidative stress levels and cell death [150]. Despite the fact that GPX4 levels are required for embryonic and neonatal mouse survival while as AD and neurodegenerative dementia are age-related progressive disorders, thus, we ablated GPX4 in adult mice and observed a 75–85% fall in GPX4 levels in several tissues including liver, brain, kidney, and lungs [57]. Interestingly, in liver tissue, we observed decreased activity of electron transport chain complexes I and IV, as well as lower ATP synthesis as well as elevated 4-hydroxylnonenal (4-HNE) abducts (a lipid peroxidation marker), enhanced astrogliosis and neuronal death in the hippocampus, and these mice died after 2 weeks demonstrating the significance of GPX4 [154]. We were inquisitive to investigate whether neurons in forebrain areas (cerebral cortex and hippocampus) that are largely impaired in AD patients are sensitive to ferroptosis in the absence of its endogenous regulator, i.e., GPX4 [18]. We established conditional ablation of GPX4 exclusively in forebrain neurons and identified significantly impaired spatial learning and memory functioning compared to age matched control mice [18]. In these mice, we found that the hippocampal neurodegeneration was accompanied by ERK activation, increased lipid peroxidation, and enhanced neuroinflammation, all of which are characteristics of the ferroptotic cascade [18]. Furthermore, we fed these mice with a diet deficient in vitamin E, a lipid soluble antioxidant with anti-ferroptosis activity, and found enhanced hippocampal neurodegeneration and behavior dysfunctions [18]. Notably, this neurodegeneration was ameliorated with liproxstatin-1 treatment [18]. Neuronal cell loss due to inactivation or knockout of GPX4 is an indicative of how excessive increase in lipid peroxidation affects neurons and leading to neurodegeneration and cognitive impairment (Fig. 3). It is particularly intriguing that distinct hallmarks of ferroptosis (iron imbalance, lipid peroxidation, and inflammation) are also key prodromal indications of AD and associated dementias are particularly remarkable [155]. Thus, ferroptosis could reflect an unappreciated pathological player in the context of neurodegenerative dementias like AD.
Oxytosis/Ferroptosis, GPX4, and Parkinson’s Disease (PD)
PD is a neurological condition characterized by bradykinesia, tremor, and postural instability as motor symptoms [156]. About 1 to 2% of adults over the age of 65 and 4% of adults over the age of 80 are affected. Every year, about 60,000 Americans are afflicted with PD and an estimated 1 million inhabitants with the disease in the USA alone, and over 10 million people worldwide [157]. Dopaminergic neurons in the midbrain’s extrapyramidal tract degenerate, resulting in PD [158]. Although the cause of its onset is not well-known, however, it is thought to be the result of interplay between genetic and environmental factors as well as buildup of α-synuclein proteins (also referred as Lewy bodies) in the peripheral, central, and autonomic nervous system [158]. Degeneration of dopamine-releasing neurons in the substantia nigra disrupts the balance of excitatory (acetylcholine) and inhibitory (dopamine) neurotransmitters in the area and at times, this disrupted balance promotes uncontrollable tremors known as dyskinesias as well as a lack of movement referred as gait freezing [159]. There is currently no cure for PD; nevertheless, treatment aims to alleviate symptoms and reduce dyskinesia. The aggregation of oxidized biomolecules linked to DA neurodegeneration implies that oxidative stress processes have a role in PD [160]. Elevated iron levels as well as oxidative stress and inflammation have been linked to afflicted areas, suggesting that ferroptosis may play a role in the pathophysiology of PD [160]. Moreover, α-synuclein has been demonstrated to have association with iron and lipid metabolism which again suggests a potential interplay between α-synuclein and ferroptosis [161, 162]. Iron buildup, striatum dopamine insufficiency, neuromelanin loss, and the emergence of accumulated α-synuclein lead to lipid peroxidation, higher oxidative stress levels, and mitochondrial dysfunction along with the loss of enzymatic antioxidants in the GSH system [163, 164] (Fig. 3). High iron concentration in the substantia nigra and globus pallidus of PD patients has been reported using magnetic resonance imaging (MRI) and these accumulated levels were linked with severity of motor impairments [165, 166]. Earlier investigations on PD in the context of ferroptosis reported PUFA peroxidation, GSH system depletion, and a decline in GPX4 activity, all of which were linked to increased oxidative stress [167]. Attenuation of dopaminergic cell death has been observed in vivo by ferrostatin-1 treatment and recently it has been found that α-synuclein aggregation triggers increase in ROS and lipid peroxidation levels in an iron-dependent fashion similar to ferroptosis [162, 168]. These increased levels in turn enhance calcium influx and consequent cell death which is significantly suppressed by either iron chelators or ferroptosis inhibitors indicating that modulation in ferroptosis could be a therapeutic target in neurodegenerative diseases [162, 168]. A recent study in 6-OHDA-induced parkinsonism model has revealed increased ROS production, Ferritin Heavy Chain 1 (FTH1) mitochondrial dysfunction, and reduction in GPX4 levels and these functionalities were alleviated in an anti-ferroptotic fashion by moxibustion treatment [169]. GPX4 being the important player in maintaining and regulating ferroptototic cell death, its distribution is associated with pathological changes in PD (Fig. 3). Explicitly, GPX4 levels have been found to be reduced in the SN of PD compared to control subjects [170]. GPX4 expression in dopaminergic nigral neurons was observed to be colocalized with neuromelanin in postmortem human brain tissue with PD when compared to control brains and it was found to be clustered within dystrophic dopaminergic axons in the putamen of PD patients [170]. Colocalization of GPX4 with neuromelanin is specifically indicative of role for GPX4 in aging of DA neurons [171]. More recently, an in vitro study using 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) has shown that MPTP decreased cell viability via increased ROS production and decreased GSH, GPX4 and Trx-1 levels and these functions were ameliorated with ferrostatin-1 and overexpression of Trx-1 [172]. Furthermore, in transgenic mice with Trx-1 overexpression, the lowered levels of GPX4 and tyrosine hydroxylase triggered by MPTP were mitigated, indicating that Trx-1 reduces ferroptosis in PD primarily by modulating GPX4 and GSH [172].
Oxytosis/Ferroptosis, GPX4, and Huntington’s Disease (HD)
HD is a fatal neurological disorder characterized by a polymorphic sequence of three CAG nucleotides in exon 1 of the IT15 gene (Huntingtin (HTT) [173]. The pathophysiological hallmark of HD is that mutant HTT (mHTT) is frequently cleaved and accumulated into cytotoxic macromolecules, resulting in neuronal loss and cellular damage [174]. This includes atrophy of brain tissues, which might start in the striatum, followed by the cerebral cortex, loss of glial cells (astrocytes and oligodendrocytes), and neuronal death [174]. Although the understanding of pathophysiology of HD is complex but evidences are suggestive of oxidative stress being one of the initial factors in the pathogenesis [175], and some therapeutic strategies against oxidative stress seems promising [176]. Recently, ferroptosis has been found to be a significant part in oxidative stress and HD. Synchrotron X-ray fluorescence analysis in R6/2 HD mouse brain has shown iron accumulation as discrete puncta in the perinuclear cytoplasm of striatal neurons compared to control mice [177]. Furthermore, MRI has revealed iron accumulation in patients with HD [178]. Studies have demonstrated deregulated levels of GSH in HD patients which affects and interferes with proper functioning of GSH uptake and its dependent enzymes like GPX4 for their action [179, 180]. A cross-sectional study in asymptomatic HD patients vs healthy subjects has revealed increased lipid peroxidation and low GSH plasma levels in HD patients indicating that oxidative stress may be associated with the disease progression [181]. Reduced GSH and GSH-S-transferase levels have been observed in the striatum, cortex, and hippocampus of 3-nitropropionic acid (3-NP)-induced HD mouse [182] and neuronal death due to 3-NP and reduced GSH levels in HD mice models were attenuated with cystamine and cysteamine supplementation [183]. Low GSH levels seem to be a characteristic of HD which may accelerate the possibility of ferroptotic cascade in HD due to failure or inactivation of ferroptosis regulating enzyme GPX4 [183] (Fig. 3). Hence, GPX4 could be a potential therapeutic target for HD in the long term.
Conclusion and Perspective
Neurodegenerative diseases continue to be challenging from both research and drug development, perspective representing a major threat to human health. Studies are constantly being carried out to find out possible therapeutic targets and over the last few years, considerable advancements have been made to understand molecular and metabolic underpinnings of newly discovered iron-dependent oxidative form of cell death called oxytosis/ferroptosis. It has been implicated in several degenerative diseases and progressively more recognized in cancer research. However, current understanding of the oxytosis/ferroptosis process is still in its early stages. Despite the key findings that have enhanced our understanding, especially in exploring the involvement of lipid peroxidation and its regulation by GPX4, our present knowledge of the oxytosis/ferroptosis process is still in its early phases. GPX4 is a critical regulator of physiologically relevant lipid signaling pathways as well as a protector against oxidative lipid degradation. We have found downregulation of GPX4 in motor neuron disorders like ALS as well as in AD results in neuronal loss, cell death, and neurodegeneration exhibiting signatures of oxytosis/ferroptosis; on the contrary, GPX4 overexpression regulated the ferroptotic cascade and attenuated cellular functions similar to oxytosis/ferroptosis inhibitors (Fer-1) (Fig. 4). Even though the exact role of oxytosis/ferroptosis in the pathophysiology of numerous neurodegenerative disorders is unknown, one crucial question for future work is whether oxytosis/ferroptosis could be used as a therapeutic intervention for neurodegenerative disorders. Despite the fact that GPX4 is considered to be the primary regulator of oxytosis/ferroptosis and appears to be the critical enzyme at the intersection of oxidative lipid homeostasis and oxytosis/ferroptosis, in-depth research into how inactivation of this essential enzyme causes extensive lipid peroxidation in the context of oxytosis/ferroptosis is needed, as this could lead to potential therapeutic interventions for neurodegenerative disorders.

The promising therapeutic significance of GPX4 as the primary regulator of ferroptosis by converting peroxides (e.g., R–OOH) to their respective alcohols using glutathione (GSH) as reducing agent, thereby preventing the generation of harmful radicals which leads to inhibition of ferroptosis. GPX4 modulates the regulation of ferroptosis-linked signaling pathways, oxidative lipid homeostasis, synaptic plasticity, neuroinflammation, mitochondrial dysfunction, neurodegeneration, and cognitive impairment in neurodegenerative diseases
Data Availability
Not applicable.
References
Stockwell BR, Angeli JPF, Bayir H, Bush AI, Conrad M, Dixon SJ et al (2017) Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171(2):273–285. https://doi.org/10.1016/j.cell.2017.09.021
Fuchs Y, Steller H (2011) Programmed cell death in animal development and disease. Cell 147(4):742–758. https://doi.org/10.1016/j.cell.2011.10.033
Thompson CB (1995) Apoptosis in the pathogenesis and treatment of disease. Science 267(5203):1456–1462. https://doi.org/10.1126/science.7878464
Bergsbaken T, Fink SL, Cookson BT (2009) Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7(2):99–109. https://doi.org/10.1038/nrmicro2070
Christofferson DE, Yuan J (2010) Necroptosis as an alternative form of programmed cell death. CurrOpin Cell Biol 22(2):263–268. https://doi.org/10.1016/j.ceb.2009.12.003
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE et al (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149(5):1060–1072. https://doi.org/10.1016/j.cell.2012.03.042
Angeli JPF, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ et al (2014) Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16(12):1180–1191. https://doi.org/10.1038/ncb3064
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X et al (2016) Ferroptosis: process and function. Cell Death Differ 23(3):369–379. https://doi.org/10.1038/cdd.2015.158
Yang WS, Stockwell BR (2016) Ferroptosis: death by lipid peroxidation. Trends Cell Biol 26(3):165–176. https://doi.org/10.1016/j.tcb.2015.10.014
Toyokuni S, Ito F, Yamashita K, Okazaki Y, Akatsuka S (2017) Iron and thiol redox signaling in cancer: an exquisite balance to escape ferroptosis. Free Radic Biol Med 108:610–626. https://doi.org/10.1016/j.freeradbiomed.2017.04.024
Guiney SJ, Adlard PA, Bush AI, Finkelstein DI, Ayton S (2017) Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem Int 104:34–48. https://doi.org/10.1016/j.neuint.2017.01.004
Chen L, Na R, McLane KD, Thompson CS, Gao J, Wang X et al (2021) Overexpression of ferroptosis defense enzyme Gpx4 retards motor neuron disease of SOD1G93A mice. Sci Rep 11(1):1–13. https://doi.org/10.1038/s41598-021-92369-8
Mou Y, Wang J, Wu J, He D, Zhang C, Duan C et al (2019) Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol 12(1):1–16. https://doi.org/10.1186/s13045-019-0720-y
Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ et al (2007) RAS–RAF–MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447(7146):865–869. https://doi.org/10.1038/nature05859
Yang WS, Stockwell BR (2008) Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 15(3):234–245. https://doi.org/10.1016/j.chembiol.2008.02.010
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P et al (2018) Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25(3):486–541. https://doi.org/10.1038/s41418-017-0012-4
Chen L, Hambright WS, Na R, Ran Q (2015) Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J Biol Chem 290(47):28097–28106. https://doi.org/10.1074/jbc.M115.680090
Hambright WS, Fonseca RS, Chen L, Na R, Ran Q (2017) Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol 12:8–17. https://doi.org/10.1016/j.redox.2017.01.021
Li J, Jia B, Cheng Y, Song Y, Li Q, Luo C (2022) Targeting molecular mediators of ferroptosis and oxidative stress for neurological disorders. Ed H Birla Oxid Med Cell Longev 2022:1–14. https://doi.org/10.1155/2022/3999083
Chen D, Chu B, Yang X, Liu Z, Jin Y, Kon N, Rabadan R, Jiang X et al (2021) iPLA2β-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat Commun 12(1):3644. https://doi.org/10.1038/s41467-021-23902-6
Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B et al (2017) Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13(1):81–90. https://doi.org/10.1038/nchembio.2238
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M et al (2017 Jan) ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13(1):91–98. https://doi.org/10.1038/nchembio.2239
Yuan H, Li X, Zhang X, Kang R, Tang D (2016) Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun 478(3):1338–1343. https://doi.org/10.1016/j.bbrc.2016.08.124
Gao S, Zhou L, Lu J, Fang Y, Wu H, Xu W, Pan Y et al (2022) Cepharanthine attenuates early brain injury after subarachnoid hemorrhage in mice via inhibiting 15-lipoxygenase-1-mediated microglia and endothelial cell ferroptosis. Oxidative Med Cell Longev:4295208. https://doi.org/10.1155/2022/4295208
Chen X, Li J, Kang R, Klionsky DJ, Tang D (2021) Ferroptosis: machinery and regulation. Autophagy 17:2054–2081
Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, Tyurin VA, Anthonymuthu TS et al (2021) PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 171(3):628–641.e26. https://doi.org/10.1016/j.cell.2017.09.044
Li K, Wang M, Huang Z-H, Wang M, Sun W-Y, Kurihara H, Huang R-T, Wang R et al (2023) ALOX5 inhibition protects against dopaminergic neurons undergoing ferroptosis. Pharmacol Res 193:106779. https://doi.org/10.1016/j.phrs.2023.106779
Yan H, Zou T, Tuo Q, Xu S, Li H, Belaidi AA, Lei P (2021) Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther 6(1):49. https://doi.org/10.1038/s41392-020-00428-9
Chidawanyika T, Mark KMK, Supattapone S (2020) A genome-wide CRISPR/Cas9 screen reveals that riboflavin regulates hydrogen peroxide entry into HAP1 cells. MBio 11(4). https://doi.org/10.1128/mBio.01704-20
Deng L, He S, Guo N, Tian W, Zhang W, Luo L (2023) Molecular mechanisms of ferroptosis and relevance to inflammation. Inflamm Res 72(2):281–299. https://doi.org/10.1007/s00011-022-01672-1
Wang Y, Wu S, Li Q, Sun H, Wang H (2023) Pharmacological inhibition of ferroptosis as a therapeutic target for neurodegenerative diseases and strokes. Adv Sci. https://doi.org/10.1002/advs.202300325
Balihodzic A, Prinz F, Dengler MA, Calin GA, Jost PJ, Pichler M (2022) Non-coding RNAs and ferroptosis: potential implications for cancer therapy. Cell Death Differ 29(6):1094–1106. https://doi.org/10.1038/s41418-022-00998-x
Ryan SK, Zelic M, Han Y, Teeple E, Chen L, Sadeghi M, Shankara S, Guo L et al (2023) Microglia ferroptosis is regulated by SEC24B and contributes to neurodegeneration. Nat Neurosci 26(1):12–26. https://doi.org/10.1038/s41593-022-01221-3
Reichert CO, de Freitas FA, Sampaio-Silva J, Rokita-Rosa L, Barros P et al (2020) Ferroptosis mechanisms involved in neurodegenerative diseases. Int J Mol Sci 21(22):8765. https://doi.org/10.3390/ijms21228765
Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, Sagalovskiy IR, Ma A et al (2020) Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 368:85–89
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius E et al (2019) FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 575:693–698
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B et al (2019) The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575:688–692
Wei R, Zhao Y, Wang J, Yang X, Li S, Wang Y, Yang X, Fei J et al (2021) Tagitinin C induces ferroptosis through PERK-Nrf2-HO-1 signaling pathway in colorectal cancer cells. Int J Biol Sci 17:2703–2717
Schipper HM, Song W, Tavitian A, Cressatti M (2019) The sinister face of heme oxygenase-1 in brain aging and disease. Prog Neurobiol 172:40–70
Campbell MR, Karaca M, Adamski KN, Chorley BN, Wang X, Bell DA (2013) Novel hematopoietic target genes in the NRF2-mediated transcriptional pathway. Oxidative Med Cell Longev 2013:120305
Dodson M, Castro-Portuguez R, Zhang DD (2019) NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol 23:101107
Margis R, Dunand C, Teixeira FK, Margis-Pinheiro M (2008) Glutathione peroxidase family–an evolutionary overview. FEBS J 275(15):3959–3970. https://doi.org/10.1111/j.1742-4658.2008.06542.x
Ursini F, Maiorino M, Gregolin C (1985) The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim Biophys Acta BBA-Gen Subj 839(1):62–70. https://doi.org/10.1016/0304-4165(85)90182-5
Chu F-F, de Silva HR, Esworthy RS, Boteva KK, Walters CE, Roses A et al (1996) Polymorphism and chromosomal localization of the GI-form of human glutathione peroxidase (GPX2) on 14q24. 1 by insitu hybridization. Genomics 32(2):272–276. https://doi.org/10.1006/geno.1996.0115
Stoytcheva ZR, Berry MJ (2009) Transcriptional regulation of mammalian selenoprotein expression. Biochim Biophys Acta BBA-Gen Subj 1790(11):1429–1440. https://doi.org/10.1016/j.bbagen.2009.05.012
Brigelius-Flohe R, Aumann K-D, Blöcker H, Gross G, Kiess M, Klöppel KD et al (1994) Phospholipid-hydroperoxide glutathione peroxidase. Genomic DNA, cDNA, and deduced amino acid sequence. J Biol Chem 269(10):7342–7348
Pushpa-Rekha TR, Burdsall AL, Oleksa LM, Chisolm GM, Driscoll DM (1995) Rat phospholipid-hydroperoxide glutathione peroxidase: cDNA cloning and identification of multiple transcription and translation start sites. J Biol Chem 270(45):26993–26999. https://doi.org/10.1074/jbc.270.45.26993
Maiorino M, Scapin M, Ursini F, Biasolo M, Bosello V, Flohé L (2003) Distinct promoters determine alternative transcription of gpx-4 into phospholipid-hydroperoxide glutathione peroxidase variants. J Biol Chem 278(36):34286–34290. https://doi.org/10.1074/jbc.M305327200
Brigelius-Flohé R, Maiorino M (2013) Glutathione peroxidases. Biochim Biophys Acta BBA-Gen Subj 1830(5):3289–3303. https://doi.org/10.1016/j.bbagen.2012.11.020
Maiorino M, Roveri A, Ursini F (2018) GPx4: from prevention of lipid peroxidation to spermatogenesis and back. In: Glutathione. CRC Press, pp. 111–127
Ursini F, Heim S, Kiess M, Maiorino M, Roveri A, Wissing J et al (1999) Dual function of the selenoprotein PHGPx during sperm maturation. Science 285(5432):13936. https://doi.org/10.1126/science.285.5432.1393
Bosello-Travain V, Conrad M, Cozza G, Negro A, Quartesan S, Rossetto M et al (2013) Protein disulfide isomerase and glutathione are alternative substrates in the one Cys catalytic cycle of glutathione peroxidase 7. Biochim Biophys Acta BBA-Gen Subj 1830(6):3846–3857. https://doi.org/10.1016/j.bbagen.2013.02.017
Borchert A, Kalms J, Roth SR, Rademacher M, Schmidt A, Holzhutter H-G et al (2018) Crystal structure and functional characterization of selenocysteine-containing glutathione peroxidase 4 suggests an alternative mechanism of peroxide reduction. Biochim Biophys Acta BBA-Mol Cell Biol Lipids 1863(9):1095–1107. https://doi.org/10.1016/j.bbalip.2018.06.006
Tosatto SC, Bosello V, Fogolari F, Mauri P, Roveri A, Toppo S et al (2008) The catalytic site of glutathione peroxidases. Antioxid Redox Signal 10(9):1515–1526. https://doi.org/10.1089/ars.2008.2055
Matsui M, Oshima M, Oshima H, Takaku K, Maruyama T, Yodoi J et al (1996) Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev Biol 178(1):179–185. https://doi.org/10.1006/dbio.1996.0208
Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG et al (2003) The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med 34(4):496–502. https://doi.org/10.1016/s0891-5849(02)01360-6
Imai H, Hirao F, Sakamoto T, Sekine K, Mizukura Y, Saito M et al (2003) Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem Biophys Res Commun 305(2):278–286. https://doi.org/10.1016/s0006-291x(03)00734-4
Shi Z-Z, Osei-Frimpong J, Kala G, Kala SV, Barrios RJ, Habib GM et al (2000) Glutathione synthesis is essential for mouse development but not for cell growth in culture. Proc Natl Acad Sci 97(10):5101–5106. https://doi.org/10.1073/pnas.97.10.5101
Chen L et al (2022) Enhanced defense against ferroptosis ameliorates cognitive impairment and reduces neurodegeneration in 5xFAD mice. Free Radic Biol Med 180(20):1–12
Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT (1989) Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 2(6):1547–1558. https://doi.org/10.1016/0896-6273(89)90043-3
Tan S, Schubert D, Maher P (2001) Oxytosis: a novel form of programmed cell death. Curr Top Med Chem 1(6):497–506. https://doi.org/10.2174/1568026013394741
Lewerenz J, Ates G, Methner A, Conrad M, Maher P (2018) Oxytosis/ferroptosis—(re-) emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system. Front Neurosci 12:214. https://doi.org/10.3389/fnins.2018.00214
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS et al (2014) Regulation of ferroptotic cancer cell death by GPX4. Cell 156(1–2):317–331. https://doi.org/10.1016/j.cell.2013.12.010
Bannai S, Kitamura E (1980) Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J Biol Chem 255(6):2372–2376
Sato H, Tamba M, Ishii T, Bannai S (1999) Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem 274(17):11455–11458. https://doi.org/10.1074/jbc.274.17.11455
Kobayashi S, Sato M, Kasakoshi T, Tsutsui T, Sugimoto M, Osaki M et al (2015) Cystathionine is a novel substrate of cystine/glutamate transporter: implications for immune function. J Biol Chem 290(14):8778–8788. https://doi.org/10.1074/jbc.M114.625053
Seibt TM, Proneth B, Conrad M (2019) Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med 133:144–152. https://doi.org/10.1016/j.freeradbiomed.2018.09.014
Ingold I, Aichler M, Yefremova E, Roveri A, Buday K, Doll S et al (2015) Expression of a catalytically inactive mutant form of glutathione peroxidase 4 (Gpx4) confers a dominant-negative effect in male fertility. J Biol Chem 290(23):14668–14678. https://doi.org/10.1074/jbc.M115.656363
Brütsch SH, Wang CC, Li L, Stender H, Neziroglu N, Richter C et al (2015) Expression of inactive glutathione peroxidase 4 leads to embryonic lethality, and inactivation of the Alox15 gene does not rescue such knock-in mice. Antioxid Redox Signal 22(4):281–293. https://doi.org/10.1089/ars.2014.5967
Maiorino M, Conrad M, Ursini F (2018) GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid Redox Signal 29(1):61–74. https://doi.org/10.1089/ars.2017.7115
Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C et al (2008) Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent-and AIF-mediated cell death. Cell Metab 8(3):237–248. https://doi.org/10.1016/j.cmet.2008.07.005
Dar NJ, Na R, Ran Q (2022) Functional deficits of 5×FAD neural stem cells are ameliorated by glutathione peroxidase 4. Cells. 11(11):1770. https://doi.org/10.3390/cells11111770.
Stockwell BR, Jiang X (2020) The chemistry and biology of ferroptosis. Cell Chem Biol 27(4):365–375. https://doi.org/10.1016/j.chembiol.2020.03.013
Koeberle SC, Kipp AP, Stuppner H, Koeberle A (2023) Ferroptosis-modulating small molecules for targeting drug-resistant cancer: challenges and opportunities in manipulating redox signaling. Med Res Rev 43(3):614–682. https://doi.org/10.1002/med.21933
Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M et al (2017) On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci 3(3):232–243. https://doi.org/10.1021/acscentsci.7b00028
Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz PS et al (2014) Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A 111:16836–16841
Jin Y, Zhuang Y, Liu M, Che J, Dong X (2021) Inhibiting ferroptosis: a novel approach for stroke therapeutics. Drug Discov Today 26:916–930
La Rosa P, Petrillo S, Fiorenza MT, Bertini ES, Piemonte F (2020) Ferroptosis in Friedreich’s ataxia: a metal-induced neurodegenerative disease. Biomolecules 10(11):1551. https://doi.org/10.3390/biom10111551
Petrillo S, D’Amico J, La Rosa P, Bertini ES, Piemonte F (2019) Targeting NRF2 for the treatment of Friedreich’s ataxia: a comparison among drugs. Int J Mol Sci 20(20):5211. https://doi.org/10.3390/ijms20205211
Buccellato FR, D’Anca M, Fenoglio C, Scarpini E, Galimberti D (2021) Role of oxidative damage in Alzheimer’s disease and neurodegeneration: from pathogenic mechanisms to biomarker discovery. Antioxidants 10(9):1353. https://doi.org/10.3390/antiox10091353
Cheng H, Wang P, Wang N, Dong W, Chen Z, Wu M, Wang Z, Yu Z et al (2023) Neuroprotection of NRF2 against ferroptosis after traumatic brain injury in mice. Antioxidants. 12(3):731. https://doi.org/10.3390/antiox12030731
Yang JH, Nguyen CD, Lee G, Na C-S (2022) Insamgobonhwan protects neuronal cells from lipid ROS and improves deficient cognitive function. Antioxidants 11(2):295. https://doi.org/10.3390/antiox11020295
Karuppagounder SS, Alin L, Chen Y, Brand D, Bourassa MW, Dietrich K, Wilkinson CM, Nadeau CA et al (2018) N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E 2 to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann Neurol 84(6):854–872. https://doi.org/10.1002/ana.25356
Costa I, Barbosa DJ, Benfeito S, Silva V, Chavarria D, Borges F, Remião F, Silva R (2023) Molecular mechanisms of ferroptosis and their involvement in brain diseases. Pharmacol Ther 244:108373. https://doi.org/10.1016/j.pharmthera.2023.108373
Hou L, Huang R, Sun F, Zhang L, Wang Q (2019) NADPH oxidase regulates paraquat and maneb-induced dopaminergic neurodegeneration through ferroptosis. Toxicology 417:64–73. https://doi.org/10.1016/j.tox.2019.02.011
Keynes RG, Karchevskaya A, Riddall D, Griffiths CH, Bellamy TC, Chan AWE, Selwood DL, Garthwaite J (2019) N 10 -carbonyl-substituted phenothiazines inhibiting lipid peroxidation and associated nitric oxide consumption powerfully protect brain tissue against oxidative stress. Chem Biol Drug Des 94(3):1680–1693. https://doi.org/10.1111/cbdd.13572
Tschuck J, Nair VP, Galhoz A, Ciceri G, Rothenaigner I, Tchieu J, Tai H-M, Stockwell BR et al (2023) Suppression of ferroptosis by vitamin A or antioxidants is essential for neuronal development. BioRxiv 2023(04):05.535746. https://doi.org/10.1101/2023.04.05.535746
Zheng H, Jiang J, Xu S, Liu W, Xie Q, Cai X, Zhang J, Liu S, Li R (2021) Nanoparticle-induced ferroptosis: detection methods, mechanisms and applications. Nanoscale 13(4):2266–2285. https://doi.org/10.1039/D0NR08478F
Zhang Y, Fan B-Y, Pang Y-L, Shen W-Y, Wang X, Zhao C-X, Li W-X, Liu C et al (2020) Neuroprotective effect of deferoxamine on erastininduced ferroptosis in primary cortical neurons. Neural Regen Res 15(8):1539. https://doi.org/10.4103/1673-5374.274344
Jiang X, Stockwell BR, Conrad M (2021) Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 22(4):266–282. https://doi.org/10.1038/s41580-020-00324-8
Gao Y, Wang T, Cheng Y, Wu Y, Zhu L, Gu Z, Wu Y, Cai L et al (2023) Melatonin ameliorates neurological deficits through MT2/IL-33/ferritin H signaling-mediated inhibition of neuroinflammation and ferroptosis after traumatic brain injury. Free Radic Biol Med 199:97–112. https://doi.org/10.1016/j.freeradbiomed.2023.02.014
Kuang F, Liu J, Tang D, Kang R (2020) Oxidative damage and antioxidant defense in ferroptosis. Front Cell. Dev Biol:8. https://doi.org/10.3389/fcell.2020.586578
Sun Y, Xia X, Basnet D, Zheng JC, Huang J, Liu J (2022) Mechanisms of ferroptosis and emerging links to the pathology of neurodegenerative diseases. Front Aging Neurosci:14. https://doi.org/10.3389/fnagi.2022.904152
Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, Merl-Pham J, Bao X et al (2020) GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci 6(1):41–53. https://doi.org/10.1021/acscentsci.9b01063
Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, Molina H, Garcia-Bermudez J et al (2020) Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol 16(12):1351–1360. https://doi.org/10.1038/s41589-020-0613-y
Duan L, Zhang Y, Yang Y, Su S, Zhou L, Lo P-C, Cai J, Qiao Y et al (2021) Baicalin inhibits ferroptosis in intracerebral hemorrhage. Front Pharmacol:12. https://doi.org/10.3389/fphar.2021.629379
Li Q, Li Q-Q, Jia J-N, Sun Q-Y, Zhou H-H, Jin W-L, Mao X-Y (2019) Baicalein exerts neuroprotective effects in FeCl3-induced posttraumatic epileptic seizures via suppressing ferroptosis. Front Pharmacol:10. https://doi.org/10.3389/fphar.2019.00638
Fischer W, Currais A, Liang Z, Pinto A, Maher P (2019) Old age-associated phenotypic screening for Alzheimer’s disease drug candidates identifies sterubin as a potent neuroprotective compound from Yerba santa. Redox Biol 21:101089. https://doi.org/10.1016/j.redox.2018.101089
Maher P, Fischer W, Liang Z, Soriano-Castell D, Pinto AFM, Rebman J, Currais A (2020) The value of herbarium collections to the discovery of novel treatments for Alzheimer’s disease, a case made with the genus Eriodictyon. Front Pharmacol:11. https://doi.org/10.3389/fphar.2020.00208
Gao Y, Li J, Wu Q, Wang S, Yang S, Li X, Chen N, Li L et al (2021) Tetrahydroxy stilbene glycoside ameliorates Alzheimer’s disease in APP/PS1 mice via glutathione peroxidase related ferroptosis. Int Immunopharmacol 99:108002. https://doi.org/10.1016/j.intimp.2021.108002
Lv H-W, Wang Q-L, Luo M, Zhu M-D, Liang H-M, Li W-J, Cai H, Zhou Z-B et al (2023) Phytochemistry and pharmacology of natural prenylated flavonoids. Arch Pharm Res 46(4):207–272. https://doi.org/10.1007/s12272-023-01443-4
Wen L, Shi D, Zhou T, Tu J, He M, Jiang Y, Yang B (2020) Identification of two novel prenylated flavonoids in mulberry leaf and their bioactivities. Food Chem 315:126236. https://doi.org/10.1016/j.foodchem.2020.126236
Zhang Y, Lu X, Tai B, Li W, Li T (2021) Ferroptosis and its multifaceted roles in cerebral stroke. Front Cell Neurosci:15. https://doi.org/10.3389/fncel.2021.615372
Takashima M, Ichihara K, Hirata Y (2019) Neuroprotective effects of Brazilian green propolis on oxytosis/ferroptosis in mouse hippocampal HT22 cells. Food Chem Toxicol 132:110669. https://doi.org/10.1016/j.fct.2019.110669
Gunesch S, Hoffmann M, Kiermeier C, Fischer W, Pinto AFM, Maurice T, Maher P, Decker M (2020) 7-O-Esters of taxifolin with pronounced and overadditive effects in neuroprotection, anti-neuroinflammation, and amelioration of short-term memory impairment in vivo. Redox Biol 29:101378. https://doi.org/10.1016/j.redox.2019.101378
Zheng K, Dong Y, Yang R, Liang Y, Wu H, He Z (2021) Regulation of ferroptosis by bioactive phytochemicals: implications for medical nutritional therapy. Pharmacol Res 168:105580. https://doi.org/10.1016/j.phrs.2021.105580
Woo JH, Shimoni Y, Yang WS, Subramaniam P, Iyer A, Nicoletti P, Rodriguez Martinez M, Lopez G, Mattioli M, Realubit R et al (2015) Elucidating compound mechanism of action by network perturbation analysis. Cell. 162:441–451
Weiwer M, Bittker JA, Lewis TA, Shimada K, Yang WS, MacPherson L, Dandapani S, Palmer M, Stockwell BR, Schreiber SL et al (2012) Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg Med Chem Lett 22:1822–1826
Eaton JK, Furst L, Ruberto RA, Moosmayer D, Hilpmann A, Ryan MJ, Zimmermann K, Cai LL, Niehues M, Badock V, Kramm A, Chen S, Hillig RC, Clemons PA, Gradl S, Montagnon C, Lazarski KE, Christian S, Bajrami B et al (2020) Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat Chem Biol 16(5):497–506. https://doi.org/10.1038/s41589-020-0501-5
Eaton JK, Ruberto RA, Kramm A, Viswanathan VS, Schreiber SL (2019) Diacylfuroxans are masked nitrile oxides that inhibit GPX4 covalently. J Am Chem Soc 141:20407–20415
Hassannia B, Wiernicki B, Ingold I, Qu F, Van Herck S, Tyurina YY, Bayir H, Abhari BA, Angeli JPF, Choi SM et al (2018) Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J Clin Invest 128:3341–3355
Robberecht W, Philips T (2013) The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 14(4):248–264. https://doi.org/10.1038/nrn3430
Hardiman O, Van Den Berg LH, Kiernan MC (2011) Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol 7(11):639–649. https://doi.org/10.1038/nrneurol.2011.153
Mitchell JD, Borasio GD (2007) Amyotrophic lateral sclerosis. Lancet 369(9578):2031–2041. https://doi.org/10.1016/S0140-6736(07)60944-1
Matus S, Medinas DB, Hetz C (2014) Common ground: stem cell approaches find shared pathways underlying ALS. Cell Stem Cell 14(6):697–699. https://doi.org/10.1016/j.stem.2014.05.001
Renton AE, Chiò A, Traynor BJ (2014) State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17(1):17–23. https://doi.org/10.1038/nn.3584
Vucic S, Rothstein JD, Kiernan MC (2014) Advances in treating amyotrophic lateral sclerosis: insights from pathophysiological studies. Trends Neurosci 37(8):433–442. https://doi.org/10.1016/j.tins.2014.05.006
Ling S-C, Polymenidou M, Cleveland DW (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79(3):416–438. https://doi.org/10.1016/j.neuron.2013.07.033
Murphy JM, Henry RG, Langmore S, Kramer JH, Miller BL, Lomen-Hoerth C (2007) Continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch Neurol 64(4):530–534. https://doi.org/10.1001/archneur.64.4.530
Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE (2005) Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 65(4):586–590. https://doi.org/10.1212/01.wnl.0000172911.39167.b6
Van Blitterswijk M, Van Es MA, Hennekam EA, Dooijes D, Van Rheenen W, Medic J et al (2012) Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet 21(17):3776–3784. https://doi.org/10.1093/hmg/dds199
Shaw PJ (2005) Molecular and cellular pathways of neurodegeneration in motor neurone disease. J Neurol Neurosurg Psychiatry 76(8):1046–1057. https://doi.org/10.1136/jnnp.2004.048652
Figueroa-Romero C, Hur J, Bender DE, Delaney CE, Cataldo MD, Smith AL et al (2012) Identification of epigenetically altered genes in sporadic amyotrophic lateral sclerosis. PLoS One 7(12):e52672. https://doi.org/10.1371/journal.pone.0052672
Kanekura K, Suzuki H, Aiso S, Matsuoka M (2009) ER stress and unfolded protein response in amyotrophic lateral sclerosis. Mol Neurobiol 39(2):81–89. https://doi.org/10.1007/s12035-009-8054-3
Hadzhieva M, Kirches E, Wilisch-Neumann A, Pachow D, Wallesch M, Schoenfeld P et al (2013) Dysregulation of iron protein expression in the G93A model of amyotrophic lateral sclerosis. Neuroscience 230:94–101. https://doi.org/10.1016/j.neuroscience.2012.11.021
Wu J, Tuo Q, Lei P (2018) Ferroptosis, a recent defined form of critical cell death in neurological disorders. J Mol Neurosci 66(2):197–206. https://doi.org/10.1007/s12031-018-1155-6
Lee JK, Shin JH, Gwag BJ, Choi E-J (2015) Iron accumulation promotes TACE-mediated TNF-α secretion and neurodegeneration in a mouse model of ALS. Neurobiol Dis 80:63–69. https://doi.org/10.1016/j.nbd.2015.05.009
Jiang X, Chen J, Bajić A, Zhang C, Song X, Carroll SL et al (2017) Quantitative real-time imaging of glutathione. Nat Commun 8(1):1–13. https://doi.org/10.1038/ncomms16087
Matsuo T, Adachi-Tominari K, Sano O, Kamei T, Nogami M, Ogi K et al (2021) Involvement of ferroptosis in human motor neuron cell death. Biochem Biophys Res Commun 566:24–29. https://doi.org/10.1016/j.bbrc.2021.05.095
Devos D, Moreau C, Kyheng M, Garçon G, Rolland AS, Blasco H et al (2019) A ferroptosis–based panel of prognostic biomarkers for amyotrophic lateral sclerosis. Sci Rep 9(1):1–6. https://doi.org/10.1038/s41598-019-39739-5
Homma T, Kobayashi S, Sato H, Fujii J (2019) Edaravone, a free radical scavenger, protects against ferroptotic cell death in vitro. Exp Cell Res 384(1):111592. https://doi.org/10.1016/j.yexcr.2019.111592
Southon A, Szostak K, Acevedo KM, Dent KA, Volitakis I, Belaidi AA et al (2020) CuII (atsm) inhibits ferroptosis: implications for treatment of neurodegenerative disease. Br J Pharmacol 177(3):656–667. https://doi.org/10.1111/bph.14881
Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD et al (1994) Motor neuron degeneration in mice that express a human Cu Zn superoxide dismutase mutation. Science 264(5166):1772–1775. https://doi.org/10.1126/science.8209258
Harper LC (2020) Alzheimer’s Association facts and figures. https://doi.org/10.1017/9781108974677.026
Comas-Herrera A. Building on hope or tackling fear? Policy responses to the growing costs of Alzheimer’s disease and other dementias. Policy Responses Grow Costs Alzheimers Dis Dement July 30 2020Whart Pension Res Counc Work Pap. 2020;(2020–19).
Querfurth HW, LaFerla FM (2010) Alzheimer’s disease. N Engl J Med 362(4):329–344. https://doi.org/10.1056/NEJMra0909142
DeTure MA, Dickson DW (2019) The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 14(1):1–18. https://doi.org/10.1186/s13024-019-0333-5
Rai SN, Tiwari N, Singh P, Mishra D, Singh AK, Hooshmandi E, Vamanu E, Singh MP (2021) Therapeutic potential of vital transcription factors in Alzheimer’s and Parkinson’s disease with particular emphasis on transcription factor EB mediated autophagy. Front Neurosci 15:777347. https://doi.org/10.3389/fnins.2021.777347
Singh AK, Rai SN, Maurya A, Mishra G, Awasthi R, Shakya A, Chellappan DK, Dua K, Vamanu E, Chaudhary SK, Singh MP (2021) Therapeutic potential of phytoconstituents in management of Alzheimer’s disease. Evidence-based complement altern med: eCAM:5578574. https://doi.org/10.1155/2021/5578574
Singh AK, Mishra G, Maurya A, Awasthi R, Kumari K, Thakur A, Rai A, Rai GK, Sharma B, Kulkarni GT, Singh SK (2019) Role of TREM2 in Alzheimer’s disease and its consequences on β- amyloid, tau and neurofibrillary tangles. Curr Alzheimer Res 16(13):1216–1229. https://doi.org/10.2174/1567205016666190903102822
Singh AK, Singh SK, Nandi MK, Mishra G, Maurya A, Rai A, Rai GK, Awasthi R, Sharma B, Kulkarni GT (2019) Berberine: a plant-derived alkaloid with therapeutic potential to combat Alzheimer’s disease. Cent Nerv Syst Agents Med Chem 19(3):154–170. https://doi.org/10.2174/1871524919666190820160053
Sfera A, Bullock K, Price A, Inderias L, Osorio C (2018) Ferrosenescence: the iron age of neurodegeneration? Mech Ageing Dev 174:63–75. https://doi.org/10.1016/j.mad.2017.11.012
Bradley-Whitman MA, Lovell MA (2015) Biomarkers of lipid peroxidation in Alzheimer disease (AD): an update. Arch Toxicol 89(7):1035–1044. https://doi.org/10.1007/s00204-015-1517-6
Wang X, Wang W, Li L, Perry G, Lee H, Zhu X (2014) Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta BBA-Mol Basis Dis 1842(8):1240–1247. https://doi.org/10.1016/j.bbadis.2013.10.015
Obulesu M, Venu R, Somashekhar R (2011) Lipid peroxidation in Alzheimer’s disease: emphasis on metal-mediated neurotoxicity. Acta Neurol Scand 124(5):295–301. https://doi.org/10.1111/j.1600-0404.2010.01483.x
Ayton S, Wang Y, Diouf I, Schneider JA, Brockman J, Morris MC et al (2020) Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Mol Psychiatry 25(11):2932–2941. https://doi.org/10.1038/s41380-019-0375-7
Derry PJ, Kent TA (2017) Correlating quantitative susceptibility mapping with cognitive decline in Alzheimer’s disease. Brain 140(8):2069–2072. https://doi.org/10.1093/brain/awx167
Lupton MK, Benyamin B, Proitsi P, Nyholt DR, Ferreira MA, Montgomery GW et al (2017) No genetic overlap between circulating iron levels and Alzheimer’s disease. J Alzheimers Dis 59(1):85–99. https://doi.org/10.3233/JAD-170027
Szewczyk B (2013) Zinc homeostasis and neurodegenerative disorders. Front Aging Neurosci 5:33. https://doi.org/10.3389/fnagi.2013.00033
Cozza G, Rossetto M, Bosello-Travain V, Maiorino M, Roveri A, Toppo S et al (2017) Glutathione peroxidase 4-catalyzed reduction of lipid hydroperoxides in membranes: the polar head of membrane phospholipids binds the enzyme and addresses the fatty acid hydroperoxide group toward the redox center. Free Radic Biol Med 112:1–11. https://doi.org/10.1016/j.freeradbiomed.2017.07.010
Ghosh D, LeVault KR, Barnett AJ, Brewer GJ (2012) A reversible early oxidized redox state that precedes macromolecular ROS damage in aging nontransgenic and 3xTg-AD mouse neurons. J Neurosci 32(17):5821–5832. https://doi.org/10.1523/JNEUROSCI.6192-11.2012
Venkateshappa C, Harish G, Mahadevan A, Bharath MS, Shankar SK (2012) Elevated oxidative stress and decreased antioxidant function in the human hippocampus and frontal cortex with increasing age: implications for neurodegeneration in Alzheimer’s disease. Neurochem Res 37(8):1601–1614. https://doi.org/10.1007/s11064-012-0755-8
Yoo M-H, Gu X, Xu X-M, Kim J-Y, Carlson BA, Patterson AD et al (2010) Delineating the role of glutathione peroxidase 4 in protecting cells against lipid hydroperoxide damage and in Alzheimer’s disease. Antioxid Redox Signal 12(7):819–827. https://doi.org/10.1089/ars.2009.2891
Yoo S-E, Chen L, Na R, Liu Y, Rios C, Van Remmen H et al (2012) Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic Biol Med 52(9):1820–1827
Yan N, Zhang J (2020) Iron metabolism, ferroptosis, and the links with Alzheimer’s disease. Front Neurosci 13:1443. https://doi.org/10.3389/fnins.2019.01443
Moustafa AA, Chakravarthy S, Phillips JR, Gupta A, Keri S, Polner B, Frank MJ, Jahanshahi M (2016) Motor symptoms in Parkinson’s disease: a unified framework. Neurosci Biobehav Rev 2016(68):727–740. https://doi.org/10.1016/j.neubiorev.2016.07.010
Chakraborty A, Brauer S, Diwan A (2020) A review of possible therapies for Parkinson’s disease. J Clin Neurosci 76:1–4. https://doi.org/10.1016/j.jocn.2020.03.047
Olanow CW, Brundin P (2013) Parkinson’s disease and alpha synuclein: is Parkinson’s disease a prion-like disorder? Mov Disord 28(1):31–40
Olanow CW, Stern MB, Sethi K (2009) The scientific and clinical basis for the treatment of Parkinson disease. Neurology 72(21 Supplement 4):S1–S136
Masaldan S, Bush AI, Devos D, Rolland AS, Moreau C (2019) Striking while the iron is hot: iron metabolism and ferroptosis in neurodegeneration. Free Radic Biol Med 133:221–233
Angelova PR, Choi ML, Berezhnov AV, Horrocks MH, Hughes CD, De S et al (2020) Alpha synuclein aggregation drives ferroptosis: an interplay of iron, calcium and lipid peroxidation. Cell Death Differ 27(10):2781–2796
Deas E, Cremades N, Angelova PR, Ludtmann MH, Yao Z, Chen S et al (2016) Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson’s disease. Antioxid Redox Signal 24(7):376–391
Dauer W, Przedborski S (2003) Parkinson’s disease: mechanisms and models. Neuron 39(6):889–909
Lees AJ, Hardy J, Revesz T (2009) Parkinson’s disease Lancet Lond Engl 373:2055–2066
Graham JM, Paley MN, Grünewald RA, Hoggard N, Griffiths PD (2000) Brain iron deposition in Parkinson’s disease imaged using the PRIME magnetic resonance sequence. Brain 123(12):2423–2431
Rossi M, Ruottinen H, Soimakallio S, Elovaara I, Dastidar P (2013) Clinical MRI for iron detection in Parkinson’s disease. Clin Imaging 37(4):631–636
Do Van B, Gouel F, Jonneaux A, Timmerman K, Gelé P, Pétrault M et al (2016) Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol Dis 94:169–178
Miotto G, Rossetto M, Di Paolo ML, Orian L, Venerando R, Roveri A et al (2020) Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol 28:101328
Huang Z, Si W, Li X, Ye S, Liu X, Ji Y et al (2021) Moxibustion protects dopaminergic neurons in Parkinson’s disease through antiferroptosis. Evid Based Complement Alternat Med 2021
Bellinger FP, Bellinger MT, Seale LA, Takemoto AS, Raman AV, Miki T et al (2011) Glutathione peroxidase 4 is associated with neuromelanin in substantia nigra and dystrophic axons in putamen of Parkinson’s brain. Mol Neurodegener 6(1):1–10
Brigelius-Flohé R (2006) Glutathione peroxidases and redox-regulated transcription factors. Biol Chem 387(10-11):1329–1335. https://doi.org/10.1515/BC.2006.166
Bai L, Yan F, Deng R, Gu R, Zhang X, Bai J (2021) Thioredoxin-1 rescues MPP+/MPTP-induced ferroptosis by increasing glutathione peroxidase 4. Mol Neurobiol 1–11
Gellera C, Meoni C, Castellotti B, Zappacosta B, Girotti F, Taroni F et al (1996) Errors in Huntington disease diagnostic test caused by trinucleotide deletion in the IT15 gene. Am J Hum Genet 59(2):475
Caron NS, Wright GE, Hayden MR. Huntington disease. 2020;
Browne SE, Beal MF (2006) Oxidative damage in Huntington’s disease pathogenesis. Antioxid Redox Signal 8(11–12):2061–2073
Velusamy T, Panneerselvam AS, Purushottam M, Anusuyadevi M, Pal PK, Jain S et al (2017) Protective effect of antioxidants on neuronal dysfunction and plasticity in Huntington’s disease. Oxidative Med Cell Longev
Chen J, Marks E, Lai B, Zhang Z, Duce JA, Lam LQ, Volitakis I, Bush AI, Hersch S, Fox JH (2013) Iron accumulates in HD neurons: protection by deferoxamine. PLoS One
Rosas HD, Chen YI, Doros G, Salat DH, Chen N, Kwong KK et al (2012) Alterations in brain transition metals in Huntington disease: an evolving and intricate story. Arch Neurol 69(7):887–893
Mi Y, Gao X, Xu H, Cui Y, Zhang Y, Gou X (2019) The emerging roles of ferroptosis in Huntington’s disease. NeuroMolecular Med 21(2):110–119
Johnson WM, Wilson-Delfosse AL, Mieyal J (2012) Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients 4(10):1399–1440
Klepac N, Relja M, Klepac R, Hećimović S, Babić T, Trkulja V (2007) Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects. J Neurol 254(12):1676–1683
Kumar P, Kalonia H, Kumar A (2010) Nitric oxide mechanism in the protective effect of antidepressants against 3-nitropropionic acid-induced cognitive deficit, glutathione and mitochondrial alterations in animal model of Huntington’s disease. Behav Pharmacol 21(3):217–230
Mao Z, Choo YS, Lesort M (2006) Cystamine and cysteamine prevent 3-NP-induced mitochondrial depolarization of Huntington’s disease knock-in striatal cells. Eur J Neurosci 23(7):1701–1710
Acknowledgements
The infrastructural facilities provided by the Department of Zoology, Aligarh Muslim University, Aligarh, and funding agencies UGC, CSIR, and ICMR, New Delhi, India, are highly acknowledged. Awards of research fellowships to NB from the CSIR, New Delhi, and SK from UGC, New Delhi, are greatly acknowledged. Furthermore, the authors are very grateful for Dr. Shakir Ahamad’s assistance with Fig. 2.
Funding
The study was supported by a Start-Up Grant from University Grants Commission (UGC) Grant No. 30-564/2021(BSR) and the Indian Council of Medical Research (ICMR) Adhoc Grant (No.5/4-5/3/13/Neuro/2022-NCD-I) to SAB.
Author information
Authors and Affiliations
Contributions
Urmilla John: literature search, preparation of figures, and writing original draft, review and editing. Nargis Bano and Sameera Khan: literature search, preparation of revised Fig. 2, writing revised draft, revision and editing. Nawab John Dar and Shahnawaz Ali Bhat: conceptualization, literature search, preparation of figures, and writing original draft, review and editing and supervision. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Dar, N.J., John, U., Bano, N. et al. Oxytosis/Ferroptosis in Neurodegeneration: the Underlying Role of Master Regulator Glutathione Peroxidase 4 (GPX4). Mol Neurobiol 61, 1507–1526 (2024). https://doi.org/10.1007/s12035-023-03646-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03646-8