
Abstract
Oxidative stress and mitochondrial damage are implicated in the evolution of neurodegenerative diseases. Increased oxidative damage in specific brain regions during aging might render the brain susceptible to degeneration. Previously, we demonstrated increased oxidative damage and lowered antioxidant function in substantia nigra during aging making it vulnerable to degeneration associated with Parkinson’s disease. To understand whether aging contributes to the vulnerability of brain regions in Alzheimer’s disease, we assessed the oxidant and antioxidant markers, glutathione (GSH) metabolic enzymes, glial fibrillary acidic protein (GFAP) expression and mitochondrial complex I (CI) activity in hippocampus (HC) and frontal cortex (FC) compared with cerebellum (CB) in human brains with increasing age (0.01–80 years). We observed significant increase in protein oxidation (HC: p = 0.01; FC: p = 0.0002) and protein nitration (HC: p = 0.001; FC: p = 0.02) and increased GFAP expression (HC: p = 0.03; FC: p = 0.001) with a decreasing trend in CI activity in HC and FC compared to CB with increasing age. These changes were associated with a decrease in antioxidant enzyme activities, such as superoxide dismutase (HC: p = 0.005), catalase (HC: p = 0.02), thioredoxin reductase (FC: p = 0.04), GSH reductase (GR) (HC: p = 0.005), glutathione-s-transferase (HC: p = 0.0001; FC: p = 0.03) and GSH (HC: p = 0.01) with age. However, these parameters were relatively unaltered in CB. We suggest that the regions HC and FC are subjected to widespread oxidative stress, loss of antioxidant function and enhanced GFAP expression during aging which might make them more susceptible to deranged physiology and selective neuronal degeneration.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In neurodegenerative diseases, the brain regions that are selectively vulnerable to neurodegeneration also exhibit increased oxidative damage and lowered antioxidant mechanisms signifying a direct correlation between oxidative stress and neuronal death. In a recent study on human postmortem brains from Parkinson’s disease (PD) subjects, we demonstrated that substantia nigra (SN) showed lower glutathione (GSH), increased protein oxidation and nitration and lipid peroxidation, compared to caudate nucleus (CD) [1]. To understand whether accumulation of oxidative stress during physiological aging might contribute to neurodegeneration in PD, we tested oxidant and antioxidant markers in SN and CD of human brains with increasing age [2]. We observed that compared to CD, SN showed significantly higher protein oxidation and nitration, lower mitochondrial function, increased astrogliosis and lowered antioxidant function. These results indicate that brain region-specific differences in the equilibrium of oxidant and antioxidant markers during aging might determine the vulnerability to degeneration in PD.
Alzheimer’s disease (AD) is an age-associated neurodegenerative disease clinically manifesting with cognitive decline and dementia [3]. The chief pathological hallmark in AD is brain atrophy and gradual loss of neurons mainly in hippocampus (HC), frontal cortex (FC) and limbic areas. Research evidences have suggested that several degenerative biochemical pathways mainly including oxidative stress and mitochondrial dysfunction could contribute to neurodegeneration in AD. Accordingly, increased oxidative damage [4, 5] and depletion of antioxidants such as glutathione (GSH) [4, 6], have been detected in the AD brain. Further, oxidative damage to neuronal proteins have been documented in the diseased human brain tissue samples [7]. Apart from oxidative damage, AD pathology is also associated with neuroinflammation and astrogliosis with upregulation of the glial marker, glial fibrillary acidic protein (GFAP) [8, 9].
Whether age-related alterations in oxidant and antioxidant markers and accumulated biochemical differences in selected regions of the human brain contribute to neurodegeneration during AD is not clear. To address this issue, we carried out in this study, a comparative evaluation of the status of oxidative damage, antioxidant function, GSH metabolic enzymes, astrogliosis and mitochondrial function with increasing age in human HC and FC compared with cerebellum (CB).
Materials and Methods
All chemicals used were of analytical grade. Bulk chemicals were obtained from Merck (Whitehouse Station, NJ, USA) and Sisco Research Laboratories Pvt. Ltd. (Mumbai, Maharashtra, India). Nitrocellulose membrane from Millipore (Billerica, MA, USA), mouse monoclonal antibody to glial fibrillary acidic protein (GFAP) (clone GA-5) from Biogenex (San Ramon, CA, USA), anti-tubulin antibody from Abcam laboratories (Cambridge, UK), horseradish peroxidase conjugated secondary antibodies from Bangalore Genei (Bangalore, Karnataka, India), anti-dinitrophenyl (DNP) antibody and protease inhibitor cocktail from Sigma (Eugene, OR, USA) were procured.
Human Tissue Samples
Brain samples were sourced from the Human Brain Tissue Repository (HBTR), Department of Neuropathology, National Institute of Mental Health and Neurosciences (NIMHANS), Bangalore, India. Postmortem brain tissues were collected with informed consent of close relatives and the Institutional Ethics Committee has approved the study protocol. The brains were from subjects who succumbed to road traffic accidents (non-alcoholics, non-diabetics, not on any medication and with no known neurological or psychiatric disorders). Demographic and clinical details of all the subjects were noted from case records. Following the death of the patients, the postmortem interval (PMI) (the elapsed time between death and the freezing of the brain samples) was also recorded. Within 1 h of death, the body was transferred to a refrigerator maintained at 2–4 °C with a recorder and uninterrupted power supply. Following autopsy, the brains were collected, sliced coronally and kept flat on salt-ice mixture (−15 to −18 °C) during dissection and then transferred in plastic zip lock bags into a box and stored at −80 °C in the HBTR. The brain areas chosen for the study were anatomically farthest from the site of injury and without distinct edema or abnormal morphology. While the major portion of the tissue was frozen for biochemical studies, small bits from various anatomical areas were fixed in buffered formalin for histopathological assessment. The samples that maintained tissue integrity were utilized for the study (data not shown). The protocol of autopsy, tissue handling and other procedures were uniformly followed for all the samples in this study similar to the earlier investigations [1, 2, 10, 11].
In the current study, three anatomical areas, hippocampus (HC) (n = 25), frontal cortex (FC) (n = 31) and cerebellum (CB) (n = 27) were analyzed from postmortem brains (Table 1). The gender distribution, age and PMI were noted in all cases. Due to the limitation in the amount of tissue available from each region, all the samples have not been used for all the assays (sample number utilized for each assay is indicated in the respective figure legend).
SDS PAGE and Western Blot
The brain samples were thawed on ice and homogenized in 1× phosphate buffered saline (PBS) containing protease inhibitor cocktail, homogenized and sonicated on ice (5 s × 5). The extracts were immediately centrifuged at 15,000×g for 10 min to remove insoluble cellular debris and the total protein in the supernatant was estimated. Equal quantities of protein (50 μg) per sample were loaded on 10 % SDS PAGE followed by western blot with anti-GFAP and anti-tubulin antibodies [1].
Estimation of Protein Carbonyls (Oxyblot) and Protein Nitration
Oxyblots were carried out as described earlier [12, 13]. Briefly, brain protein extract (4 mg/ml) was derivatized by dinitrophenyl hydrazine (DNPH) in a 20 μl reaction mixture in the presence of 12 % SDS for 20 min at room temperature. The reaction was stopped by neutralization with 2 M Tris in 30 % glycerol and 5 μl of the sample was spotted in triplicate on nitrocellulose membrane and probed with anti-DNP antibody. Non-derivatized samples did not show anti-DNP immunoreactivity confirming the specificity of the antibody (data not shown).
To detect protein nitration, protein (10 μg/sample) from different samples were spotted in triplicate onto a nitrocellulose membrane and probed with polyclonal anti-3-nitrotyrosine (3-NT) antibody. Western signals were densitometrically quantified and normalized against the respective anti-tubulin or anti-beta actin signal [1].
Estimation of Lipid Peroxidation
Lipid peroxidation was measured by estimation of malondialdehyde (MDA) by thiobarbituric acid reaction method [14]. Tissues were sonicated and centrifuged at 16,000×g (10 min) at 4 °C. 100 μl of the supernatant was added to a mixture containing 0.75 ml of acetic acid (pH 3.5, 20 % v/v), 0.1 ml SDS (8 %, w/v) and 0.75 ml of thiobarbituric acid (0.8 %, w/v) and heated in a boiling water bath for 45 min. The adducts formed were extracted into 1.5 ml of 1-butanol and centrifuged at 2,500 rpm (10 min) and their absorbance was measured at 532 nm. The amount of MDA formed was calculated using the molar extinction coefficient (241 mol cm−1).
Preparation of Mitochondria and Complex I (CI) Assay
Mitochondria from the tissue samples were freshly prepared and their purity was established by electron microscopy as described earlier [15] (data not shown) and based on the rotenone sensitivity of the CI activity [16, 17]. The CI assay was initiated by addition of aliquots of brain mitochondria to 50 mM potassium phosphate/Tris–HCl, pH 7.4, 500 μM ethylenediamine tetra acetic acid (EDTA), 1 % bovine serum albumin, 200 μM NADH, and 200 μM decylubiquinone with and without 2 μM rotenone in the presence of potassium cyanide (KCN) with 0.002 % dichloroindophenol as a secondary electron acceptor. The decrease in the absorbance at 600 nm was recorded as a measure of enzyme reaction rate at 37 °C for 10 min, and specific activity was calculated. The results were plotted as relative rotenone sensitive specific activity. In all the mitochondrial preparations, rotenone exposure caused ≥95 % inhibition of NADH dehydrogenase activity (rotenone-sensitive activity) (data not shown) indicating highest purity of mitochondria with negligible contamination of cytosolic content.
Catalase Assay
Catalase activity was assayed by the method described previously [18]. The enzyme activity was expressed as μmol H2O2 consumed/min/mg protein (e = 43.6 mM−1 cm−1). Reaction mixture containing 15 μg protein (sample) was mixed with 900 μl phosphate buffer (0.1 M, pH 7.0) and 50 μl of H2O2 (8.8 mM) and the decrease in absorbance at 240 nm was followed for 5 min.
Superoxide Dismutase (SOD) Assay
SOD activity was assayed using its inhibitory action on quercetin oxidation based on the method described earlier with minor modifications [19]. The final reaction mixture contained 30 mM Tris–HCl (pH 9.1), 0.5 mM EDTA, 50 mM TEMED, 0.05 mM quercetin and 10 μl of brain extract supernatant containing 10 μg of protein. The reaction was monitored at 406 nm for 10 min. One unit of SOD activity was defined as the amount of enzyme (per mg protein) that inhibits quercetin oxidation reaction by 50 % of maximal value.
Glutathione-s-transferase (GST) Assay
GST was assayed by the 1-chloro 2-4-dinitro benzene (CDNB) method [20]. To 1 ml reaction mixture containing phosphate buffer (0.1 M, pH 6.5; 0.5 mM EDTA), CDNB (1.5 mM) and 50 μl GSH (1 mM), 30 μg protein (sample) was added and the increase in absorbance at 340 nm was monitored for 5 min. The enzyme activity was expressed as n moles of S-2,4, dinitrophenyl glutathione formed/min/mg protein (MEC = 9.6 mM−1 cm−1).
Glutathione Reductase (GR)
Solubilized brain protein extract (100 μg) was assayed at 25 °C in 0.1 M Tris–HCl (pH 8.1) and 0.2 mM NADPH and the reaction was initiated by the addition of 1 mM GSSG. The enzyme activity was measured by monitoring the oxidation of NADPH, spectrophotometrically at 340 nm, as described earlier [21].
Glutathione Peroxidase (GPx) Assay
GPx activity was determined by t-butyl hydroperoxide (tbHP) method [22]. The reaction mixture containing 150 μg protein (sample), 0.1 M phosphate buffer, 0.5 mM EDTA, 100 μl Glutathione reductase (0.24 U), 100 μl GSH (1 mM), 100 μl NADPH (0.15 mM) was incubated at 37 °C for 3 min and the reaction was initiated by the addition of 100 μl tbHP (0.12 mM). Change in absorbance at 340 nm was monitored for 5 min spectrophotometrically and the activity was expressed as n moles of NADPH oxidized/min/mg protein (MEC = 6.22 mM−1 cm−1).
Estimation of Total Glutathione (GSH + GSSG)
Total glutathione estimations were carried out by the 5, 5′ dithio-bis-2-nitro benzoic acid recycling method as described earlier [23]. All estimations were conducted in triplicate and total glutathione concentrations were normalized per mg protein.
Gamma-glutamyl Cysteine Ligase (γ-GCL) Assay
γ-GCL enzyme activity was measured by the method previously described [24]. Briefly, brain samples were homogenized and sonicated in 1× PBS and centrifuged at 9,500×g for 10 min at 4 °C. The supernatant (40 μg) was added to a reaction cocktail containing 100 mM Tris–HCl (pH 8.0), 150 mM KCl, 5 mM Na2ATP, 2 mM phosphoenol pyruvate, 10 mM l-glutamate, 20 mM MgCl2, 2 mM Na2EDTA and Pyruvate kinase/Lactate dehydrogenase mix (17 U each) and reaction kinetics was monitored at 340 nm for 10 min. Reaction was initiated by the addition of 10 mM l-alpha aminobutyrate. Assays run in the absence of l-alpha aminobutyrate served as the controls. Enzyme activity was normalized per mg protein.
Statistical Analysis
In case of the different anatomical areas of the human brains used in this study, for most ages, there was only one sample available. Hence, at least three different extracts/mitochondrial preparations from each sample were independently assayed such that n ≥ 3 corresponds to at least three independent experiments. Quantitative data were expressed as mean ± SD and plotted in a scattered plot and “Pearson’s correlation of linear regression” was applied to determine the “r” value. The significance of the regression was indicated by the “p” value, based on the number of samples in each group. A trend-line was plotted to visually indicate the correlation of the assay parameter with increasing age. In all the experiments, quantitative data with p < 0.05 were considered to be statistically significant.
Results
Biochemical studies in AD have demonstrated significant oxidative stress, nitrosative stress and mitochondrial dysfunction [25–28]. To investigate whether enhanced oxidative damage during physiological aging makes anatomical brain regions HC and FC selectively vulnerable to AD, we carried out the analyses of different oxidant and antioxidant markers in aging human brains. First, we quantitated the level of oxidative damage of proteins in HC (n = 17) and FC (n = 24) compared to CB (n = 20). We noted increased protein oxidation (protein carbonylation in tissue extracts as determined by oxyblot) in HC and FC with increasing age while it was relatively unaltered in CB (HC: r = 0.58, p = 0.01; FC: r = 0.68, p = 0.0002; CB: r = 0.26; p = 0.15) (Fig. 1). Similarly, protein nitration (determined by total protein 3-NT modification) showed an increase with increasing age both in HC and FC but was unaltered in CB (HC: r = 0.72, p = 0.001; FC: r = 0.43, p = 0.02; CB: r = 0.25, p = 0.15) (Fig. 2).

Analysis of protein oxidation in postmortem human brains with increasing age (HC, FC, CB). Total protein extract after DNP-derivatization (~10 μg) in HC (n = 17), FC (n = 24) and CB (n = 20) were spotted on nitrocellulose membrane in triplicate followed by anti-DNP western blot (Oxyblot). Following densitometric analysis, the average for each sample was plotted as mean ± SD followed by regression analysis. The r and p values for each region are shown. Representative anti-DNP blot (all the samples were spotted in a single blot but for continuity during visual comparison, the images were cut in between two sets of triplicates and arranged horizontally next to each other) and tubulin western blot of HC tissue extracts with increasing age (a) and the quantitative plots of anti-DNP signal (normalized with tubulin signal) in HC, FC and CB (b) are shown

Analysis of protein nitration (3-NT) in postmortem human brains with increasing age (HC, FC, CB). Total protein extracts (~100 μg) from HC (n = 17), FC (n = 24) and CB (n = 20) were spotted on nitrocellulose membrane in triplicate followed by anti-3NT western blot. Following densitometric analysis, the average for each sample was plotted as mean ± SD followed by regression analysis. The r and p values for each region are shown. Representative anti-3-NT blot (all the samples were spotted in a single blot but for continuity during visual comparison, the images were cut in between two sets of triplicates and arranged horizontally next to each other), β-actin western and slot blot of HC extracts; 3-NT and tubulin blot of CB tissue extracts with increasing age (a); the quantitative plots of anti-3-NT signal (normalized with β-actin or tubulin signal) in HC, FC and CB (b) are shown
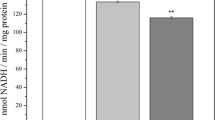
Since oxidative damage is directly related to mitochondrial dysfunction, we evaluated the mitochondrial complex I (CI) activity in HC, FC and CB during normal physiological aging. CI is one of the most severely affected mitochondrial enzymes during aging [29] and inhibition of its activity significantly influences the mitochondrial health [30]. The CI activity was relatively decreased with increasing age in HC compared to FC and CB (HC: r = −0.53, p = 0.07; FC: r = −0.25, p = 0.14; CB: r = 0.17, p = 0.24) (Fig. 3a) but was not statistically significant. Similar to mitochondrial damage, neuroinflammation with microglial activation and reactive gliosis are evident during AD with enhanced expression of the astroglial marker GFAP [9, 10, 31]. We observed age-dependent increase in GFAP expression in HC and FC but not in CB (HC: r = 0.49, p = 0.03; FC: r = 0.76, p = 0.001; CB: r = −0.15, p = 0.27) (Fig. 3b, c). These data indicate that distinct biochemical and physiological changes occur in HC and FC compared to CB which reflect region-specific vulnerability with aging and neurodegeneration. Next, we tested whether these regional differences could be due to altered antioxidant function in these regions.

Quantitative analysis of mitochondrial complex I (CI) activity and GFAP in postmortem human HC, FC and CB with increasing age. a Mitochondria were prepared from different samples [HC (n = 17), FC (n = 24) and CB (n = 20)] followed by CI assay. The average rotenone-sensitive CI activity for each sample was plotted as mean ± SD followed by regression analysis. The r and p values for each region are shown in. b A representative anti-GFAP western blot of three FC samples. c, d Total extracts (50 μg) from different samples were spotted on nitrocellulose membrane with increasing age in triplicate followed by anti-GFAP western blot (all the samples were spotted in a single blot but for continuity during visual comparison, the images were cut in between two sets of triplicates and arranged horizontally next to each other). Following densitometric analysis, the average for each sample was plotted as mean ± SD followed by regression analysis. The r and p values for each region are shown. Age-dependent alterations in GFAP signal (normalized with the respective tubulin signal) in HC, FC and CB are also shown
In the HC, there was a significant decrease in SOD activity with increasing age while in FC and CB, the activity was relatively unaltered (HC: r = −0.76, p = 0.005; FC: r = −0.04, p = 0.43; CB: r = 0.29; p = 0.12) (Fig. 4a). Catalase activity showed ≥2 fold increase in CB samples compared to FC and HC. However, catalase activity was decreased in HC with increasing age while it was unaltered in CB (HC: r = −0.51, p = 0.02; CB: r = −0.31; p = 0.10) (Fig. 4b). On the other hand, the catalase activity was increased in an age-dependent manner in FC (FC: r = 0.46, p = 0.03). GSH depletion has been implicated in AD [5–7, 32]. The total GSH content was significantly higher in FC compared to HC and CB. While the GSH levels were unaltered in FC and CB with increasing age (FC: r = −0.30, p = 0.09; CB: r = −0.02; p = 0.50), it was significantly lowered in HC (r = −0.58; p = 0.01) (Fig. 4c). The decreased GSH content in HC could be due to lowered synthesis. To investigate this, we assayed the activity of GCL (the rate-limiting enzyme in GSH synthesis) which was relatively lower in HC compared to FC and CB but was not significantly lowered with age (HC: r = −0.54, p = 0.08; FC: r = −0.02, p = 0.45; CB: r = 0.09; p = 0.30) (Fig. 4d). On the other hand, the activity of the antioxidant enzyme TrX was unaltered with increasing age in HC and CB (HC: r = −0.11, p = 0.33; CB: r = −0.33, p = 0.09) while it was decreased in FC (r = −0.42, p = 0.04) (Fig. 4e).

Age-associated changes in the antioxidant activities in human postmortem brains (HC, FC, CB). Total extracts from HC, FC and CB samples were utilized for different antioxidant assays and the average for each sample was plotted as mean ± SD followed by regression analysis. The r and p values for each region are shown. a The scatter plot of SOD activity with increasing age in HC (n = 18), FC (n = 22) and CB (n = 20) is shown. b The comparison of age-dependent catalase activity in HC (n = 18), FC (n = 22) and CB (n = 20) is shown. c and d The comparison of age-dependent total GSH content and GCL activity in HC (n = 18), FC (n = 22) and CB (n = 20), respectively are shown. e The scatter plot of TrxR activity with increasing age in HC (n = 18), FC (n = 22) and CB (n = 20) is shown
We observed that the activity of GPx was unaltered with increasing age in HC and FC while it was decreased in CB (HC: r = 0.08, p = 0.37; FC: r = −0.08, p = 0.39; CB: r = −0.49, p = 0.04) (Fig. 5a). On the other hand, GR activity was significantly decreased with increasing age in HC and CB while it was relatively stable in FC (HC: r = −0.61, p = 0.005; FC: r = −0.21, p = 0.18; CB: r = −0.43; p = 0.04) (Fig. 5b). Interestingly, the most stable GSH metabolic enzyme GST was significantly lowered with increasing age in HC while it was unchanged in CB and increased in FC (HC: r = −0.78, p = 0.0001; FC: r = 0.43, p = 0.03; CB: r = 0.16; p = 0.30) (Fig. 5c, d). A summary of these finding is given in Table 2.

Analysis of GSH-associated enzymes in postmortem human brains with increasing age (HC, FC, CB). Total extracts from HC, FC and CB samples were utilized for different assays and the average for each sample was plotted as mean ± SD followed by regression analysis. The r and p values for each region are shown. a shows the GPx activity in HC (n = 18), FC (n = 20) and CB (n = 19) with increasing age. b shows the GR activity in the HC (n = 18), FC (n = 21) and CB (n = 19) while c and d correspond to the GST activity in HC (n = 18) and FC (n = 20) and CB (n = 16) respectively
Discussion
Oxidative damage during AD has been linked with neurodegeneration [33]. Butterfield et al. [34] reported elevated protein oxidation in HC from brains of subjects with mild cognitive impairment (MCI) compared to age-matched controls. MCI is a transitional state between normal cognitive status during aging and early dementia or AD and most MCI subjects eventually develop AD. Using redox proteomics, oxidatively modified proteins such as alpha-enolase, glutamine synthetase, pyruvate kinase M2 and peptidyl-prolylcis/trans isomerase 1 were identified in MCI brains which indicated the functional involvement of energy metabolism, synaptic plasticity and mitogenesis in MCI [34]. The current study has provided a mechanistic explanation for selective vulnerability of HC and FC in MCI and AD by demonstrating age-dependent increase in protein carbonyls (Fig. 1). Protein nitration is an important post-translational modification associated with aging and AD [35]. In another study, Butterfield et al. [36] reported elevated protein nitration based on slot blot and immunohistochemical analyses in the inferior parietal lobe and HC of post-mortem brains of MCI subjects. Nitration of total proteins in HC and FC also displayed similar trend with age-dependent increase compared to CB (Fig. 2).
Lipid peroxidation was elevated in HC and pyriform cortex of AD samples compared to controls [37]. The oxidative changes were significantly increased in the medial temporal lobe, where histopathologic alterations are most severe. Padurariu et al. [38] reported increased lipid peroxidation in the serum of the MCI and AD patients, compared to age-matched control group. In a recent proteomics study, Reed et al. [39] identified protein-bound hydroxynonenal (HNE) in early AD (EAD), which is a transitional stage between MCI and late-stage AD. Among these, six HNE-bound proteins that play important cellular roles in antioxidant defense (Mn-SOD), neuronal communication, neurite outgrowth (dihydropyriminidase-related protein 2) and energy metabolism were identified to be comparable among MCI, EAD and late-stage AD.
Elevated oxidative damage in AD and MCI could be, to some extent due to altered protein levels and activities of antioxidant enzymes in HC and other regions [40]. Lovell et al. [40] demonstrated elevated SOD, catalase, GPx and GR activities in HC in cases of AD compared to controls. The antioxidant activities were significantly higher in those regions which showed higher lipid peroxidation indicating compensatory increase in antioxidant activity in response to increased oxidative stress. On the contrary, Padurariu et al. [38] reported significant decrease in the activities of SOD and GPx in the serum of MCI and AD patients. The current study clearly indicates age-dependent decline in the activities of SOD and catalase in HC which could contribute to the lowered antioxidant load in neurodegeneration (Fig. 4). Bermejo et al. [41] demonstrated decreased GSH levels and GSH/GSSG ratio in AD and MCI patients compared to controls. This could be contributed by lower GSH in HC during physiological aging as observed in the current study (Fig. 4). The total GSH content by itself might provide significant information on the oxidative status in the brain. The distribution of brain GSH in different neuroanatomical areas is different and this is evident in normal brains (as shown in Fig. 4c). In a related study, Mandal et al. [4] reported in vivo detection of GSH by non-invasive imaging in different neuroanatomical brain regions of healthy subjects and patients with MCI and AD. Accordingly, healthy young brains displayed higher GSH content and a specific GSH distribution pattern. The mean GSH content and distribution varied between male and female healthy subjects when compared between left frontal cortex (LFC) and right frontal cortex (RFC). In case of a high GSH content in left parietal cortex of a young male, the LFC region also displayed high GSH and vice versa. The difference in total GSH content between healthy young female control and female AD patients in RFC and difference in GSH content between healthy young male control and male AD patients in LFC region was statistically significant. These data indicate that the distribution of antioxidants in the human brain is associated with gender, age, disease condition and anatomical area.
Apart from GSH, HC also displayed decreased activities of GSH-related enzymes GST and GR indicating a compromise of GSH metabolism with implications for oxidative damage (Figs. 4, 5). On the other hand, FC also showed lower activities of GST and TrxR which could contribute to oxidative stress (Figs. 4, 5).
CI activity showed a decreasing trend in HC with increasing age (Fig. 3a), indicating mitochondrial dysfunction due to the cumulative effect of lowered antioxidant and increased oxidative damage. CI inhibition is closely associated with oxidative damage, aging and neurodegeneration. Previous studies in isolated mitochondria have demonstrated that CI activity was specifically susceptible to oxidative damage [42, 43]. Depletion of the antioxidant GSH leading to oxidative stress, selectively inhibited CI activity in dopaminergic neurons [13, 44, 45]. Decreased CI activity in the SN of PD brains compared to controls, the hallmark of PD pathology could be induced by oxidative damage [46]. Inactivation of CI activity by different toxins and inhibitors in the brain is associated with neurodegeneration indicating the susceptibility of the complex [47]. CI activity is the most severely affected mitochondrial enzyme during age-associated oxidative stress [29]. In synaptic mitochondria, CI exerts a major control over mitochondrial metabolism such that a decrease in its activity by 25 % significantly affected ATP synthesis and mitochondrial bioenergetics. On the other hand, inhibition of CIII and CIV up to ~80 % was required to show similar effect [30]. The inhibition of CI shows selectivity in different anatomical areas. In a recent study [1], we demonstrated that CI activity was inhibited in the SN of human postmortem brains but not in other regions such as striatum and FC and this was directly associated with oxidative damage.
We observed that most of the activities investigated in the current study were unchanged with increasing age in CB indicating that these effects were neuro-anatomical site-specific to HC and FC. The resistance of cerebellar neurons to oxidative damage in the human brain could be due to the elevated antioxidant mechanisms. In an expression study [48], compared to HC, CB showed significant up-regulation of two inducible proteins heme oxygenase-1 and manganese SOD-2 which are critically involved in the cellular defense against endogenous or exogenous oxidative damage. Recently, Ciavardelli et al. [49] studied the temporal and region specific proteomic alterations in a triple transgenic AD mouse model. As an internal control, CB, a neuroanatomical structure not affected by AD, displayed upregulation of proteins involved in carbohydrate metabolism and protein catabolism. One such protein Glyoxalase 1 (GLO1, a component of the glyoxalase system that detoxifies α-ketoaldehydes) which prevents the formation of advanced glycation end products that induce oxidative stress and reduce the proteolysis of carbonylated MAP-tau, was found to be specifically upregulated in CB. The study also showed that proteins with chaperone function showed elevated expression, which could contribute to the resistance of cerebellar neurons against neurodegeneration. In the current study, we observed that CB revealed least oxidative damage (Figs. 1, 2) and minimal variation in the antioxidant activities (Fig. 4). CB also appears to have other protective mechanisms which make it the least affected region in diseases like stroke and AD. Wu et al. [50] demonstrated that an intrinsic survival pathway that is regulated by low level stimulation of N-methyl-d-aspartate (NMDA) could protect against death of cerebellar neurons. A study on the postmortem human brain samples from AD patients showed that frontal and temporal cortices showed 6–7 folds higher levels of total tau protein than the cerebellar cortex [51]. Causevic et al. [52] established that, the β-amyloid precursor protein (APP) and tau proteins involved in AD pathology were highly reduced in CB compared to entorhinal cortex and HC. All these observations indicate that, among all other regions of the human brain, CB is the most resistant to oxidative damage, neurodegerenation and AD pathology.
Increased GFAP expression is observed following stress, infection and aging as a compensatory response with reactive astrocytosis and gliosis [53, 54]. Cell culture studies have shown that the age of the astrocyte and the expression levels of GFAP has a significantly negative impact on the viability and neurite outgrowth of the neurons in the vicinity [55]. Other studies [56–59] have also revealed increased GFAP expression in HC with physiological implications for cognitive and memory functions and neurodegeneration during aging. It is therefore possible that increased GFAP in HC and FC as observed in the present study (Fig. 3b, c) might influence the neuronal activity making it more susceptible to age-associated neurodegeneration and cognitive impairment.
Most of the studies related to the oxidant and antioxidant markers in human brains have been carried out in samples of AD and other conditions [5]. But, there are very few studies carried out in non-diseased brains in anatomical areas relevant to AD with increasing age. In that respect, the present study is different and has clearly indicated that age-dependent increase in oxidative markers and lowered antioxidant function are crucial in predisposing regions such as FC and HC to degenerative changes. Further, it also indicates that the inter-regional variations among different anatomical areas in the human brain add to the complexity of brain function during normal physiology, aging and pathology.
In conclusion, our study showed a distinct increase in oxidative damage and GFAP expression, decreased antioxidant response and a non-statistically significant trend towards decreasing activity of CI in HC region of human brain with increasing age. However, the associated biochemical events following GFAP expression need further evaluation to elucidate its physiological significance. Consistent with our previous study [2] the current study also showed that increased protein oxidation and nitration are linked with aging. But, whether all proteins are oxidized/nitrated to the same extent and by the same source and whether these are random or organized as hierarchical events require further investigation. The identification of specific oxidation/nitration events that affect protein function in aging and pathologies could be potential biomarkers in diagnostics and prognostics.
Abbreviations
- HC:
-
Hippocampus
- FC:
-
Frontal cortex
- CB:
-
Cerebellum
- AD:
-
Alzheimer’s disease
- PMI:
-
Postmortem interval
- MCI:
-
Mild cognitive impairment
- SOD:
-
Superoxide dismutase
- GSH:
-
Glutathione
- GR:
-
Glutathione reductase
- GPx:
-
Glutathione peroxidase
- GST:
-
Glutathione-s-transferase
- GFAP:
-
Glial fibrillay acidic protein
- GCL:
-
Glutamylcysteine ligase
- ThrR:
-
Thioredoxin reductase
References
Mythri RB, Venkateshappa C, Harish G, Mahadevan A, Muthane UB, Yasha TC, Srinivas Bharath MM, Shankar SK (2011) Evaluation of markers of oxidative stress, antioxidant function and astrocytic proliferation in the striatum and frontal cortex of Parkinson’s disease brains. Neurochem Res 36(8):1452–1463. doi:10.1007/s11064-011-0471-9
Venkateshappa C, Harish G, Mythri RB, Mahadevan A, Srinivas Bharath MM, Shankar SK (2011) Increased oxidative damage and decreased antioxidant function in aging human substantia nigra compared to striatum: implications for Parkinson’s disease. Neurochem Res. doi:10.1007/s11064-011-0619-7
Harman D (2006) Alzheimer’s disease pathogenesis: role of aging. Ann N Y Acad Sci 1067:454–460. doi:10.1196/annals.1354.065
Mandal PK, Tripathi M, Sugunan S (2012) Brain oxidative stress: detection and mapping of anti-oxidant marker ‘Glutathione’ in different brain regions of healthy male/female, MCI and Alzheimer patients using non-invasive magnetic resonance spectroscopy. Biochem Biophys Res Commun 417(1):43–48. doi:10.1016/j.bbrc.2011.11.047
Ansari MA, Scheff SW (2010) Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol 69(2):155–167. doi:10.1097/NEN.0b013e3181cb5af4
Pocernich CB, Butterfield DA (2011) Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochim Biophys Acta. doi:10.1016/j.bbadis.2011.10.003
Pamplona R, Dalfo E, Ayala V, Bellmunt MJ, Prat J, Ferrer I, Portero-Otin M (2005) Proteins in human brain cortex are modified by oxidation, glycoxidation, and lipoxidation. Effects of Alzheimer disease and identification of lipoxidation targets. J Biol Chem 280(22):21522–21530. doi:10.1074/jbc.M502255200
Korolainen MA, Auriola S, Nyman TA, Alafuzoff I, Pirttila T (2005) Proteomic analysis of glial fibrillary acidic protein in Alzheimer’s disease and aging brain. Neurobiol Dis 20(3):858–870. doi:10.1016/j.nbd.2005.05.021
Eikelenboom P, Rozemuller AJ, Hoozemans JJ, Veerhuis R, van Gool WA (2000) Neuroinflammation and Alzheimer disease: clinical and therapeutic implications. Alzheimer Dis Assoc Disord 14(Suppl 1):S54–S61
Harish G, Venkateshappa C, Mahadevan A, Pruthi N, Bharath MMS, Shankar SK (2011) Effect of storage time, postmortem interval, agonal state, and gender on the postmortem preservation of glial fibrillary acidic protein and oxidatively damaged proteins in human brains. Biopreserv Biobank. doi:10.1089/bio.2011.0033
Harish G, Venkateshappa C, Mahadevan A, Pruthi N, Srinivas Bharath MM, Shankar SK (2011) Glutathione metabolism is modulated by postmortem interval, gender difference and agonal state in postmortem human brains. Neurochem Int. doi:10.1016/j.neuint.2011.08.024
Chandana R, Mythri RB, Mahadevan A, Shankar SK, Srinivas Bharath MM (2009) Biochemical analysis of protein stability in human brain collected at different post-mortem intervals. Indian J Med Res 129(2):189–199
Jagatha B, Mythri RB, Vali S, Bharath MM (2008) Curcumin treatment alleviates the effects of glutathione depletion in vitro and in vivo: therapeutic implications for Parkinson’s disease explained via in silico studies. Free Radic Biol Med 44(5):907–917. doi:10.1016/j.freeradbiomed.2007.11.011
Ohkawa H, Ohishi N, Yagi K (1979) Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 95(2):351–358
Frasca JM, Parks VR (1965) A routine techbique for double staining ultrathin sections using uranyl and lead salts. J Cell Biol 25:157–161
Mythri RB, Jagatha B, Pradhan N, Andersen J, Bharath MM (2007) Mitochondrial complex I inhibition in Parkinson’s disease: how can curcumin protect mitochondria? Antioxid Redox Signal 9(3):399–408. doi:10.1089/ars.2007.9.ft-25
Trounce IA, Kim YL, Jun AS, Wallace DC (1996) Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol 264:484–509
Aebi H (1984) Catalase in vitro. Methods Enzymol 105:121–126
Bagnyukova TV, Storey KB, Lushchak VI (2003) Induction of oxidative stress in Rana ridibunda during recovery from winter hibernation. J Therm Biol 28:21–28
Guthenberg C, Alin P, Mannervik B (1985) Glutathione transferase from rat testis. Methods Enzymol 113:507–510
Carlberg I, Mannervik B (1985) Glutathione reductase. Methods Enzymol 113:484–490
Flohe L, Gunzler WA (1984) Assays of glutathione peroxidase. Methods Enzymol 105:114–121
Tietze F (1969) Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem 27(3):502–522
Seelig GF, Meister A (1985) Glutathione biosynthesis; gamma-glutamylcysteine synthetase from rat kidney. Methods Enzymol 113:379–390
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443(7113):787–795. doi:10.1038/nature05292
Mao P (1812) Reddy PH (2011) Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: implications for early intervention and therapeutics. Biochim Biophys Acta 11:1359–1370. doi:10.1016/j.bbadis.2011.08.005
Muller WE, Eckert A, Kurz C, Eckert GP, Leuner K (2010) Mitochondrial dysfunction: common final pathway in brain aging and Alzheimer’s disease–therapeutic aspects. Mol Neurobiol 41(2–3):159–171. doi:10.1007/s12035-010-8141-5
Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G (1997) Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J Neurosci 17(8):2653–2657
Lenaz G, Bovina C, Castelluccio C, Fato R, Formiggini G, Genova ML, Marchetti M, Pich MM, Pallotti F, Parenti Castelli G, Biagini G (1997) Mitochondrial complex I defects in aging. Mol Cell Biochem 174(1–2):329–333
Davey GP, Peuchen S, Clark JB (1998) Energy thresholds in brain mitochondria. Potential involvement in neurodegeneration. J Biol Chem 273(21):12753–12757
Lee YJ, Han SB, Nam SY, Oh KW, Hong JT (2010) Inflammation and Alzheimer’s disease. Arch Pharm Res 33(10):1539–1556. doi:10.1007/s12272-010-1006-7
Liu H, Wang H, Shenvi S, Hagen TM, Liu RM (2004) Glutathione metabolism during aging and in Alzheimer disease. Ann N Y Acad Sci 1019:346–349. doi:10.1196/annals.1297.059
Bonda DJ, Wang X, Perry G, Nunomura A, Tabaton M, Zhu X, Smith MA (2010) Oxidative stress in Alzheimer disease: a possibility for prevention. Neuropharmacology 59(4–5):290–294. doi:10.1016/j.neuropharm.2010.04.005
Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB, Markesbery WR (2006) Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer’s disease. Neurobiol Dis 22(2):223–232. doi:10.1016/j.nbd.2005.11.002
Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA (2006) Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol Dis 22(1):76–87. doi:10.1016/j.nbd.2005.10.004
Butterfield DA, Reed TT, Perluigi M, De Marco C, Coccia R, Keller JN, Markesbery WR, Sultana R (2007) Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res 1148:243–248. doi:10.1016/j.brainres.2007.02.084
Lovell MA, Ehmann WD, Butler SM, Markesbery WR (1995) Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer’s disease. Neurology 45(8):1594–1601
Padurariu M, Ciobica A, Hritcu L, Stoica B, Bild W, Stefanescu C (2010) Changes of some oxidative stress markers in the serum of patients with mild cognitive impairment and Alzheimer’s disease. Neurosci Lett 469(1):6–10. doi:10.1016/j.neulet.2009.11.033
Reed TT, Pierce WM, Markesbery WR, Butterfield DA (2009) Proteomic identification of HNE-bound proteins in early Alzheimer disease: Insights into the role of lipid peroxidation in the progression of AD. Brain Res 1274:66–76. doi:10.1016/j.brainres.2009.04.009
Sultana R, Piroddi M, Galli F, Butterfield DA (2008) Protein levels and activity of some antioxidant enzymes in hippocampus of subjects with amnestic mild cognitive impairment. Neurochem Res 33(12):2540–2546. doi:10.1007/s11064-008-9593-0
Bermejo P, Martin-Aragon S, Benedi J, Susin C, Felici E, Gil P, Ribera JM, Villar AM (2008) Peripheral levels of glutathione and protein oxidation as markers in the development of Alzheimer’s disease from Mild Cognitive Impairment. Free Radic Res 42(2):162–170. doi:10.1080/10715760701861373
Hillered L, Chan PH (1988) Effects of arachidonic acid on respiratory activities in isolated brain mitochondria. J Neurosci Res 19(1):94–100. doi:10.1002/jnr.490190113
Zhang Y, Marcillat O, Giulivi C, Ernster L, Davies KJ (1990) The oxidative inactivation of mitochondrial electron transport chain components and ATPase. J Biol Chem 265(27):16330–16336
Chinta SJ, Andersen JK (2006) Reversible inhibition of mitochondrial complex I activity following chronic dopaminergic glutathione depletion in vitro: implications for Parkinson’s disease. Free Radic Biol Med 41(9):1442–1448. doi:10.1016/j.freeradbiomed.2006.08.002
Murchison D, Griffith WH (2007) Calcium buffering systems and calcium signaling in aged rat basal forebrain neurons. Aging Cell 6(3):297–305. doi:10.1111/j.1474-9726.2007.00293.x
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem 54(3):823–827
Schapira AH (2010) Complex I: inhibitors, inhibition and neurodegeneration. Exp Neurol 224(2):331–335. doi:10.1016/j.expneurol.2010.03.028
Colombrita C, Calabrese V, Stella AM, Mattei F, Alkon DL, Scapagnini G (2003) Regional rat brain distribution of heme oxygenase-1 and manganese superoxide dismutase mRNA: relevance of redox homeostasis in the aging processes. Exp Biol Med (Maywood) 228(5):517–524
Ciavardelli D, Silvestri E, Del Viscovo A, Bomba M, De Gregorio D, Moreno M, Di Ilio C, Goglia F, Canzoniero LM, Sensi SL (2010) Alterations of brain and cerebellar proteomes linked to Abeta and tau pathology in a female triple-transgenic murine model of Alzheimer’s disease. Cell Death Dis 1:e90. doi:10.1038/cddis.2010.68
Wu X, Jiang X, Marini AM, Lipsky RH (2005) Delineating and understanding cerebellar neuroprotective pathways: potential implication for protecting the cortex. Ann N Y Acad Sci 1053:39–47. doi:10.1196/annals.1344.004
Khatoon S, Grundke-Iqbal I, Iqbal K (1994) Levels of normal and abnormally phosphorylated tau in different cellular and regional compartments of Alzheimer disease and control brains. FEBS Lett 351(1):80–84
Causevic M, Farooq U, Lovestone S, Killick R (2010) Beta-Amyloid precursor protein and tau protein levels are differently regulated in human cerebellum compared to brain regions vulnerable to Alzheimer’s type neurodegeneration. Neurosci Lett 485(3):162–166. doi:10.1016/j.neulet.2010.08.088
Williams AJ, Wei HH, Dave JR, Tortella FC (2007) Acute and delayed neuroinflammatory response following experimental penetrating ballistic brain injury in the rat. J Neuroinflammation 4:17. doi:10.1186/1742-2094-4-17
Zlotnik I (1968) The reaction of astrocytes to acute virus infections of the central nervous system. Br J Exp Pathol 49(6):555–564
Rozovsky I, Wei M, Morgan TE, Finch CE (2005) Reversible age impairments in neurite outgrowth by manipulations of astrocytic GFAP. Neurobiol Aging 26(5):705–715. doi:10.1016/j.neurobiolaging.2004.06.009
Hayakawa N, Kato H, Araki T (2007) Age-related changes of astorocytes, oligodendrocytes and microglia in the mouse hippocampal CA1 sector. Mech Ageing Dev 128(4):311–316. doi:10.1016/j.mad.2007.01.005
Porchet R, Probst A, Bouras C, Draberova E, Draber P, Riederer BM (2003) Analysis of glial acidic fibrillary protein in the human entorhinal cortex during aging and in Alzheimer’s disease. Proteomics 3(8):1476–1485. doi:10.1002/pmic.200300456
Sandhir R, Onyszchuk G, Berman NE (2008) Exacerbated glial response in the aged mouse hippocampus following controlled cortical impact injury. Exp Neurol 213(2):372–380. doi:10.1016/j.expneurol.2008.06.013
Ross GW, O’Callaghan JP, Sharp DS, Petrovitch H, Miller DB, Abbott RD, Nelson J, Launer LJ, Foley DJ, Burchfiel CM, Hardman J, White LR (2003) Quantification of regional glial fibrillary acidic protein levels in Alzheimer’s disease. Acta Neurol Scand 107(5):318–323
Acknowledgments
The authors thank the Human Brain Tissue Repository (HBTR), NIMHANS, Bangalore, India, for providing the human brain tissue samples required for the study. This study was financially supported by a grant from the Indian Council of Medical Research (ICMR IRIS ID No. 2009-07710) (to MMSB). VC gratefully acknowledges the financial support from Sri Siddhartha Medical College, Tumkur, India. GH is supported by a senior research fellowship from ICMR, India. The authors gratefully acknowledge the donors and their relatives for the kind gift of human brains for neurobiological studies.
Conflict of interest
The authors declare that there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Venkateshappa, C., Harish, G., Mahadevan, A. et al. Elevated Oxidative Stress and Decreased Antioxidant Function in the Human Hippocampus and Frontal Cortex with Increasing Age: Implications for Neurodegeneration in Alzheimer’s Disease. Neurochem Res 37, 1601–1614 (2012). https://doi.org/10.1007/s11064-012-0755-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-012-0755-8